
Abstract
Bone marrow-derived mesenchymal stem cells (MSCs) improve cardiac function after ischemia/reperfusion injury, in part, due to the release of cytoprotective paracrine factors. Toll-like receptor 4 (TLR4) is expressed in MSCs and regulates the expression of cytoprotective factors, cytokines, and chemokines. Lipopolysaccharide (LPS) stimulation of TLR4 activates two distinct signaling pathways that are either MyD88 dependent or MyD88 independent/TIR-domain-containing adapter-inducing interferon-β (TRIF) dependent. While it was reported previously that LPS treatment improved MSC-mediated cardioprotection, the mechanism underlying such improved effect remains unknown. To study the role of MyD88 signaling in MSC cardioprotective activity, wild type (WT) and MyD88−/− MSCs were treated with LPS (200 ng/mL) for 24 h. WT and MyD88−/− MSCs with or without LPS pretreatment were infused into the coronary circulation of isolated mouse hearts (Langendorff model) and then subjected to ischemia (25 min) and reperfusion (50 min). Saline served as a negative control. Both untreated and LPS-pretreated WT MSCs significantly improved postischemic recovery of myocardial function of isolated mouse hearts, as evidenced by improved left ventricular developed pressure and ventricular contractility assessment (ie, the rate of left ventricle pressure change over time, ± dp/dt). LPS-pretreated WT MSCs conferred better cardiac function recovery than untreated MSCs; however, such effect of LPS was abolished when using MyD88−/− MSCs. In addition, LPS stimulated stat3 activity in WT MSCs, but not MyD88−/− MSCs. stat3 small interfering RNA abolished the effect of LPS in improving the cardioprotection of WT MSCs. In conclusion, this study demonstrates that LPS improves MSC-mediated cardioprotection by MyD88-dependent activation of stat3.
Introduction
Myocardial ischemic injury and infarction are the leading causes of death and disability worldwide. Ischemic injury leads to scar formation, electric uncoupling, morphologic structure changes, and ventricular remodeling, which have great impact on the quality of life [1]. Multiple stem cells, such as hematopoietic stem cells (HSCs), embryonic stem cells, induced pluripotent stem cells, and mesenchymal stem cells (MSCs) have been used as therapeutic reagents for treating myocardial ischemic injury and infarction [2]. Among all the stem cell types, MSCs have multiple advantages for use in repairing the infarcted myocardium: MSCs are safe; can be easily isolated and amplified from patients' bone marrow; are immunologically tolerated as transplants; and possess multilineage differentiation potential [3].
Intramyocardial injection of MSCs reduces inflammation, fibrosis, infarct size, and ventricular remodeling, and thereby improves cardiac function [4 –7]. Although the exact mechanism underlying the improved benefits is not yet fully elucidated, strong evidence indicated that MSCs displayed beneficial cardioprotection partly through the release of soluble paracrine factors, which reduced inflammation, decreased apoptosis, and inhibited cardiac remodeling [8,9].
Despite these favorable attributes, MSC-mediated protection is modest and has limited duration [10 –14]. Various clinical trials have demonstrated that, while effective, the delivery of MSCs to ischemic myocardium resulted in only modest benefits. Abdel-Latif et al. [15] reported that bone marrow cell transplantation resulted in only modest improvements in physiologic and anatomic parameters in patients with both acute myocardial infarction (MI) and chronic ischemic heart disease. Moreover, although transplanted MSCs survived in infarcted myocardium for as long as 6 months, no persistent benefit was observed [16]. Therefore, there is a critical need to optimize MSC-conferred protection and identify the exact paracrine factor(s) that mediate MSC-related therapeutic benefits.
Toll-like receptor 4 (TLR4) is highly expressed in MSCs and regulates MSC function [17]. Upon activation by TLR4 ligands (eg, lipopolysaccharides [LPS]), TLR4 activates two distinct signaling pathways that are either MyD88 dependent or MyD88 independent/TIR-domain-containing adapter-inducing interferon-β (TRIF) dependent to initiate the downstream signal transducers that then produce a variety of paracrine factors. Although it was reported previously that LPS preconditioning improved MSC-mediated cardioprotection in a rat model of acute MI [18], the underlying mechanism remains unknown. In this study, we hypothesize that, preconditioning MSCs with LPS improves cardioprotection by MyD88-dependent mechanism.
Materials and Methods
Reagents
All chemicals used in this study were purchased from Sigma Company (St. Louis, MO).
Animals
Normal adult male C57BL/6J mice and MyD88−/− mice (25–30 g, 8–9 weeks old, B6.129P2(SJL)-Myd88tm1Defr /J) were obtained from Jackson Labs, fed a standard diet, and acclimated in a quiet quarantine room for 1 week before the experiments. The animal protocol was reviewed and approved by the Institutional Animal Care and Use Committee of Indiana University. All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication No. 85-23, revised 1996).
