
Abstract
Doxycycline (DOX), an antibacterial drug, has been widely used in the inducible gene expression system. However, its effect was largely ignored when studying functions of the inducible transgene. By using a DOX-inducible Tet-ON system, we identified that DOX alone dramatically promoted dopaminergic (DA) neuron differentiation from human pluripotent stem cells (hPSCs), whereas the studied gene had no significant effects after considering the confounding factor DOX. These findings suggest that the effect of DOX should be taken into consideration when it is used in the inducible system especially during DA neuron differentiation from hPSCs. Meanwhile, it also suggests that DOX can be used as an efficient and inexpensive molecule to increase DA neuron differentiation efficacy from hPSCs for cell therapy.
Background
Doxycycline (DOX), a synthetic antibiotic of tetracyclines, has been widely used as antibacterial drug for the treatment of many infectious diseases [1]. Beyond its antimicrobial activity, DOX is also widely used as a controllable and noninvasive mediator of inducible gene expression system in biomedical research with little consideration on its confounding effects on mammalian cells [2 –8].
DOX has been extensively reported to exert a variety of actions on mammalian cells. DOX achieves its antibiotic effect through binding to the 30S subunit of bacterial ribosomes and inhibiting protein synthesis [1]. This inhibitory effect has also been observed in the impairment of mitochondrial function in eukaryotic cells [9]. Specifically, DOX was found to inhibit matrix metalloproteinase (MMP) and enhance chondrogenesis in vitro and cartilage repair in vivo [10 –12]. DOX was also shown to induce apoptosis, inhibit proliferation, or change the metabolic phenotype of different types of cancer cells and normal cells [13 –16].
Unlike somatic and cancer cells, human pluripotent stem cells (hPSCs) including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) are capable of self-renewing indefinitely and differentiating into any cell type originated from the three germ layers [17]. Human ESC (hESC)/iPSC-based cell models have been extensively used for studying disease pathogenesis, cell replacement therapy, and developmental biology [17,18]. Recently, it was shown that DOX could dramatically improve the survival and self-renewal ability of hPSCs [19,20], an effect distinct from that of somatic cells. In other findings, DOX has been demonstrated to protect neural stem cells and neurons from damage by anti-inflammatory effect or inhibition of MMPs [21 –25]. Moreover, DOX could also increase adult neurogenesis in mouse brain [26]. Based on the aforementioned findings, we were interested to know whether DOX affects neuron differentiation from hPSCs and determine whether the DOX-inducible system has any nonspecific effect.
In this study, we examined the effect of DOX (0.1 and 1 μg/mL) during dopaminergic (DA) neuron differentiation from hPSCs. In addition, the effect of DOX (1 μg/mL) in a lentivirus-based Tet-On system was determined. The Tet-On system is used to induce the expression of a male-specific gene, SRY, during early DA neuron differentiation. We found that DOX at a commonly used working concentration (1 μg/mL) could significantly increase the DA neuron differentiation from both hESCs and iPSCs by enhancing pathway activity including WNT signaling and Hedgehog signaling. We also showed that this enhancement effect on DA neuron differentiation was dose dependent, and displayed clonal variation. Based on these findings, we further demonstrated that DOX could indeed confound the experimental results and may lead to false conclusion. However, this confounding effect of DOX could be eliminated by including additional controls.
Materials and Methods
Cell and cell culture
hES cell line H1 (WA01) and human iPS cell line iBC1.2 (generated in this laboratory and reported previously [27]) were maintained on Matrigel-coated plates/dishes (Corning) in mTeSR medium (STEMCELL) before differentiation. Upon 80% confluency, cells were passaged at ratio 1:4 using Dispase (STEMCELL) enzyme.
HE-4, HE-5, HR-1, IE-2, and IE-4 cell lines were generated by single cell cloning method from parental human iPSCs (hiPSCs) or hESCs transduced with enhanced green fluorescent protein (EGFP)/sex-determining region of Y-chromosome (SRY)-EGFP lentivirus. HE was EGFP-only-positive hESCs. HR was SRY-positive hESCs. IE was EGFP-only-positive hiPSCs. In brief, Accutase (BD Corning) was used to dissociate the sorted EGFP-positive cells. Then 100–300 cells per 6 well were seeded with CloneR (STEMCELL). After cell colony emerged, random manual pick-up was carried out. Individual colonies were maintained using mTeSR medium. Only colonies with EGFP signals were used in this study.
In this study, we denoted cells generated by single cell cloning as “single clonal.” Comparatively, their parental cells (H1/iBC1.2) and parental cells transduced with lentivirus (H1+EGFP, H1+SRY) were denoted as “polyclonal” (equivalent to heterogeneous).
