
Abstract
Auditory signals are processed in multiple central nervous system structures, including the auditory cortex (AC). Development of stem cell biology provides the opportunity to identify neural stem cells (NSCs) in the central nervous system. However, it is unclear whether NSCs exist in the AC. The aim of this study is to determine the existence of NSCs in the postnatal mouse AC. To accomplish this aim, postnatal mouse AC tissues were dissected and dissociated into singular cells and small cell clumps, which were suspended in the culture medium to observe neurosphere formation. The spheres were examined by quantitative real-time polymerase chain reaction and immunofluorescence to determine expression of NSC genes and proteins. In addition, AC-spheres were cultured in the presence or absence of astrocyte-conditioned medium (ACM) to study neural differentiation. The results show that AC-derived cells were able to proliferate to form neurospheres, which expressed multiple NSC genes and proteins, including SOX2 and NESTIN. AC-derived NSCs (AC-NSCs) differentiated into cells expressing neuronal and glial cell markers. However, the neuronal generation rate is low in the culture medium containing nerve growth factor, ∼8%. To stimulate neuronal generation, AC-NSCs were cultured in the culture medium containing ACM. In the presence of ACM, ∼29% AC-NSCs differentiated into cells expressing neuronal marker class III β-tubulin (TUJ1). It was observed that the length of neurites of AC-NSC-derived neurons in the ACM group was significantly longer than that of the control group. In addition, synaptic protein immunostaining showed significantly higher expression of synaptic proteins in the ACM group. These results suggest that ACM is able to stimulate neuronal differentiation, extension of neurites, and expression of synaptic proteins. Identifying AC-NSCs and determining effects of ACM on NSC differentiation will be important for the auditory research and other neural systems.
Introduction
In the mammalian auditory system, sound is captured by the outer ear, conducted through the middle ear, and perceived by the inner ear. In the inner ear, sound waves are transformed into auditory signals by sensory hair cells, which in turn depolarize spiral ganglion neurons and initiate action potentials. The auditory signals are transferred from the inner ear to the brainstem via the cochlear nerve and then processed by multiple central nervous system structures, including trapezoid body, superior olivary complex, lateral lemniscus, inferior colliculi, medial geniculate nucleus, and eventually auditory cortex (AC) [1,2]. The auditory system is vulnerable to a number of insults, including ototoxic drugs, genetic disorders, aging, trauma, and infections. Degeneration of the mammalian auditory system is usually irreversible, which causes permanent hearing disorders [3,4]. In addition, head trauma to the AC area causes cortical deafness, which results in damage to the cells of the AC area to lead to unawareness of sounds [5]. Several approaches have been investigated both in clinics and laboratory to treat hearing loss, including hearing aids, bone-anchored hearing aids, cochlear implants, brainstem implants, midbrain implants, gene therapy, as well as stem cell-based cell replacement that is explored in this study [6 –11]. Stem cell-based replacement may prove to be an efficient approach to substitute damaged cell types, including neurons in the AC area [12]. It is noted that several critical issues are required to be addressed before application of stem cells to the treatment of hearing disorders, including identification of tissue-specific stem cells.
A general feature of stem cells is the ability to self-renew and differentiate into several cell types [13,14]. Many types of stem cells, including embryonic stem cells, neural stem cells (NSCs), mesenchymal stem cells, and hematopoietic stem cells have been studied previously [15 –18]. These stem cells have shown the ability to proliferate and differentiate into function cell types both in vitro and in vivo [15,17 –19]. NSCs usually present in the developing nervous system, which are able to differentiate into neurons, astrocytes, and oligodendrocytes. In the adult brain, NSCs are found in two NSC niches: subventricular zone and subgranular zone [20 –23]. Adult NSCs in these two stem cell niches are able to enter the cell cycle to proliferate, as well as differentiate into transient amplifying progenitors, neuroblasts, and eventually functional neural cell types. In the in vitro studies, NSCs are able to proliferate to form typical neurospheres in the suspension culture medium [24]. Therefore, suspension culture methods are usually applied to identify and isolate NSCs from cultures of nervous tissues [16,21,25].
In the auditory system, stem/progenitor cells have been identified in the auditory hair cell epithelium, spiral ganglion, and cochlear nucleus [16,26 –29]. These auditory stem/progenitor cells show limited capabilities in proliferation and differentiation into auditory cell types. However, it is unclear (i) whether NSCs exist in the AC, and (ii) whether there is a NSC niche in the AC. This study focused on identification/isolation of NSCs in the postnatal mouse AC. The differentiation ability of potential AC-derived NSCs (AC-NSCs) was also evaluated in this report.