Preparation of mouse bone marrow MSCs
A single-step purification method using plastic adherence was used as previously described [19]. Briefly, MSCs were harvested from bilateral femurs by removing the epiphyses and flushing the shaft with complete medium (IMDM with 10% fetal bovine serum and 1% pen-strep; ThermoFisher). Cells were disaggregated by vigorous pipetting using a syringe with a 23-gauge needle and passed through a 30 micron nylon mesh to remove remaining clumps of tissue. Cells were washed by adding a complete medium and centrifuged at 300 rcf for 5 min at room temperature. The cell pellets were resuspended with the complete medium and cultured in 25 cm2 flasks (ThermoFisher) at 37°C in 5% CO2. After 48 h, nonadherent cells in suspension were discarded. Fresh medium was added and replaced every 3 days thereafter. At 90% confluence, cells were trypsinized by the addition of 0.25% trypsin-EDTA (ThermoFisher) and replated in 75 cm2 flasks. After three passages, cell surface markers were examined using Flow Cytometry analysis. MSCs were positive for CD44 [20] and negative for CD45, CD11b, CD90, and CD117 [19,21]. All experiments used cells between passages 4–10.
Small interfering RNA transfection
Small interfering RNA (siRNA) transfection was performed as previously described [20]. Briefly, siRNA that specifically targeted mouse stat3 was designed with the software provided by Dharmacon siDESIGN center (Dharmacon Research, CO). A siRNA sequence (5′-CAGCACAACCUUCGAAGAA-3′) corresponding to residues 517–535 of the coding region of mouse stat3 was selected. MSCs from the same starting cell isolation were transfected with the aforementioned specific stat3 siRNA at a concentration of 100 nM. MSCs transfected with scramble siRNA were used as a negative control (ThermoFisher). Transfection was performed with Lipofectamine 2000 (ThermoFisher) as per the manufacturers' instructions. Briefly, 24 h before siRNA transfection, cells were plated in six-well plates at 5 × 104 cells/well/2 mL. On culture day 2, cells were washed with Opti-MEM media (ThermoFisher) and the lipofectamine-siRNA complex (100 nM) was then added. One day after transfection (day 3 in culture), the lipofectamine-siRNA complex was washed out and the complete IMDM medium with or without 200 ng/mL LPS was added to the cells and incubated for an additional 1 day. The cell number was counted with the aid of the TC10™ Automated Cell Counter (Bio-Rad, Hercules, CA) and cell extracts were prepared for western blot analysis.
Isolated mouse heart perfusion (Langendorff model)
Mice were anesthetized (Pentobarbital, 60 mg/kg, i.p.) and heparinized (500U, i.p.). The heart was rapidly excised and placed in 4°C modified KH solution containing the following (in mM): 119 NaCl, 4.7 KCl, 2.5 CaCl2, 0.5 EDTA, 25 NaHCO3, 1.2 KHPO4, 1.2 MgSO4, 2 Na pyruvate, 10 HEPES, and 11 dextrose. The aorta was cannulated and the heart was perfused under constant pressure (75 mmHg) with oxygenated (95% O2–5% CO2) KH solution at 37.5°C. A left atrial resection was performed before inserting a water-filled wrap balloon through the atrium into the ventricle. The balloon was adjusted to mean end diastolic pressure (EDP) of 8 mmHg. The hearts were allowed to equilibrate for 15 min before infusing vehicle or MSCs and paced at 420 beats/min. During equilibration, MSCs were trypsinized, collected, and counted. One million viable cells were isolated and suspended in 1 mL of KH solution (37°C). Over the course of 1 min immediately before ischemia, the MSC solution was infused into the coronary circulation. A three stopcock placed above the aortic root was used to create global ischemia. During global ischemia the hearts were placed in a 37°C degassed organ bath. Left ventricular pressure (LVP) was continuously recorded using a PowerLab 8 preamplifier/digitizer (AD Instruments, Inc.). The values of left ventricular developed pressure (LVDP), +dp/dt, and −dp/dt were calculated with the aid of PowerLab software. At the end of reperfusion, the hearts were removed from the Langendorff device and snap-frozen in liquid nitrogen for future analysis.
Western blotting
The cells transfected with either stat3 siRNA or scramble siRNA were stimulated with 200 ng/mL LPS for 24 h. Western blot analysis was performed to measure stat3 activity. Cells were lysed in cold RIPA buffer containing protease cocktail and phosphatase inhibitor cocktail 2 (Sigma). Lysates were centrifuged at 16,000 rcf for 15 min. The protein extracts (8–10 μg/lane) were electrophoresed on a 4%–12% Bis-Tris gel (ThermoFisher) and transferred to a nitrocellulose membrane. The membranes were blocked in 5% nonfat milk solution for 2 h and incubated with primary antibodies for p-stat3, T-stat3, and GAPDH (Cell Signaling Technology). After three washes with phosphate-buffered saline (PBS) containing 1% Tween 20, membranes were incubated in 5% nonfat milk solution containing horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (ThermoFisher). Signal detection was performed using SuperSignal West Pico chemiluminescent substrate (Pierce). Films were scanned and band density was analyzed with ImageJ software (NIH) and normalized to GAPDH.