In vitro differentiation of hPSCs into DA neurons
In vitro differentiation was achieved following a previously published protocol [28] with minor modification.
In brief, hPSCs were dissociated using Accutase (BD Corning) and plated onto Matrigel-coated plates at a density of 50,000–70,000/cm2. After ∼2 days of culture in mTeSR when cells reached no less than 90% confluency, mTeSR was replaced by differentiation medium including floor plate (FP) induction medium [knockout serum replacement (KSR) medium: knockout Dulbecco's modified Eagle's medium (DMEM; Gibco) containing 15% KSR (Gibco), 1% GlutaMax (100 × ; Gibco), 1% MEM nonessential amino acid (NEAA; 100 × ; Gibco), 100 μM 2-Mercaptoethanol; N2 medium: DMEM/F12 (Gibco) containing 1% N2 (100 × ; Gibco), 1% GlutaMax (100 × ; Gibco), 1% MEM NEAA (100 × ; Gibco), 100 μM 2-Mercaptoethanol] for 11 days and neural differentiation medium [B27 medium: neural basal medium (Gibco) containing 1% B27 (50 × ; Gibco), 1% GlutaMax (100 × ; Gibco)] for 9 days before passaging. At day 20 (D20), cells were passed using Accutase onto Matrigel-coated plates at density of 300,000–400,000/cm2 and cultured in neural differentiation medium thereafter.
Specific temporal addition of induction factors is given in Fig. 1A that includes 100 nM LDN193189 (Stemgent), 10 μM SB431542 (Tocris), 100 ng/mL sonic hedgehog (SHH) C25II (R&D), 2 μM Purmorphamine (Stemgent), 100 ng/mL FGF8 (Invitrogen), 3 μM CHIR99021 (Selleckchem), 1 ng/mL TGF-β3 (R&D), 20 ng/mL BDNF (Peprotech), 20 ng/mL GDNF (Peprotech), 10 μM DAPT (Tocris), 1 μM dibutyryl cAMP (Sigma), and 0.2 mM ascorbic acid (STEMCELL).

Characterization of midbrain DA neuron differentiation.
Plasmid construction and virus production
Lentiviral plasmids FUW-tetO-FLAG-hSRY-IRES-EGFP, FUW-tetO-EGFP, and FUW-m2rtTA (Fig. 3A) were gifts from Dr. Tatsuo Kido (UCSF). Lentiviruses were packaged in HEK293T cells. In brief, 5 million HEK293T cells were seeded onto 10 cm dishes the day before Lipofectamine 2000 (Invitrogen) transfection. Supernatant was collected after 48- and 72-h transfection. Lentiviruses were concentrated using Pierce Concentrators (150K MWCO; Thermo). Concentrated viruses were stored at −80°C. Lentiviruses infection ability were determined on HEK293T cells with increasing amounts of virus.
Using p3XFLAG-hSRY (gift from Prof. Chris Lau's Laboratory, UCSF) as template, its mutant p3XFLAG-mutSRY [8 bp deletion in SRY complementary DNA (cDNA) that resulted in null protein] was obtained with designed primer (NEBaseChanger, F: CGATGATTACAGTCCAGC, R: TACGCTTAACATAGCAGAAG) using Q5 Site-Directed Mutagenesis Kit (NEB). Using appropriate primers (F: CGGGTACCaccatggactacaaagacc, R: GACTCTCGAGCTACAGCTTTGTCCAGTG), sequence of FLAG-hSRY and FLAG-mutSRY was then obtained from p3XFLAG-hSRY and p3XFLAG-mutSRY, respectively. Subsequently, both FLAG-hSRY and FLAG-mutSRY were cloned into the adeno associated virus (AAV) vector individually. AAVs were packaged in HEK293T cells.
In brief, 5 million HEK293T cells were seeded on 10 cm dishes the day before jetPRIME (Polyplus) transfection. After 72-h transfection, cells were lysed in phosphate-buffered saline (PBS) using repeated freeze-and-thaw method. After centrifugation (200g, 5 min), viruses in the supernatant was precipitated using PEG-it solution (System Biosciences) following the protocol. Precipitated viruses were suspended in PBS and stored at −80°C. AAV infection ability was tittered in HepG2 cells.
Fluorescence-activated cell sorting
Cells infected with Lentivirus were first treated with DOX (1 μg/mL) for 24 h to induce EGFP expression. EGFP-positive cells were sorted out using BD fluorescence-activated cell sorting (FACS) Aria II Cell Sorter. The sorted cells were amplified using feeder-free hPSC culture method without DOX addition.