Materials and Methods
Isolation and culture of postnatal mouse AC
This study was carried out in accordance with the recommendations of guidelines of Wayne State University Institutional Animal Care and Use Committee (protocol no. IACUC-18-01-0387). Postnatal day 3 (P3) Swiss Webster mice were decapitated, and the brain tissues were isolated and rinsed in cold 0.1 M phosphate-buffered saline (PBS) solution. As the previous publication described [30], the posterior intersection of cerebral hemispheres was located and identified as the auditory pathway region start line (Fig. 1A, B), whereas the auditory pathway region end line was determined as 1.2 mm anterior to the start line. The 1.2 mm-thick section of P3 mouse brain was placed in the culture dish containing cold 0.1 M PBS (Fig. 1B, the coronal image), in which the cerebral cortex region was determined within the white dot line. The cortex midline was identified and divided into 10 equally divided sections starting from point A (the anterior longitudinal cerebral fissure). The AC area, which was in the superior part of the temporal lobe, was identified within the 4th section along the cortex midline, ∼3.4th–4.0th of the above mentioned 10 sections. The section of the AC area was determined according to a previous publication [31]. To prevent contamination from surrounding tissues, the auditory cortex tissue (ACT) was harvested within the anatomical AC area (the blue box area in Fig. 1B). Therefore, tissues of ∼0.5 mm in diameter were usually collected as shown in the rectangle area in the coronal image. Following ACT harvest, the edges of collected tissues were trimmed to further prevent contamination from surrounding brain tissues (the red box area in Fig. 1B).

Identification and dissection of postnatal mouse AC.
Dissociation was performed using 0.025% Trypsin (Invitrogen) and mechanical trituration. Dissociated cells were cultured in the suspension culture medium, which contained Dulbecco's modified Eagle medium (DMEM)/F12, 1% N2, 2% B27, epidermal growth factor (20 ng/mL), fibroblast growth factor 2 (20 ng/mL), and 0.1% penicillin–streptomycin (all from Invitrogen) in a 37°C incubator supplied with 5% CO2. Six independent batches of AC primary cultures were performed in this study.
Generation of AC-derived neurospheres
AC primary cultures were observed daily using a phase contrast microscope (Leica). After 5–6 days, cells were collected and treated by TrypLE (Invitrogen) at 37°C, centrifuged to remove TrypLE, resuspended in the fresh suspension medium, gently triturated, and maintained in the incubator for the formation of spherical structures. Fresh suspension culture medium was added every 2–3 days. AC-derived spherical structures that had been passaged at least three times for over 15 days were included in the following experiments (Fig. 1C).
Proliferation assays
To evaluate cell proliferation, AC-derived spheres (ACSs) were dissociated, cultured, and passaged in the suspension medium as described above. During tertiary culture, 5-ethynyl-2-deoxyuridine (EdU, 200 ng/mL; Sigma) was added to the culture medium. After 48 h, cells were fixed in 4% paraformaldehyde containing PBS at room temperature for 15 min and blocked in 5% donkey serum (Jackson Immunoresearch) containing 0.2% Triton X-100 (Sigma) for 30 min. Samples were incubated in Click-iT reaction buffer, CuSO4, Alexa Fluor 555, and reaction buffer additive (Invitrogen) for 30 min. Nuclei were labeled by 4,6-diamidino-2-phenylindole (DAPI, universal nucleus marker; Invitrogen). Samples were observed and imaged using Leica 3000B epifluorescence microscope and/or SPE confocal microscope.
To calculate the doubling time of ACSs, the cell number of ACSs was determined on the first day of the primary, secondary, and tertiary cultures. The doubling time calculation was performed using our previously published methods [27,32]. In brief, the doubling time is (t1 − t0)/log2(c1 − c0). c1 = cell number at time point t1, c0 = cell number at time point t0.
To evaluate whether a singular ACS can proliferate to form spheres, dissociated ACTs were serially diluted to obtain singular cells, which were plated into 96-well plates (targeting one cell per well). The 96-well plates were observed on the cell dissociation day and the day after to ensure only one singular cell in each studied well. The wells containing one singular ACT cells were observed for 18 days.