RNA sample preparation and RNA-sequencing assay
Wild-type (WT) or MyD88−/− MSCs were plated at 5 × 104 cells/well/mL for 24 h and then treated with LPS (200 ng/mL) for another 24 h. Total RNA was extracted before and after LPS treatment, following a standard protocol [25]. Experiments were conducted in triplicate.
Standard methods were used for RNA sequencing (RNA-seq) library construction, EZBead preparation, and Next-Gen sequencing, based on the Life Technologies SOLiD 5500xl system. Briefly, 2 μg of total RNA per sample was used for library preparation. The ribosomal RNA (rRNA) was first depleted using the standard protocol of RiboMinus Eukaryote Kit for RNA-Seq (Ambion), and rRNA-depleted RNA was concentrated using a PureLink RNA Micro Kit (Invitrogen) with 1 volume of lysis buffer and 2.5 volumes of 100% ethanol. After rRNA depletion, a whole transcriptome library was prepared and barcoded per sample using the standard protocol of SOLiD Total RNA-seq Kit (ThermoFisher). Each barcoded library was quantified by quantitative PCR (qPCR) using SOLiD Library TaqMan qPCR Module (ThermoFisher) and pooled in equal molarity. EZBead preparation, bead library amplification, and bead enrichment were then conducted using the Life Technologies EZ Bead E80 System. Finally, sequencing by ligation was performed using a standard single-read, 5′-3′ strand-specific sequencing procedure (75 nt-read) on SOLiD 5500xl in the Center for Medical Genomics at IU. Each SOLiD 5500xl lane can sequence ∼8 billion nucleotides, which is equivalent to >100 million 75-bp reads, over 80% of which were mappable to the reference genome.
Bioinformatics analysis for RNA-seq data
RNA-seq data analysis included the following steps: quality assessment, sequence alignment, and alternative splicing analysis. The RNA-seq data can be accessed through the Gene Expression Omnibus (
Data processing and quality assessment
We used SOLiD Instrument Control Software and SOLiD Experiment Tracking System software for read quality recalibration. Each sequence read was scanned for low-quality regions, and if a five-base sliding window had an average quality score <20, the read was truncated at that position. Any read <35 bases was discarded. Our experience suggests that this strategy effectively eliminates low-quality reads, while retaining high-quality regions [22 –24].
Sequence alignment
We used BFAST (
Differential expression analysis
The edgeR package [27] was used to identify genes differentially expressed between control and LPS groups. False discovery rate (FDR) was calculated within edgeR using Benjamini and Hochberg correction [28]. Analysis was limited to those genes with ≥1 count per million in at least two of the samples in one of the conditions.
Heat map analysis
Heat map of the differentially expressed genes induced by LPS in WT MSC was generated with the R package ComplexHeatmap [29].
Pathway and regulatory analysis
Qiagen ingenuity pathway analysis was used to identify pathways that are significantly enriched in differentially expressed (FDR ≤0.05) genes. Upstream regulator analysis uses the curated knowledge base to identify molecules that could possibly be responsible for the observed changes in gene expression, based on a z score indicating whether the activity of a specific regulatory factor is stimulated or inhibited. A detailed definition of z-score can be found in the Qiagen white paper (
stat3 binding site prediction
Putative stat3 binding sites were predicted based on the promoter sequences (−1,500 bp to +500 bp from the transcription start sites) of the genes to be evaluated. Position-specific score matrices were used to calculate the possibility of a specific transcription factor (stat3) binding at one genomic locus, as described previously [30]. The highest matching score during this scanning was used to evaluate the binding potential of the stat3 in the promoter region.
Statistical analysis on phenotype data
All reported values were mean ± standard error of the mean, and P < 0.05 was considered statistically significant. LVDP, +dp/dt, and −dp/dt were as percentage of baseline during equilibrium. Data were compared using one-way or two-way analysis of variance with post hoc Bonferroni.
Results
LPS improved MSC-mediated cardioprotection in WT, but not MyD88−/− MSCs in an isolated mouse Langendorff heart preparation
To examine whether LPS improved MSC-conferred protection, plastic-adherent cells from mouse bone marrow from WT and MyD88−/− mice were isolated and propagated under normal cell culture conditions (5% CO2 at 37°C, 90% humidity) [8]. These cells were negative for the HSC marker CD45, as well as CD11b, CD106, CD117, and CD90, and were positive for stem cell antigen-1 and the MSC marker CD44 [8]. We have also demonstrated previously that purified stromal cells were able to differentiate into adipocytes and osteoblasts, suggesting that these cells indeed had MSC characteristics [8]. As shown in Fig. 1, isolated Langendorff mouse hearts infused with LPS-pretreated MSCs demonstrated improved functional recovery compared to untreated MSC and vehicle groups. In comparison with the baseline, postischemic LVDP, +dp/dt, and −dp/dt were markedly decreased, but EDP was greatly elevated in all treatment groups (Fig. 1). Intracoronary infusion of WT MSCs and WT MSCs + LPS significantly enhanced postischemic recovery of myocardial function as exhibited by LVDP, +dp/dt, and −dp/dt compared to vehicle (P < 0.05). The WT MSCs + LPS group displayed an even better recovery than untreated WT MSCs from ischemic injury (Fig. 1). In contrast, LPS did not improve the cardioprotective effect of MyD88−/− MSCs. Although MyD88−/− MSCs exhibited a similar cardioprotection effect as untreated WT MSCs, LPS failed to further potentiate their cardioprotective effect. Notably, the value of EDP, an index of heart injury, progressively increased to a relatively higher level after ischemia in all groups, indicating significant cardiac injury (Figs. 1G and 2F). All hearts receiving cell infusion demonstrated smaller EDP compared to vehicle (Figs. 1G and 2F). Taken together, LPS significantly improved MSC-mediated cardioprotection in WT, but not in MyD88−/− MSCs.