RNA extraction, cDNA synthesis, and quantitative real-time polymerase chain reaction
Total RNA was extracted using TRIzol Reagent (Invitrogen) as stated by the manufacturer's protocol. Final RNA concentration was measured by Nanodrop 2000 (Thermo). Reverse transcription was performed using a MasterMix kit (Takara) following the standard manual. Quantitative polymerase chain reaction (qPCR) was performed using a Universal SYBR Green MasterMix (Takara) on QuantStudio 7 Flex real-time PCR system (Applied Biosystems). Gene expression was normalized to GAPDH.
Primers used were as follows: ASCL1 (F: CTAAAGATGCAGGTTGTGCG, R: GGAGCTTCTCGACTTCACCA), DDC (F: ATTGTCAAAGGAGCAGCATGT, R: AGGAAGCCCTGGAGAGAGAC), FOXA1 (F: GCCTGAGTTCATGTTGCTGA, R: CTGTGAAGATGGAAGGGCAT), FOXA2 (F: CATGTTGCTCACGGAGGAGT, R: TTTAAACTGCCATGCACTCG), LMX1A (F: TTCTGCTGATCTTGCTGCTG, R: AGAGACAGGGCTGAGTGTCC), NEUROG2 (F: ATCCGAGCAGCACTAACACG, R: GCTGAGGCACAGTTAGAGCC), OTX2 (F: GCTGTTGTTGCTGTTGTTGG, R: AGAGGAGGTGGCACTGAAAA), SRY (F: AAGATGCTGCCGAAGAATTG, R: TAAGTGGCCTAGCTGGTGCT), and TH (F: CACGAAGTACTCCAGGTGGG, R: CGAGCTGTGAAGGTGTTTGA).
RNA sequencing and data analysis
Total RNA was isolated using TRIzol Reagent (Invitrogen) plus RecoverAll™ kit (Invitrogen). Extracted total RNA was quantified using a NanoDrop2000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). Qualified RNA samples were sent out to messenger RNA (mRNA) library construction and sequencing that were performed by Novogene. The sequencing tags were aligned by Bowtie 2 [29]. mRNA expression was defined as RPKM (reads per kilobase per million mapped reads). Differentially expressed genes (DEGs) were prioritized by DEGseq [30] and further filtered in with fold change ≥2 and P ≤ 0.05. Heatmap analysis was performed using R.
Immunostaining
Cells from different stages of differentiation were subjected to immunostaining with first fixed by 4% paraformaldehyde (PFA) for 20 min at room temperature (RT). To stain nuclear protein, cells were permeabilized with 0.1% Triton X-100 for 45 min at RT. After blocking with 1% bovine serum albumin at RT for 45 min, cells were incubated with primary antibodies [anti-FOXA2 (1:30; Abcam), anti-LMX1 (1:2,000; Millipore), anti-Ki-67 (1:50; BD), anti-TH (1:1,000; Pel-Freez), anti-Tuj1 (1:500; Sigma), anti-MAP2 (1:100; Sigma), anti-TBR1 (1:200; Abcam), and anti-FOXG1 (1:100; Abcam)] in 4°C for overnight. After that, cells were incubated with selected Alexa secondary antibodies for 1 h at RT. Finally, cells were incubated with Hoechst 33342 for 10 min at RT. Between each step, cells were washed at least twice with PBS. Cells were mounted for imaging.
Protein extraction and western blotting
Collected cells were lysed using RIPA buffer (Thermo) according to the standard protocol. Final protein concentration was measured using BCA assay kit (Thermo). Protein samples were first normalized to the same concentration. Western blotting was performed following standard protocol using primary antibodies including anti-FOXA2 (1:5,000; Abcam), anti-FLAG (1:8,000; Sigma), anti-SRY (1:2,000, gift from UCSF), and anti-GAPDH (1:5,000, CST). GAPDH was used as the internal control.
Detection of dopamine by high performance liquid chromatography (HPLC)
Conditioned culture medium was collected after overnight exposure to D48 differentiated neurons. Acetonitrile was used to precipitate proteins out of the collected medium. After that, the supernatant was filtered and subsequently analyzed using Agilent 6460 Triple Quadrupole LC/MS (liquid chromatography–mass spectrometry) system. The dopamine standard was purchased from Sigma. The culture medium conditioned in empty well was used as negative control.
Proliferation analysis
Cells were seeded in 96-well plate with same cell number. Differentiation (DOX group vs. control group) was initiated upon >90% confluence. Cell variability was determined using cell-counting kit-8 (DOJINDO) from D3 to D7.