Induction of AC-derived cells and astrocyte-conditioned medium preparation
After suspension culture for 15 days, AC-derived spherical structures were dissociated with TrypLE and seeded into the neural differentiation medium containing 50% DMEM/F12, 48% neurobasal medium, 1% fetal bovine serum (FBS; Hyclone), 55 nM 2-mercaptoethanol, 0.1% penicillin-streptomycin, and 20 ng/mL nerve growth factor (NGF; Invitrogen) on 0.1% gelatin-coated culture wells. This culture experiment aimed to guide neural differentiation.
Astrocyte-conditioned medium (ACM) was obtained as we previously reported [16,33]. In brief, the cortex tissues of newborn Swiss Webster pups were dissected and dissociated by trypsin and trituration. Dissociated cells were cultured in 98% DMEM/High glucose, 1% FBS, and 0.1% penicillin–streptomycin for 20–30 min for the attachment of potential astrocytes, followed by a full medium change to remove other cell types. Cells were passaged for 2–3 times, and the conditioned medium was collected and concentrated by 30 kDa molecular weight cutoff tubes (Millipore). Concentrated ACM was filtered by a 0.22 μm syringe filter. ACM was added to the neural differentiation medium at a ratio of 1:10.
AC-derived cells were cultured in the presence or absence of ACM for 4–6 days. At the end of the experiment, cell samples were harvested for the following gene expression evaluation or fixed for immunofluorescence examination.
Gene expression study
Total RNAs of the ACT, AC-NSCs, and AC-NSC-derived neurons (ACNs) were extracted by an RNeasy Mini Kit, followed by cDNA conversion using a QuantiTect Reverse Transcription Kit according to manufacturers' protocols (all from Qiagen). A Bio-Rad CFX system was used with SYBR Green Supermix (n = 3; Bio-Rad). The mean of quantification cycle (Cq) was analyzed by the regression model of the Bio-Rad CFX Manager software. The expression of the Gapdh gene was used as a reference to calculate the relative expression levels of studied genes [17,34,35]. The primers for quantitative real-time polymerase chain reaction (PCR) were listed in Supplementary Table S1.
Immunofluorescence
AC-NSCs and ACNs were fixed by 4% paraformaldehyde containing PBS, followed by the treatment of 5% donkey serum containing 0.2% Triton X-100 for 30 min at room temperature. Samples were incubated in primary antibodies at 4°C overnight, followed by corresponding secondary antibodies incubation at room temperature for 1–2 h. Primary antibodies used in this study were shown in Supplementary Table S2. Secondary antibodies included Alexa Fluor 405, 488, 549, and 647 conjugated donkey anti-mouse, goat, rabbit, or chicken antibodies (1:500; Jackson Immunoresearch). DAPI, the universal nucleus marker, was used to label all nuclei in the sample. Samples were observed and imaged by Leica 3000B epifluorescence microscope and/or Leica SPE confocal microscope.
Quantitative study and statistical analysis
In this research, samples for statistical analyses were collected from six independent primary culture experiments. Cells, neurite outgrowth, and synaptogenesis were counted and analyzed by the Cell Counter plugin module, linear measurement tool, and particle analyze module of the ImageJ software (NIH), as we previously reported [17,36]. The number of positive-labeling cells and the number of DAPI-positive cells were counted by the Cell Counter plugin module of the ImageJ software.
In general, n = 8–10 cell cultures per group were randomly selected from six independent experiments for each quantitative study, and the means are compared between different treatment conditions. For the quantification of immunofluorescence staining study, cell culture preparations were randomly collected from six independent experiments (n = 8 cell cultures per group). The percentage of positive cells was number of positive cells/number of DAPI-positive cells × 100%. For the quantitative study of neurite outgrowth, the length of three randomly selected neurites were collected from each cell culture preparations in the control and ACM groups (n = 8 cell cultures in each group from six independent experiments, respectively).
For the quantification study of synaptogenesis, SV2 puncta and SYNAPSIN/PSD93 colocalization images were collected (n = 10 cell cultures per group). The number and area (0.4–3 μm2) of synaptic puncta along the neuron–neuron connections were quantified by the particle analyze feature using our previously reported methods [16,17,33]. Normality test was performed by Statistical Package for the Social Sciences (SPSS). Student's t-test was applied for the analysis of the two-group study. P < 0.05 was considered as the criterion of statistical significance in this study.