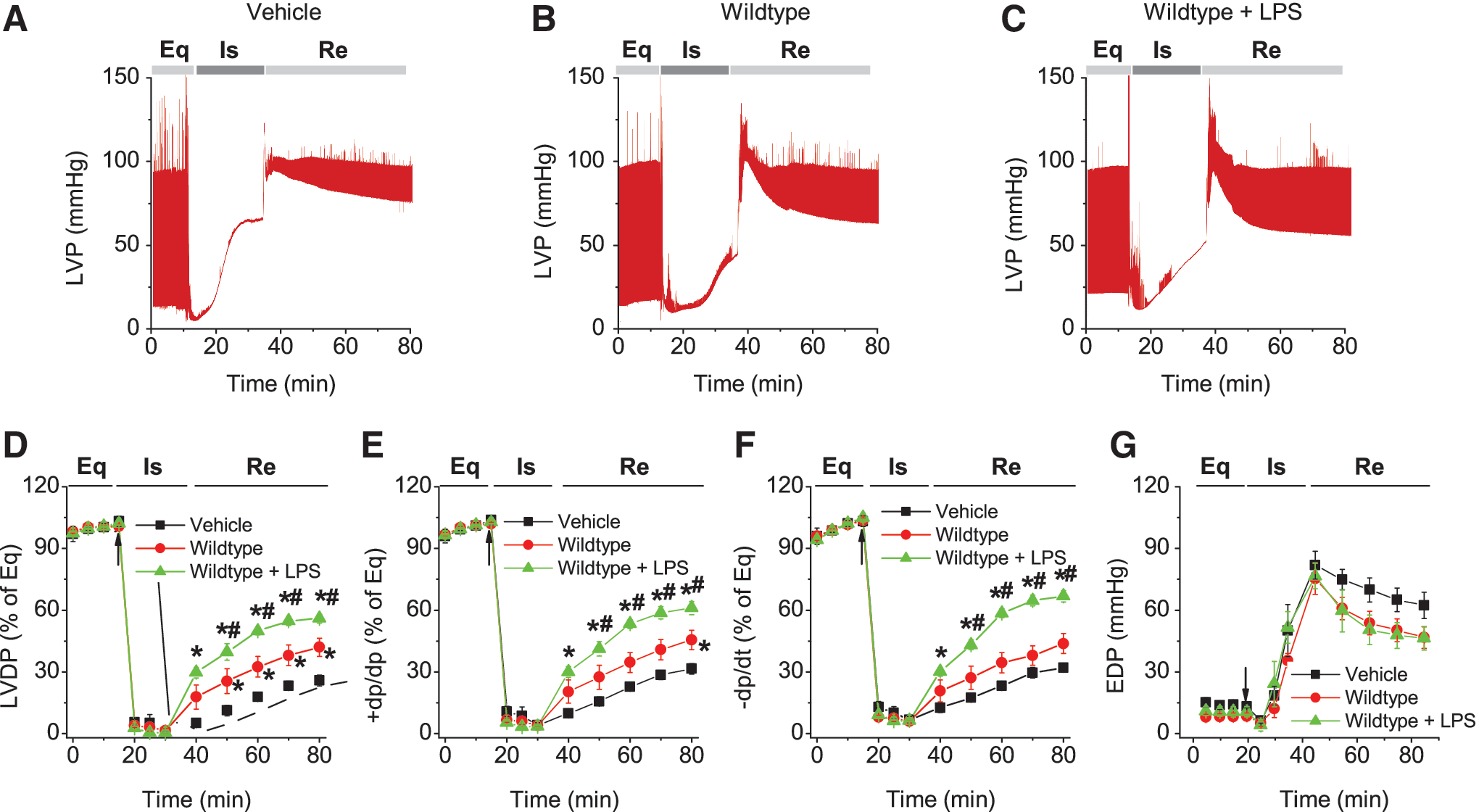
LPS improved the cardioprotection derived from WT MSCs. WT or MyD88−/− MSCs, with or without LPS pretreatment, were infused into the coronary circulation before global ischemia.

LPS failed to improve the cardioprotection of MyD88−/− MSCs. MyD88−/− MSC, with or without LPS pretreatment, were infused into the coronary circulation.