D7 differentiated sample were dissociated using Accutase (BD Corning) and seeded into 96-well plate with 8,000 cells/well up to 3 days (progenitor culture medium was used for culture). Proliferation rate was measured using cell-counting kit-8 (DOJINDO) according to the manufacturer's protocol. The untreated cells served as the negative control, whereas the EGF+bFGF-treated cells served as the positive control.
KEGG and GO enrichment analysis
DEGs were subjected to KEGG pathway and GO enrichment analysis using DAVID with default parameters. All human genes were used as the background.
Gene set enrichment analysis
Normalized gene expression data were subjected to gene set enrichment analysis (GSEA). GSEA was performed using gene sets from Molecular Signature Database. Since the sample size in our study is 2 versus 2, log2_Ratio_of_Classes was used as the correlation metric. GSEA output estimated P value, normalized enrichment score, and false discovery rate as indicators of degree of enrichment for each gene set.
Statistical analysis
All data are represented as mean ± standard error of three or more independent replicates. The unpaired Student's t-test and analysis of variance was adopted to investigate associations. Linear regression was used to evaluate the linear correlation between two variables. P ≤ 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 5 for windows.
Results
Directed midbrain DA neuron differentiation from hPSCs in vitro
To enable DA neuron differentiation from hPSC, we first validated a published protocol [28] that consists of two stages including FP induction and neural differentiation (Fig. 1A). After 11 days of FP induction, cells were extremely compact as previously reported [28]. Neurites could be observed after ∼25–30 days of induction (14–19 days postdifferentiation) (Fig. 1A).
By quantitative reverse transcription PCR (RT-qPCR), the dynamic upregulation of selected DA markers (ASCL1, DDC, FOXA1, NEUROG2, LMX1A, and TH) during the differentiation process confirmed the induction of DA neurons (Fig. 1B).
In addition to transcript analysis, immunostaining was also performed and the results showed the positive staining of DA markers FOXA2, LMX1 at the progenitor stage (Fig. 1C) and TH at the neuronal stage (Fig. 1D). Pan-neuron markers MAP2 and Tuj1 were also detected in the differentiated neurons (Fig. 1D). Unexpectedly, positive staining of non-midbrain markers FOXG1 and TBR1 also occurred in a small number of the differentiated neurons (Supplementary Fig. S1). This result further confirmed the impurity of the differentiated DA neurons that was consistent with previous publications [28,31]. Proliferation marker Ki-67 was highly expressed in the progenitor stage, whereas its expression diminished in the postmitotic neuronal stage (Fig. 1C, D).
In vitro properties of FP-derived DA neurons were also investigated. The neurons generated by this method could be cultured for at least up to 80 days postdifferentiation (data not shown). DA (neurotransmitter) was detectable in the culture medium that further confirmed the generation of DA neurons (Fig. 1E). Altogether, these data suggest that the published FP-based DA neuron differentiation protocol is reproducible with expected results.
DOX induces DA neuron differentiation from normal hPSCs
To investigate the effect of DOX during DA neuron differentiation from hPSCs, the cells differentiated under normal condition was considered as control, whereas the same cells cultured in media supplemented with DOX was regarded as treatment (Fig. 2A). Paired samples were harvested at progenitor stage (D7–D11) at the same time (Fig. 2A). We analyzed the expression of DA markers FOXA1, FOXA2, LMX1A, and OTX2 by qPCR, and surprisingly found that their expression at the progenitor stage was significantly increased by DOX (1 μg/mL) treatment during FP induction for both iPSC (Fig. 2B) and ESC (Fig. 2C). Furthermore, the effect of DOX diminished dramatically when the concentration was reduced to 0.1 μg/mL, indicating that this DOX effect was dose dependent (Fig. 2C). Beyond the early DA neuron differentiation, we also found out that DOX treatment along differentiation process could significantly increase DA neuronal markers including LMX1A, FOXA2, DAT, DDC, and TH (Supplementary Fig. S2).