Results
Identification and characterization of AC-NSCs in vitro
The NSC suspension medium has been used to isolate and maintain NSCs in primary cultures in previous reports [16,21], which was used to culture AC cells to identify potential NSCs in this study. In the primary culture, singular cells and small cell clumps were observed after dissociation of the P3 AC (Fig. 2A), which formed spherical structures in 3–5 days in the suspension culture medium. To test the ability of secondary sphere formation, primary spheres were dissociated to obtain singular cells and small cell clumps. It was found that these singular cells and small cell clumps grew into secondary spherical structures in the suspension culture medium in 3–5 days (Fig. 2B). Similarly, singular cells and small cell clumps were obtained following dissociation of secondary spheres, which aggregated to tertiary spheres in the suspension culture medium in 3–5 days (Fig. 2C). It usually takes at least 15 days to identify NSC spheres by this suspension culture method.
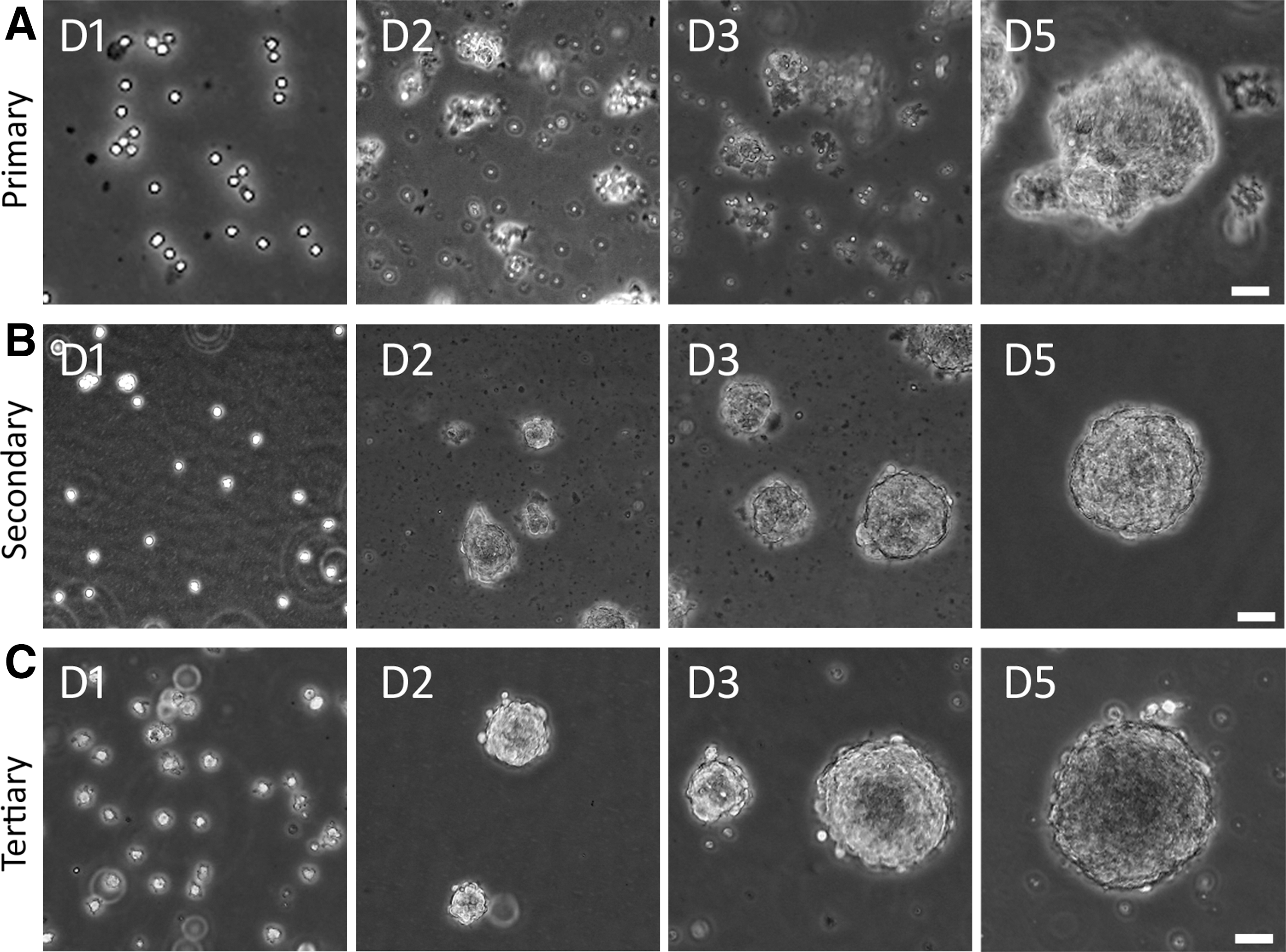
Identification of ACSs from P3 mouse AC.