MyD88−/− MSCs exhibited a slower rate of proliferation and did not activate stat3 in response to LPS
We reported previously that increased stat3 activity improved MSC-mediated cardioprotection in a paracrine manner [8]. To further examine the crosstalk between MyD88-dependent signaling and stat3 activity, WT and MyD88−/− MSCs were stimulated with LPS. As shown in Fig. 3A, B, LPS significantly activated stat3 in WT MSCs, but not in MyD88−/− MSCs. In addition, MyD88−/− MSCs proliferated at a much slower rate, compared to WT cells (Fig. 3C). LPS at 200 ng/mL did not change the rate of proliferation in either WT or MyD88−/− cells (data not shown).
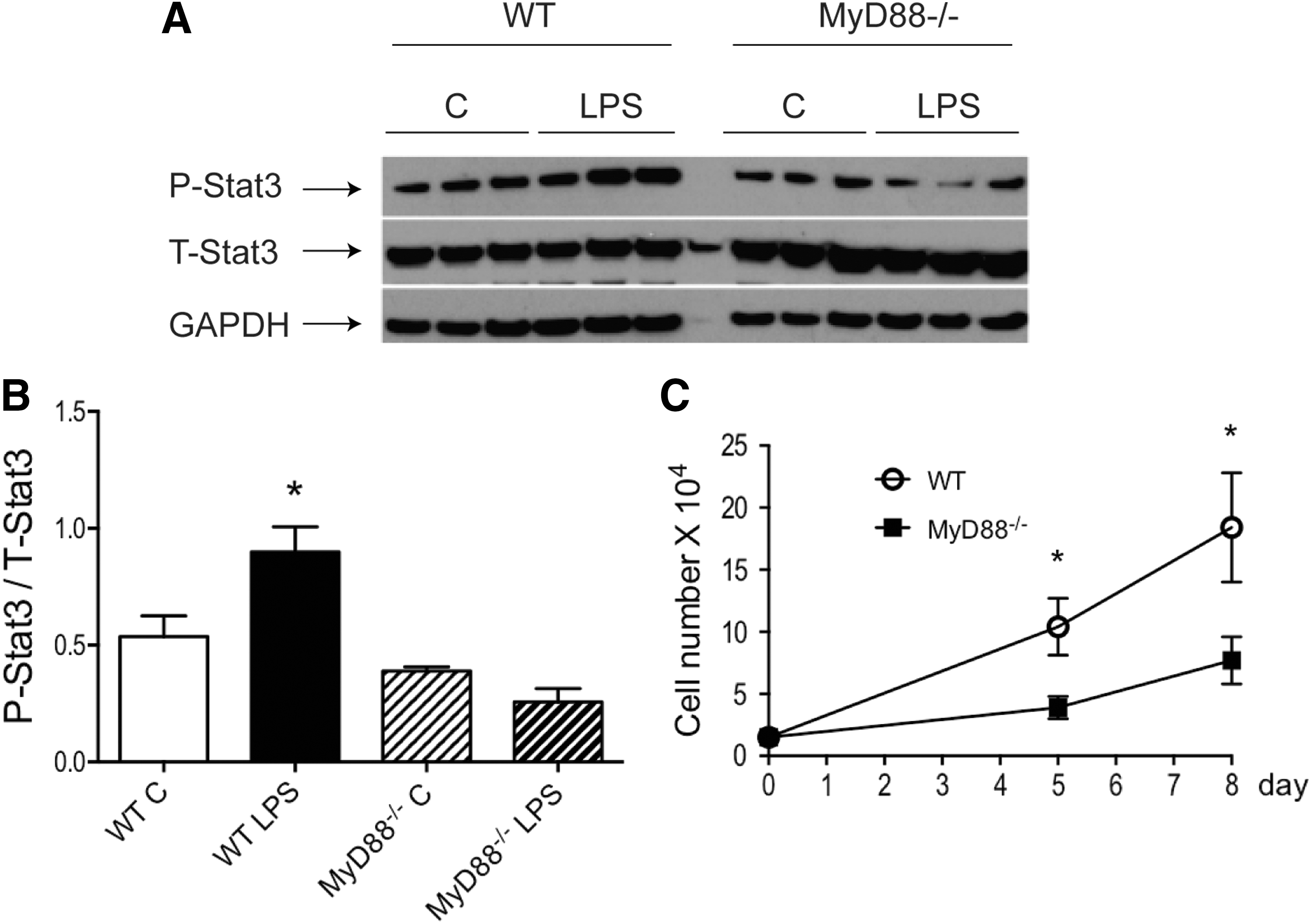
MyD88−/− MSCs did not activate stat3 in response to LPS stimulation and exhibited a slower rate of proliferation. WT and MyD88−/− MSCs were stimulated with or without LPS (200 ng/mL) for 24 h. Phosphorylated stat3 levels in protein extracts from all groups were analyzed by western blot.
LPS stimulation changed the global gene expression in MSC
To investigate LPS-induced transcriptome changes in mouse MSC, RNA-seq was conducted before and after LPS treatment [8]. WT or MyD88−/− MSCs were plated at 5 × 104 cells/well in six-well plates for 24 h and further treated with LPS (200 ng/mL) for another 24 h. Experiments were conducted in triplicates. LPS was found to regulate global gene expression of WT MSCs.