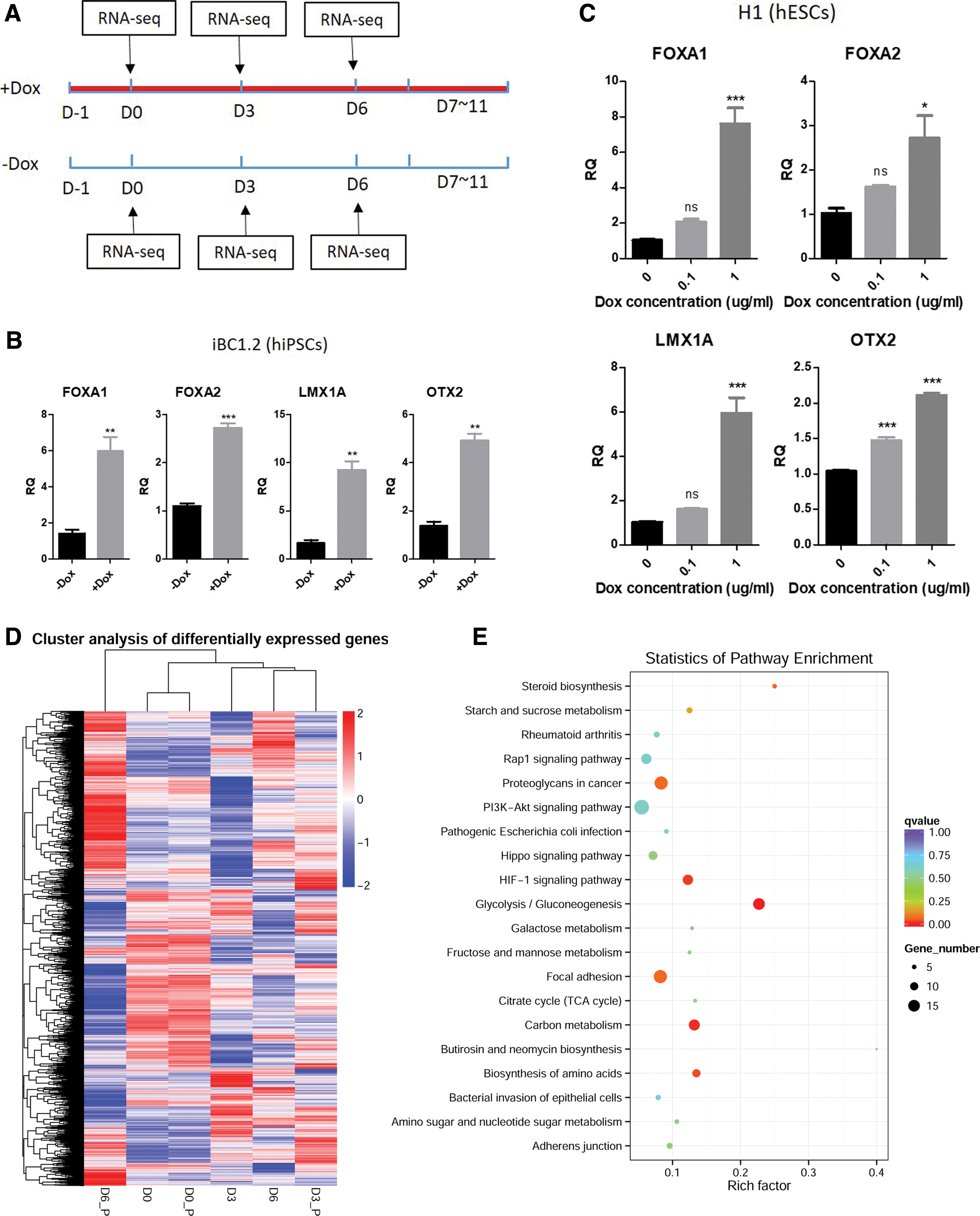
Effects of DOX on early DA neuron differentiation from normal hPSCs.
To evaluate the extent to which DOX enhances early DA neuron differentiation and analyze the altered pathways by DOX, paired samples treated with DOX or vehicle control from D0, D3, and D6 were harvested and total RNA was extracted for RNA sequencing (Fig. 2A). We analyzed the DEGs in the paired samples at each time point. The different expression pattern of the union of DEGs from all time points can represent the identity of each sample. The heatmap showed that number of DEGs at D0 (D0_P vs. D0) was less than DEGs at D3 (D3_P vs. D3) or D6 (D6_P vs. D6) (Fig. 2D). From the unsupervised hierarchical clustering result (column dendrogram), we observed that DOX-treated D3 sample (D3_P) clustered closer to the more differentiated D6 sample without treating DOX (D6). However, DOX-treated D6 sample (D6_P) was in an independent cluster of higher order (Fig. 2D). These results suggest that DOX treatment promotes or accelerates the differentiation toward DA fate. Pathway enrichment analysis of the DEGs at undifferentiated stage (D0) showed that glycolysis and PI3K-AKT signaling were among the top 20 enriched pathways (Fig. 2E), which was consistent with previous findings [16,19].