NSCs are able to proliferate and express NSC genes/proteins, including SOX2 and NESTIN. The proliferation of AC-derived cells was suggested by the above-mentioned primary, secondary, and tertiary sphere formation. To further confirm cell proliferation, a complementary method, the EdU incorporation study was used, as EdU incorporates into DNA during cell proliferation (Fig. 3A). It was observed that 31.72% ± 2.82% (n = 8 cell cultures; mean ± standard error) cells in tertiary spheres incorporated EdU after 48 h culture, indicating that cells entered the S-phase of the cell cycle. The data suggest that AC-derived cells are able to enter the cell cycle to proliferate. To better describe cell proliferation, the doubling time of NSCs was calculated, which was 36.48 ± 2.64 h (mean ± standard error; Supplementary Fig. S1). In the single-cell culture of ACT, after serial dilution, singular cells were observed in 4 wells of the 96-well plate (one cell per well). These four single cells can grow into a small sphere on day 9 and expand into a large sphere at day 18 (Supplementary Fig. S2). These complementary approaches suggest the proliferation ability of ACT-derived cells.
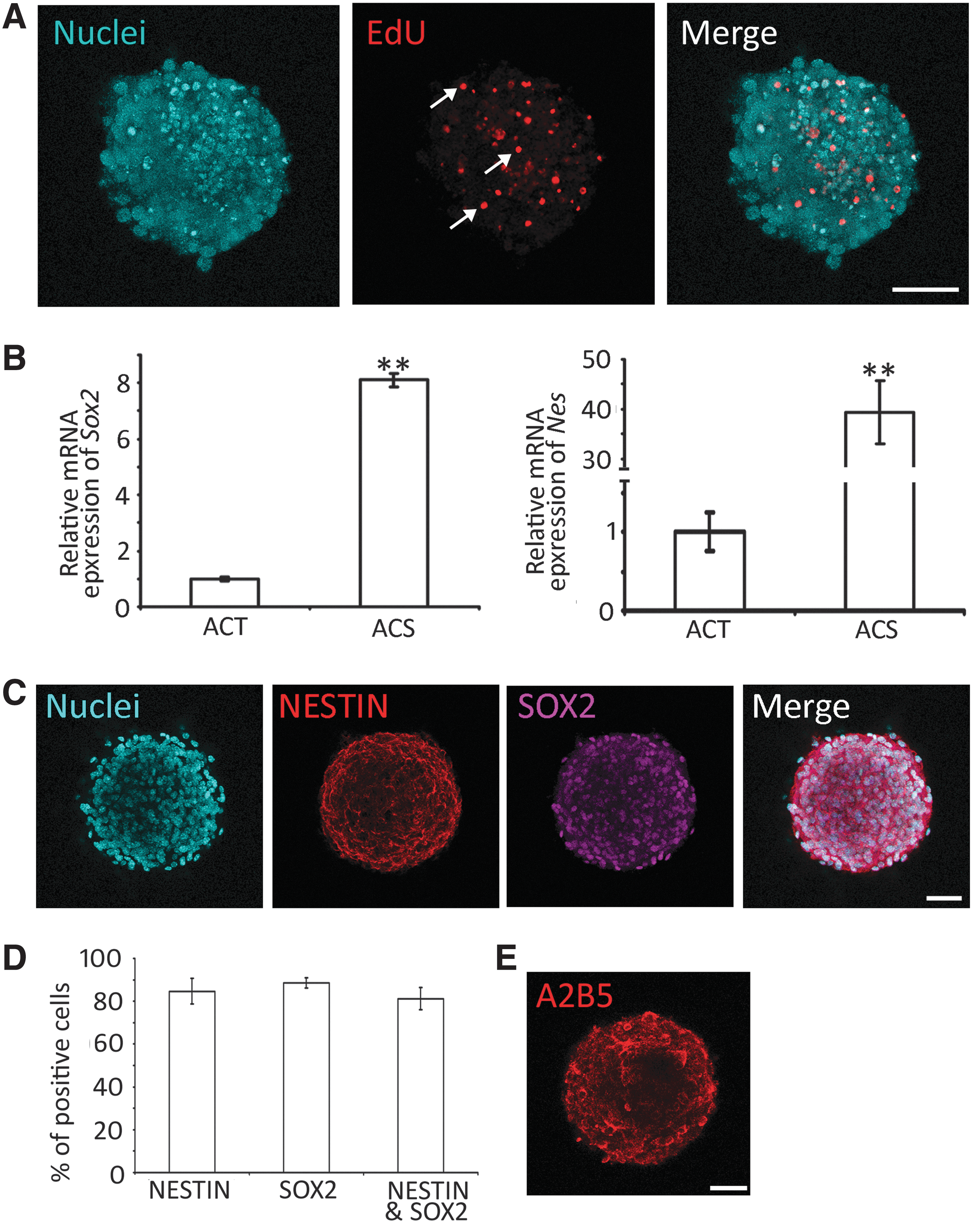
ACSs express NSC genes and proteins.