As shown in Supplementary Table S1, the expression levels of 295 genes were significantly altered after LPS treatment (FDR <0.05). The expression levels of the 295 genes in all the four experimental conditions were demonstrated in Fig. 4. Further pathway analysis suggested that 62 of the 295 genes were related to inflammatory response. Specifically, the P values for 47 subcategories of inflammatory response were significantly enriched (Supplementary Table S2) with P values between 1.4 × 10−3 and 4.4 × 10−9. This is consistent with the current knowledge that MSCs regulate immune and inflammatory responses, and facilitate the repair of damaged tissues, including cardiac tissues.
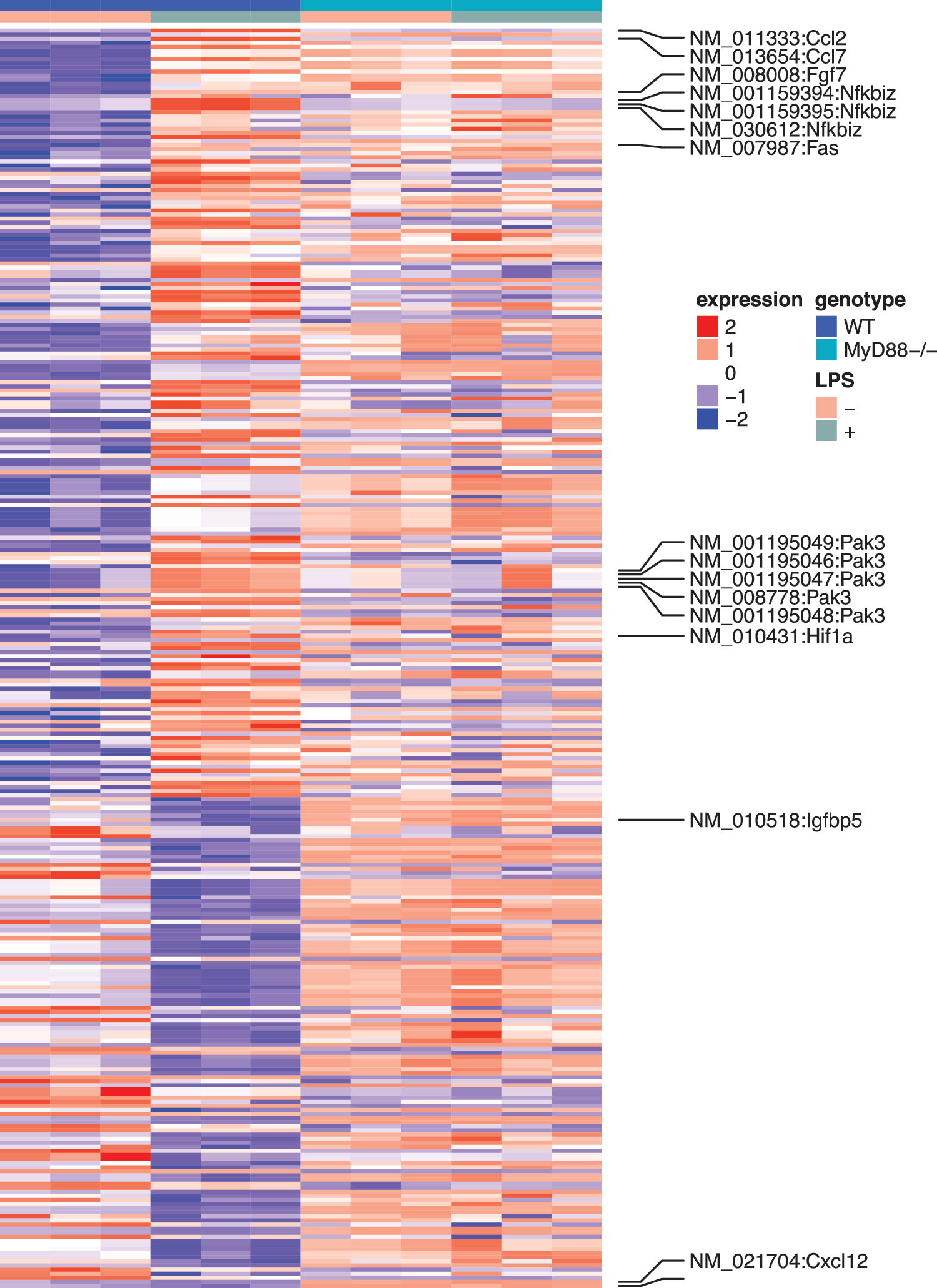
Heat map of the LPS induced differentially expressed genes in WT and MyD88−/− MSCs. The rows are genes and the columns are samples. The paracrine factors and intracellular signaling molecules were labeled.