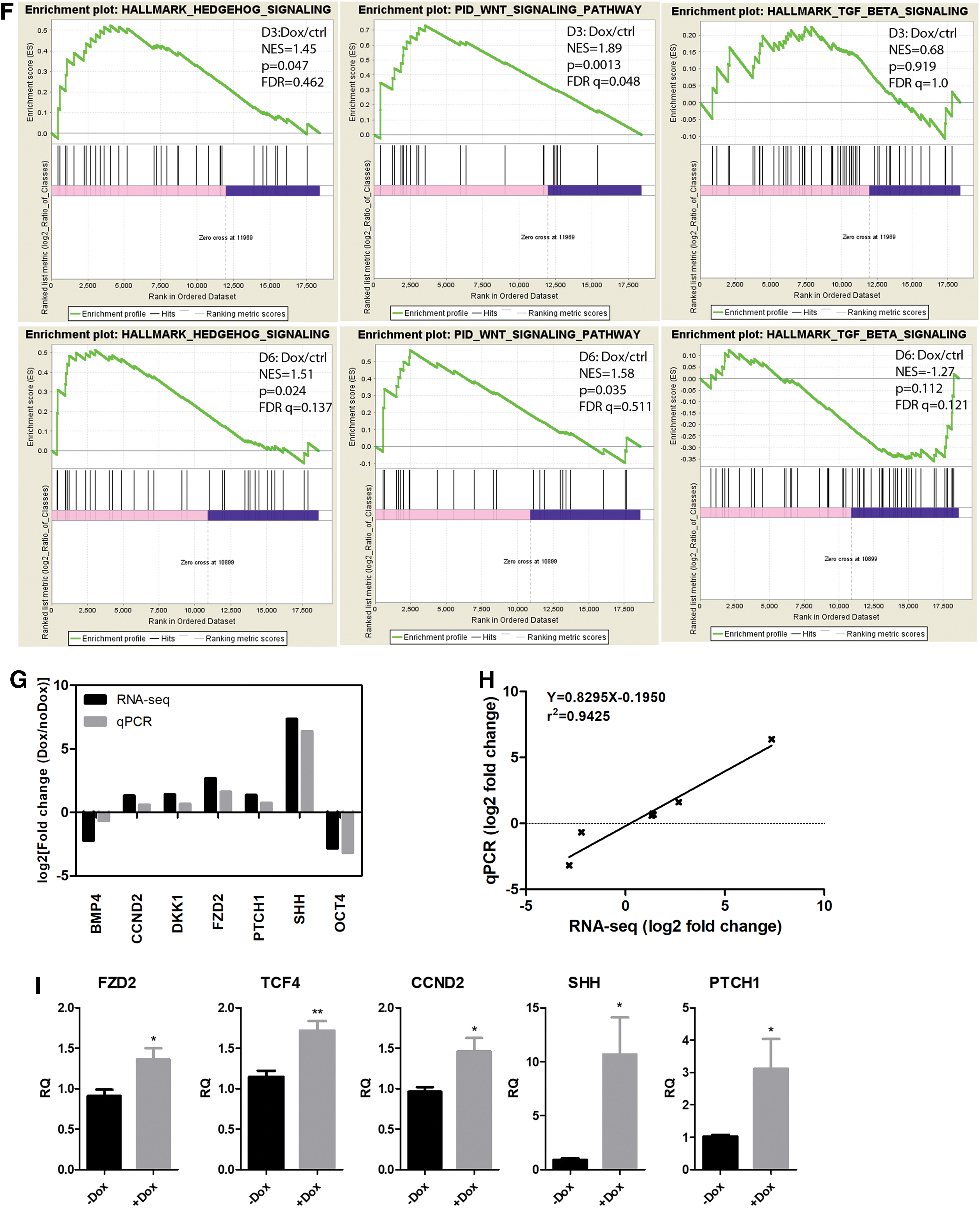
To evaluate the enhancement/suppression of DA fate specification pathways by DOX, GSEA was performed on expression data from D3 and D6 (DOX vs. ctrl). GSEA results showed that gene set of WNT signaling and Hedgehog signaling pathways were enriched both at D3 and D6 in DOX-treated group, whereas gene set of TGF-beta signaling showed no significant difference between DOX and control (Fig. 2F).
To confirm that data from RNA-seq was reliable, we selected several important genes for qPCR validation. For each gene, the fold change obtained from RNA-seq was comparable with that from qPCR (Fig. 2G). The correlation efficiency between qPCR and RNA-seq based on fold change was as high as 0.9425 (Fig. 2H). The enhancement of WNT signaling and Hedgehog signaling by DOX was further validated in ESC differentiated samples by qPCR of important regulators and downstream targets (Fig. 2I).
All these data indicated that DOX could promote DA progenitor fate specification both from ESCs and iPSCs partly through the WNT and Hedgehog signaling pathways.