Next, we examined the expression of NSC genes/proteins. In real-time PCR, the ACT expressed NSC genes, including Sox2 and Nes (encoding NESTIN). However, tertiary spheres expressed much higher levels of Sox2 and Nes (Fig. 3B). In the protein expression study, tertiary spheres were immunostained with a number of NSC-specific antibodies. It was observed that 88.59% ± 2.33% and 84.68% ± 6.05% (mean ± standard error) cells expressed SOX2 and NESTIN, respectively, whereas 81.17% ± 5.16% cells were immunostained by both SOX2 and NESTIN antibodies (Fig. 3C, D). In addition to the expression of NESTIN and SOX2, the spheres also expressed A2B5 (oligodendrocyte precursor; Fig. 3E), but not TUJ1 (neuron marker), NeuN (mature neuron marker), or CD45 (hematopoietic marker; Supplementary Fig. S3).
Taken together, since AC-derived cells proliferated to form spherical structures and expressed NSC genes and proteins, they were termed AC-NSCs in this study.
Differentiation of AC-NSCs
NSCs are able to differentiate into neural cell types, including neurons, astrocytes, and oligodendrocytes [20 –23]. Since neurotrophins are critical for the development and differentiation of neural cell types [37 –39], AC-NSCs were exposed to neural differentiation culture medium containing NGF to stimulate neural differentiation. It was found that AC-NSCs attached and spread in the NGF-containing medium. Cells with neuronal morphology were observed using light microscope (Fig. 4A). To further characterize cell phenotype, gene expression of differentiated AC-NSCs was explored. In real-time PCR, the ACT, AC-NSC, and ACN expressed the similar level of the Neurod1 gene. The ACT and ACN expressed higher levels of Tubb3 (encoding Class III β-tubulin, recognized by TUJ1 antibodies) and Psd93 than the AC-NSC (Fig. 4B).

Differentiation of AC-NSCs.
To further determine protein expression of AC-NSC-derived cells, multiple-labeling immunofluorescence was explored. It was observed that AC-NSC-derived cells were immunostained by neuronal markers TUJ1, NEFL, and NEUROD1, as well as glial cell protein GFAP (Figs. 4C and 5A). Oligodendrocyte protein MOG (Myelin oligodendrocyte glycoprotein) immunostaining was hardly detected. To initiate evaluation of the function of the neurons, it was also observed that AC-NSC-derived cells were immunostained by sodium channel protein (Na-V) (Fig. 4C). As AC-NSC-derived TUJ1-expressing cells expressed multiple neuronal markers, they were termed as ACNs in this study. In the quantitative study, the percentage of TUJ1+ and GFAP+ cells were 7.68% ± 0.34% and 87.65% ± 1.14%, respectively (mean ± standard error; Fig. 5B, C). Very few cells were labeled by the oligodendrocyte antibody MOG, which was excluded from the quantitative study. It was also observed that a couple of cells were labeled by both GFAP and TUJ1 antibodies (Fig. 5A). A previous study showed that some glial protein-expressing cells have the ability to differentiate into neurons [40], which was also observed in this report. These GFAP-TUJ1 double-labeled cells were rarely found in this study.

ACM promotes neural differentiation of AC-NSCs.