We observed increases in the stat3 activity. Five genes known to be activated by stat3 all showed increased expression levels. These included chemokine (C-C motif) ligand 2 (CCL2), hypoxia inducible factor 1 alpha subunit (HIF1A), schlafen 2 (which is known to be altered by LPS), guanylate binding protein 2, and nuclear factor of κ light polypeptide gene enhancer in B cell inhibitor, ζ (NFKBIZ). In addition, among the four genes that are known to be inhibited by stat3, three showed decreased expression levels, including insulin-like growth factor binding protein 5, transcription factor 4, and hyaluronan synthase 2. Given the known functions of these stat3 target genes in proinflammation and tissue growth, we hypothesize that LPS alters the global gene expression profiles of MSCs, in part, through stat3 pathways.
Interestingly, most LPS-induced changes were abolished in MyD88−/− MSCs. Among the 295 genes whose expression levels were altered in WT MSCs, only eight were significantly different in MyD88−/− animals. These eight genes were noncoding genes, seven of which were small nucleolar RNAs and one was a small Cajal body-specific RNA. This result suggests that LPS-induced changes in WT MSCs were largely abolished in the MyD88−/− MSCs.
stat3 ablation abolished the effect of LPS in improving MSC-mediated cardioprotection following ischemia/reperfusion (I/R) injury
To examine the role of stat3 in MSC-conferred cardioprotective effect, hearts were randomized into three groups: saline, MSCs + LPS + scramble siRNA, and MSCs + LPS + stat3 siRNA. After establishing the mouse I/R model, MSCs (1 × 105 cells in 1 mL of PBS) were infused into the coronary circulation before global ischemia. The representative recording traces of LVP are shown in Fig. 5A–C. As shown in Fig. 5, LPS pretreatment improved the cardioprotection of scramble siRNA-transfected cells, but not stat3-ablated cells. Heart infused with LPS-pretreated, scramble siRNA-transfected cells shows better postischemic functional recovery, compared to vehicle and stat3-ablated cells, as evidenced by improved LVDP, +dp/dt, and −dp/dt. The postischemic functional recovery of the stat3 siRNA group was down to a level of the vehicle group (Fig. 5). Among the three groups, no differences were observed in EDP, suggesting similar I/R injury (Fig. 5G).

Ablation of stat3 abolished the effect of LPS in improving MSC-mediated protection. MSCs with or without stat3 siRNA transfection were infused into the coronary circulation.
Discussion
We herein report that LPS pretreatment improved MSC-mediated cardioprotection in mouse models of cardiac I/R injury. However, such LPS effects were abolished when using MSCs isolated from MyD88−/− mice. We also demonstrated that MyD88−/− MSCs proliferated much more slowly and failed to activate stat3 in response to LPS stimulation compared to WT MSCs. The ablation of stat3 abolished the protective effect of WT MSCs in isolated mouse heart I/R model. Most LPS-induced global gene expression changes were abolished in MyD88−/− MSCs. Collectively, this experimental evidence strongly supports the conclusion that LPS improves MSC-mediated cardioprotection by MyD88-dependent activation of stat3.
Ischemic heart disease occurs in ∼40% of the population older than 40 years and is the leading cause of death worldwide. During ischemia, the heart is deprived of oxygen and nutrients, leading to apoptosis and necrosis of cardiomyocytes and endothelial cells [31,32]. Subsequent restoration of blood flow results in additional damage to the myocardium, which is known as I/R injury [32 –34]. A more effective therapy is urgently needed and MSCs are emerging as a promising therapeutic agent for heart repair. In recent years, many clinical trials have been performed, accompanied by the improvement of LVDP, EF, and FS. However, such beneficial effects were modest and only lasted 6 months [35,36]; at 5 years follow-up, the helpful effects were no longer significant [37].
Researchers have proposed many hypotheses to explain mechanisms of MSC-enhanced cardiac function, including trans-differentiation, electrophysiological coupling, and paracrine protection [9]. However, clinical trials showed that retention of transplanted cells was low in the heart, most of which were lost through the vasculature and accumulated in lung, spleen, and pancreas; as a consequence, few cells were found engrafted in the heart [38]. In addition, survival of transplanted MSCs in the inflammatory environment of infarcted myocardium was low, as typically 90% of cells died within 1 week [39]. Thus, the number of transplanted cells was insufficient to explain significant cardiac function improvement after ischemic injury. A mechanism of paracrine factors was reported by many researchers to explain MSC-mediated heart function enhancement in the short term [9]. MSCs can secrete a broad variety of cytokines, chemokines, and growth factors, which promote neovascularization, cardiac metabolism, and cardiac regeneration. These paracrine factors could exert cardioprotection through the following mechanisms: (1) increasing blood flow within ischemic myocardium to alleviate cardiomyocyte necrosis and apoptosis; (2) reducing the distance between the ischemic tissue area and the nearest blood vessels through angiogenesis; (3) enhancing the permeability of blood vessel to plasma and micromolecules [40]; and (4) recruiting MSCs and HSCs of the circulating blood stem cells to the injured myocardium.