DOX induces DA neuron differentiation from hPSCs transduced with Tet-On lentivector
To examine the effect of DOX when being applied during Tet-On induction, hPSCs were transduced with a Tet-On lentivector expressing only EGFP. DOX was added to the medium during hPSCs differentiation (Fig. 3A). Single cell-derived iPSC clones (IE-2 and IE-4) or ESC H1 clones (HE4 and HE-5) with Tet-On EGFP vector are given in Fig. 3B. In IE-2 and IE-4, DOX (1 μg/mL) treatment during FP induction significantly increased the expression of DA progenitor markers, as measured by both RT-qPCR and western blot (Fig. 3C, D). In HE-4 and HE-5, DOX treatment also increased the DA progenitor markers; however, clonal variation exists for some of the markers (Fig. 3E). These data support that DOX could affect DA differentiation when hPSC is genetically modified with Tet-On lentivector.

Effects of DOX on early DA neuron differentiation from hPSCs transfected with inducible lentivirus vector.
DOX masks the genuine function of the inducible target gene
There are many studies adding DOX to the differentiation culture without proper control to exclude the effect of DOX [7,32 –34]. To test whether the effect of DOX will confound the interpretation on the function of a gene to be studied, we first chose a male-specific gene, SRY, which showed no significant effect on DA neuron differentiation in an independent study in our laboratory. In that study, overexpression of SRY was achieved by utilizing AAV delivery system. Transgene expression by AAV remained high (as indicated by GFP expression) even at the progenitor stage (Fig. 4A). Although SRY remained highly expressed during FP induction, we observed no significant changes on DA marker expression at progenitor stage among the groups of vector control (GFP), SRY overexpression (SRY), and a mutant form of SRY (MUT) (Fig. 4B).

Confounding effect of DOX on the inducible target gene function during early DA neuron differentiation.
Then, we used a DOX-inducible Tet-On system to express SRY during early DA neuron differentiation (Fig. 3A). A single cell-derived ESC clone (HR-1) was used in this study (Fig. 4C). The result indicated that DOX-induced SRY overexpression during FP induction significantly increased expression of DA progenitor markers, compared with “no DOX” control (Fig. 4D). Without considering the effect of DOX, one could conclude that SRY overexpression induces DA neuron differentiation. However, it is difficult to discriminate the effect of SRY from that of DOX from this experiment.
To study the effect of SRY, it is critical to include proper controls. As single cell-derived ESC clones showed clonal variation in response to DOX treatment, we used polyclonal ESC here. After FACS sorting, EGFP-expressing (H1+EGFP) and SRY/EGFP-expressing (H1+SRY) polyclonal cells were used for the experiment (Figs. 3A and 4E, F). The qPCR and western blot confirmed SRY expression when induced by DOX (Fig. 4G). When both cultures were subjected to DA neuron differentiation in the presence of DOX, we found no statistic significant differences for LMX1A between them, but decreasing trend could be observed for FOXA1, FOXA2, and OTX2 in H1+SRY with DOX group compared with H1+EGFP with DOX group (Fig. 4H). The decreasing trend observed in Fig. 4H (H1+EGFP vs. H1+SRY, with DOX) may be the result of the synergistic effect of SRY and DOX or the nonspecific effect of DOX.
These data demonstrated that the enhancement of DA neuron differentiation (Fig. 4D) could be falsely attributed to the function of SRY, if no proper controls are included. In summary, we suggest that DOX should be carefully used in Tet-inducible experiments that might lead to false-positive results and misleading conclusions.
Discussion
DOX, an antibacterial drug, has long been known to exert effects on different cell types including somatic cell lines, cancer cells, and chondrocytes [11,13 –16]. However, few studies examined its effect on hPSCs, cells with properties of infinitive self-renewal and pluripotency. Only one group reports that DOX can increase the proliferation of hPSCs through activating PI3K-AKT pathway [19,20]. In addition, previous studies have shown that DOX is able to increase neurogenesis in vivo, and used as a drug to treat stroke by inhibiting microglia activation [23 –26].
Among the different types of neurons, midbrain DA neurons play important roles in the regulation of voluntary movement, emotion, and reward [35]. The degeneration of DA neurons is the cause of Parkinson's disease (PD). With the emerging usage of stem cells, DA neurons differentiated from hPSCs have been widely considered for cell transplantation in the treatment of PD [36,37]. Meanwhile, the in vitro differentiation of DA neuron enables the study of regulatory factors during DA neuron development [38 –40] and the recapitulation of disease pathogenesis [41 –43]. In related research, DOX-inducible system was widely applied because of its controllable mediating ability. Taken together, we extended to study the DOX effect on DA fate specification in vitro and its potential confounding ability on biomedical research in the system of DA differentiation from hPSCs.