Stimulation of the neuronal differentiation of AC-NSCs
ACM is known to stimulate synapse formation in the central nervous system and stem cell-derived neurons [16,33,41 –43]. However, it is unclear whether ACM is able to affect the cell fate determination of NSCs. To address this issue, the same batch of AC-NSCs were dissociated and seeded into two groups of cultures, the first group (control group) contained the control culture medium (the neural differentiation medium) in the absence of ACM, while the second group (ACM group) contained the control culture medium and ACM. It was observed that AC-NSCs differentiated into a number of neuron-like cells expressing TUJ1 in the presence of ACM (Fig. 5A). The quantitative study showed that 28.79% ± 1.64% and 7.68% ± 0.34% (mean ± standard error) AC-NSCs differentiated into TUJ1-expressing cells in the ACM and control groups, respectively (Fig. 5B), which was statistically significant (P < 0.01, Student's t-test, n = 8 cell cultures per group). This study suggests that ACM is able to stimulate neuronal differentiation of AC-NSCs.
Since the proportion of TUJ1-expressing cells was increased, the percentage of GFAP-expressing cells was investigated. It was observed that a number of AC-NSCs differentiated into glial-like cells expressing GFAP (Fig. 5A). In the quantitative study, 68.51% ± 2.54% and 87.65% ± 1.14% (mean ± standard error) AC-NSCs differentiated into GFAP-expressing cells in the ACM and control groups, respectively (Fig. 5C). The statistical analysis showed significant difference between the ACM and control groups (P < 0.01, Student's t-test, n = 8 cell cultures per group). This study suggests that ACM increases the neuronal differentiation of AC-NSCs at the cost of decreased number of GFAP-expressing cells.
ACM stimulates neurite extension and expression of synaptic protein of ACNs
A significant feature of neurons is their neurite outgrowth. To examine whether ACM plays a role in neurite extension of ACNs, AC-NSCs were cultured in the presence or absence of ACM in the neural differentiation medium (Fig. 6A). It was observed that significantly longer neurite outgrowth was observed in the ACM group (159.31 ± 6.06 μm), whereas it was 80.87 ± 3.04 μm in the control group (mean ± standard error; Fig. 6B). Statistical analysis showed significant difference (P < 0.01, Student's t-test, n = 8 cell cultures per group), suggesting that ACM is able to promote the extension of neurites of ACNs.

ACM promotes neurite outgrowth of ACNs.
SV2 immunostaining was used to determine whether ACM stimulated the expression of synaptic proteins of ACNs. In the control group, very few SV2 puncta were observed in the soma and neurites of ACNs (Fig. 7A, B). However, a number of SV2 puncta were identified in the presence of ACM (Fig. 7C, D). Statistical analysis showed significant differences in the SV2 expression between the ACM and control groups (P < 0.01, Student's t-test, n = 10 cell cultures per group).

ACM stimulates the synaptogenesis of ACNs by SV2 immunostaining.
Pre- and postsynaptic protein immunostaining was investigated to further characterize expression of synaptic proteins of ACNs. It was found that presynaptic protein SYNAPSIN and postsynaptic protein PSD93 were observed in ACNs of the control and ACM groups (Fig. 8A, B). In addition, significantly more SYNAPSIN and PSD93 colocalization puncta were identified in the ACM group (Fig. 8C, D; P < 0.01, Student's t-test, n = 10 cell cultures per group).

ACM stimulates synaptogenesis of ACNs by pre- and postsynaptic protein immunostaining.
Taken together, ACM is able to stimulate extension of neurites and expression of synaptic proteins in ACNs.
Discussion
In this study, AC-NSCs were isolated from postnatal mouse AC and maintained in the suspension culture. These AC-NSCs are able to enter the cell cycle to proliferate and form NSC spheres in vitro. AC-NSCs express NSC genes and proteins SOX2 and NESTIN. These AC-NSCs are able to differentiate into neural cell types, including neurons and glial cells. Interestingly, it is found that ACM is able to stimulate neuronal differentiation and neurite extension of AC-NSCs. ACM is also observed to enhance expression of synaptic proteins of ACNs.
The self-renewal and differentiation ability of stem cells has attracted efforts to identify and guide them into hearing cell types. For instance, pluripotent embryonic stem cells have been explored to differentiate into sensory hair cells and spiral ganglion neurons [11,44 –48]. Tissue-specific stem cells, including hair cell progenitor cells and spiral ganglion-derived NSCs have been identified and induced into cells expressing hair cell or spiral ganglion genes and proteins [16,32,33,46, and 49 –52]. However, the existence of NSCs in the AC has not been determined previously.