TLR4 is a type I transmembrane glycoprotein that is expressed in MSCs [41]. Upon stimulation by its ligand LPS, TLR4 activates two distinct signaling pathways that are MyD88 dependent or TRIF dependent, ultimately leading to the activation of transcription factors. LPS has an antiapoptotic effect in neonatal mouse myocytes exposed to hypoxia, but this effect of LPS was neutralized in MyD88 KO myocytes [42]. In this study, we demonstrated that LPS enhanced WT-MSC-mediated postischemic recovery after I/R injury, but not MyD88−/− MSC-mediated recovery. In addition, LPS stimulated a gene expression profile in WT cells, whereas most LPS-induced changes were abolished in MyD88−/− MSCs. Such findings indicate that TLR4 activates transcription factors through a MyD88-dependent signaling pathway, which is consistent with the results of Zhu et al. [42].
stat3 is a transcription factor that conducts a fundamental role in physiological homeostasis. Deregulated stat3 signaling is sufficient to induce dilated cardiomyopathy and adverse remodeling after MI [43,44]. stat3 is a multifaceted molecule in the heart and is responsible for communication between cardiomyocytes and cardiac fibroblasts, modulation of the microenvironment, and regulation of cardiac inflammation, namely participating in cardiomyocyte growth, survival, sarcomere architecture, energetics, and metabolism [43 –45]. In this study, we found that LPS can markedly improve MSC-mediated cardioprotection in isolated hearts of a mouse I/R model and that stat3 ablation in MSCs abolished these beneficial effects (Fig. 4). Because the experimental period was too short for neoangiogenesis to occur, such findings may suggest that stat3 modulates MSC-mediated protection by regulating the release of paracrine factors.
We reported previously that stat3 ablation in MSCs resulted in a decreased expression of the angiogenic factor vascular endothelial growth factor (VEGF), upregulation of the proinflammatory chemokine RANTES, and attenuated MSC-mediated cardioprotection [8]. It is possible that stat3 could exert its beneficial effect, at least in part, through the expression of paracrine factors and intracellular signaling molecules. In Table 1, the gene expression level of paracrine factors, including CCL2/monocyte chemotactic protein (MCP)1, CCL7, fibroblast growth factor (FGF)7, CCL9, Hif1a, NFKBIZ, ler3, Fas, and Pak3, was found to be enhanced by LPS induction in WT MSCs, while the expression of Cxcl12/SDF-1, lgf1, and lgfbp5 was reduced. Among the upregulated paracrine factors, CCL2/MCP1 had the highest, 4.99 log2-fold-change, followed by CCL7, which was enhanced 4.04 log2-fold. However, in the Myd88−/− MSCs, the effect of LPS on the global gene expression was abolished. CCL2 and CCL7 are both chemokines, which are a subgroup of cytokines with the specific function of chemoattraction (chemotactic cytokines) [46]. It is already known that chemokines interfere with key events following MI: they modulate the inflammatory response, the molecular and cellular composition of the scar, and the implicit remodeling of the ventricle and heart function [2]. CCL2 is referred to MCP1 or small inducible cytokine A2, which can recruit monocytes, memory T cells, and dendritic cells to the sites of inflammation produced by either tissue injury or infection. It was reported that in animal models, CCL2 is upregulated after MI, and is important in angiogenesis and collateralization both in vivo and in vitro [2]. CCL7, also known as MCP3, has been found to be crucially involved both in MSCs homing to the ischemic myocardium as well as in their intramyocardial migration and survival. Moreover, it was reported in a rat model that, overexpression of CCL7 induced homing of repeatedly administrated MSCs 1 month after MI, which in turn improved heart remodeling and preserved heart function [47].
LPS Induces Differential Gene Expression of Paracrine Factors and Intracellular Signaling Molecules in WT MSCs, But Not in MyD88−/− MSCs
FDR, false discovery rate; LPS, lipopolysaccharides; MSC, mesenchymal stem cell; TNF, tumor necrosis factor; WT, wild type.
The expression of FGF7 and NFKBIZ is enhanced 2.23 and 2 log2-fold by LPS, respectively. The FGF family is a highly complex family of growth factors. In the adult, FGFs are important in wound healing, tissue repair, metabolism, and homeostasis [48]. FGF7, also known as keratinocyte growth factor, is expressed specifically in mesenchyme. Treatment of injured epithelia with FGF7 results in an improved wound-healing response. The mitogenic and cytoprotective properties of FGF7 are already being put to advantageous use in the clinic [49].
Nuclear factor kappa beta inhibitor zeta is a protein encoded by Nfkbiz gene, and it functions as a transcriptional regulator of genes encoding intermediates of inflammation. The significantly increased expression of Nfkbiz was observed in cells subjected to I/R both in vivo and in vitro [50].
In summary, all these observations support the notion that the MyD88-dependent pathway(s) promote MSC-mediated cardioprotection through a MyD88/stat3-dependent, transcriptional induction of protective paracrine factors.
Footnotes
Acknowledgments
The work was supported by NIH R01CA213466 and the Startup funds provided by the Department of Medical and Molecular genetics, Indiana University.
Author Disclosure Statement
No competing financial interests exist.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.