In our study, we surprisingly found that DOX supplementation could dramatically advance DA fate specification when using unmodified hPSCs and Tet-On lentivector-integrated hPSC for transgene expression. This effect was dose dependent. We further found clonal variation among the different single cell-derived ESC clones in response to DOX treatment. This variation might be because of the heterogeneity of hPSCs or the different integration sites of the lentivector [44 –47]. We also found that DOX treatment could increase cell survival during early DA neuron differentiation partially by suppressing the apoptosis pathway (Supplementary Fig. S3 and S4A), and promote cell proliferation of DA neuron progenitors (Supplementary Fig. S4B) that was consistent with previous finding [19]. The increasing effect by DOX was also observed on differentiated DA neurons (Supplementary Fig. S2) making DOX a promising drug for in vitro DA neuron production.
Of interest, further systematic analysis showed that glycolysis pathway (that is known to be affected by DOX treatment in HEK293T cells [16]) and PI3K-AKT signaling pathway (that is reported to be activated upon DOX addition in PSCs [19]) were both significantly enriched in the DOX treatment group at pluripotent stage (D0). It is intriguing to find whether pathways (TGF-beta/WNT/SHH signaling) that are known to be essential for DA neuron differentiation [28] were affected by DOX. By comparing DOX treatment with control group at D3 and D6, we found that gene sets of WNT and Hedgehog signaling pathways were largely enriched at both D3 and D6 by GSEA. However, gene set of TGF-beta signaling was not significantly associated with DOX either at D3 or D6. Further validation in independent hES cell line was conducted by qPCR of WNT/Hedgehog pathway regulators and targets. Indeed, DOX has been reported to activate Wnt signaling during bone repair [48].
The enhancing effect of DOX on DA fate determination, on the one hand, makes it a promising chemical that could be applied to increase the DA neuron generation efficacy for cell therapy. On the other hand, it also warns against the confounding effect of DOX when used in DA neuron-related biomedical research that utilized hPSC as a model.
To further confirm its potential confounding effect, we applied a DOX-inducible Tet-On lentivirus system to study the effect of the sex determination gene SRY during early DA neuron differentiation. The genuine effect of SRY on DA neuron specification was confirmed as neither increasing nor decreasing using an AAV expression system in which DOX was not involved. However DOX-induced SRY overexpression of HR-1 (single clone) by the Tet-On system increased DA neuron specification without considering DOX. After eliminating DOX confounding effect by comparing H1+EGFP and H1+SRY (polyclonal, with DOX), the increasing effect of SRY shifted to no effect or decreasing effect on DA progenitor formation.
Of interest, in our study we found that DOX-promoting effect in H1+SRY was gone when compared with H1+EGFP or HR-1 cells (data not shown). There are several possible reasons. First, DOX effect was clonal variable (HE-5 vs. HE-4) on transduced hESCs. This could explain the difference of DOX effect on HR-1 and H1+SRY. Second, SRY might counteract DOX effect. SRY, as a sex-determining gene, was reported to suppress Wnt signaling pathway [49 –51]. The counteracting effect of SRY to DOX can explain the difference of DOX effect on H1+EGFP and H1+SRY. However, the hypothesis on SRY counteracting DOX effect during hESC DA neuron differentiation required further confirmation. The reason why and to what extent SRY counteracting DOX effect also needs further investigation. In this part, we revealed that DOX could indeed mask the genuine effect of SRY during early DA neuron differentiation.
This is the first study to report DOX effect on DA neuron differentiation process that renders DOX as a promising chemical in stem cell therapy. Moreover, we first demonstrated that the effect of DOX on differentiation can indeed lead to false scientific conclusion on studied genes. Cautions should be taken by researchers in applying DOX to biomedical research.
Finally, as our work only confined to studying the effect of DOX on DA fate specification from hPSCs, additional studies of DOX's effect on other lineage/cell type specification are required.
Footnotes
Acknowledgments
The authors thank Dr. Kai Kei Miu for help with LC-MS analysis. This work was supported by the Hong Kong General Research Fund (Project No. 14169717) of the Research Grants Council, CUHK Special Budget for Key Laboratories Approved by the State Ministries to support the MOE Key Laboratory for Regenerative Medicine (CUHK-Jinan University); One-off Funding for Joint Lab/Research Collaboration (MOE Key Laboratory for Regenerative Medicine, Project 3132969; CUHK-CAS GIBH Joint Research Laboratory on Stem Cell and Regenerative Medicine, Project 3132966 and 4930732).
Author Disclosure Statement
The authors declare no competing financial interests.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.