These AC-NSCs are able to enter the cell cycle to proliferate, which is supported by the complementary approaches of cell population doubling time and EdU incorporation assay. Moreover, AC-NSCs are able to form secondary and tertiary neurospheres in the suspension culture. AC-NSCs have been maintained in the suspension culture medium for at least 10 passages. These data suggest the proliferation ability of AC-NSCs.
In the real-time PCR gene expression study, AC-NSCs express higher levels of NSC genes Sox2 and Nes, whereas the ACT expresses very low levels of these NSC genes. We found that expression of Sox2 was higher in the neurospheres, whereas levels of Nes were remarkably higher in the neurospheres. Higher expression of NSC genes in the neurosphere is likely due to high percentage of NSCs in vitro. The difference for the increased levels of Sox2 versus Nes is still obscure. One possibility is likely due to the nature of these genes/proteins: Nes encodes an intermediate filament protein Nestin that is widely distributed in the cell, whereas Sox2 encodes a transcription factor Sox2. Further studies may be needed to elucidate this in the future study.
In the protein expression study, immunofluorescence reveals that AC-NSCs express multiple NSC proteins SOX2 and NESTIN. NESTIN is an intermediate filament protein and SOX2 is a transcription factor expressed in the nucleus. The combination of these two proteins is usually used to indicate the presence of NSCs [16,17,33]. The quantification study of NESTIN and SOX2 was performed in AC-NSCs. Taken together, these data indicate the identification/isolation of NSCs from AC tissues in vitro.
AC-NSCs are able to differentiate into several neural cell types, including neurons and astrocytes. In the gene expression study, AC-NSCs express very low levels of neuronal gene Tubb3 and Psd93, whereas induced cells express higher levels of these genes. This study indicates the neural differentiation ability of AC-NSCs. However, the neuronal differentiation rate is relatively low ∼8% in NGF-containing medium. To stimulate neuronal differentiation of AC-NSCs, several approaches have been tested. A proper microenvironment may be required for the differentiation of NSCs. In the nervous system, astrocytes are the neighboring cells of neurons. It is speculated that astrocyte-released cytokines and growth factors may play a role in the differentiation of AC-NSCs. Therefore, ACM was collected from the astrocyte culture and was added to the neural differentiation medium to evaluate its effect on neural differentiation. Indeed, neuronal differentiation rate is significantly increased (∼29%) in the ACM group, which suggests its stimulation effects on neuronal differentiation. Meanwhile, it is also observed that astrocyte differentiation rates are ∼88% and 69% in the ACM and control groups, respectively, suggesting that the proportion of GFAP-expressing cells decreases as the neuronal differentiation increases. In addition, our new data showed that ACNs express sodium channel protein Na-V, suggesting the function of the neurons at least at the protein expression level.
ACM has been reported to stimulate synapse formation in the central nervous system as well as in stem cell-derived neurons [16,41]. Therefore, the synaptogenic ability of ACM was investigated in ACNs. It is observed that SV2 puncta staining is significantly increased in the ACM group, which suggests the synaptogenic effects of ACM. Similarly, the presynaptic protein SYNAPSIN and postsynaptic protein PSD93 immunostaining as well as confocal microscope-based colocalization studies show significantly more puncta in the ACM group. These observations suggest the synaptogenesis ability of ACM, which is consistent with previous reports [16,33,41]. Interestingly, ACM was found to enhance the neurite extension of ACNs, ∼159 and 81 μm in the ACM and control groups, respectively (P < 0.01, Student's t-test), which has not been reported previously and may deserve an independent study in the future.
In summary, AC-NSCs are identified in this study, which possess several key NSC features, including self-renewal, formation of neural spheres, expression of NSC genes/proteins, and differentiation into neural cell types. ACM is able to stimulate neural differentiation and expression of synaptic proteins of ACNs. Future studies will include determination of proper stem cell niche that are required for the maintenance and differentiation of AC-NSCs. The in vitro identification of NSCs from the ACT may stimulate the related in vivo work, which may indicate the possibility to regenerate or replace auditory neurons in the case when neurons are degenerated or dysfunctional in hearing loss in the future. Understanding AC-NSC and its stem cell niche will not only be important for the auditory system but also be helpful for other neural systems.
Footnotes
Acknowledgments
The authors thank Dr. Bingqing Hao and Li Tian for valuable comments and technical support as well as DSHB and NeuroMab for antibodies. This study is supported by NIH R01 grant to ZH (R01DC013275).
Author Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.