
Abstract
Engraftment of oligodendrocyte progenitor cells (OPCs), which form myelinating oligodendrocytes, has the potential to treat demyelinating diseases such as multiple sclerosis. However, conventional strategies for generating oligodendrocytes have mainly focused on direct differentiation into forebrain- or spinal cord–restricted oligodendrocytes without establishing or amplifying stem/progenitor cells. Taking advantage of a recently established culture system, we generated expandable EN1- and GBX2-positive glial-restricted progenitor-like cells (GPLCs) near the anterior hindbrain. These cells expressed PDGFRα, CD9, S100β, and SOX10 and mostly differentiated into GFAP-positive astrocytes and MBP-positive oligodendrocytes. RNA-seq analysis revealed that the transcriptome of GPLCs was similar to that of O4-positive OPCs, but distinct from that of rosette-type neural stem cells. Notably, engrafted GPLCs not only differentiated into GFAP-positive astrocytes but also myelinated the brains of adult shiverer mice 8 weeks after transplantation. Our strategy for establishing anterior hindbrain-specific GPLCs with gliogenic potency will facilitate their use in the treatment of demyelinating diseases and studies of the molecular mechanisms underlying glial development in the hindbrain.
Introduction
In recent years, remarkable progress has taken place in stem cell research aimed at unraveling the details of human development and treating incurable diseases [1 –3]. Neural stem cells (NSCs), which give rise to neurons and glial cells in the central nervous system (CNS), are considered promising candidates for treatment of neurodegenerative diseases such as Alzheimer's, Parkinson's, and Huntington's [4 –7].
In light of the risks of immune responses to allogeneic transplants, NSCs derived from induced pluripotent stem cells (iPSCs) are considered to be ideal sources of material for cell therapy [8].
However, the restricted fate specification of NSC populations and their gradual transition from neurogenesis to gliogenesis have hampered their preclinical and clinical applications for the treatment of demyelinating diseases [9,10]. Moreover, NSCs and their progenies are heterogeneous depending on the brain region and developmental stage [11 –13], making it necessary to establish positionally specified NSCs or glial cells as regenerative medicine to alleviate regionally occurring diseases such as multiple system atrophy (MSA, MSA-P with striatonigral degeneration, and MSA-C resulting from olivopontocerebellar atrophy) [14,15] and as in vitro model for drug evaluation to understand progression in diseases that exhibit region-specific susceptibility, including Parkinson's disease and cerebellar ataxia [16 –18].
In this regard, previous studies showed that region-specific NSCs or their progeny can be induced by dose-dependent activation of WNT signaling [19 –21]. Even though regional identity can be further confirmed based on products of neural differentiation (eg, tyrosine hydroxylase neurons in midbrain; serotonin neurons in hindbrain), it is not fully understood whether glial cells have positionally distinct identities [22].
Moreover, previously established NSC lines, including primitive NSCs (pNSCs) and rosette type NSCs (rNSCs), have limited potential to differentiate into oligodendrocytes and remain in the immature O4-positive stage rather than the MBP-positive mature stage [23 –25]. Accordingly, it is necessary to establish more gliogenic progenitor cells committed to mature oligodendrocytes to treat demyelinating diseases and understand the pathogenesis of glial development [10,26,27].
In this context, Wang et al. developed stepwise protocols for the establishment of glial-restricted progenitor cells (GPCs) and PDGFRα+ oligodendrocyte progenitor cells (OPCs) from OLIG2+/NKX2.2+ pre-OPCs.
However, questions persist about the cellular characteristics of GPCs and PDGFRα+ OPCs, including how long these cells can be propagated, how they acquire glial characteristics, and how their regional markers change [28,29]; this is especially true when their clinical applications and disease modeling are taken into consideration. Furthermore, despite the importance of understanding cerebellar development and diseases, including cerebellar ataxia and MSA [30,31], differentiation of human pluripotent stem cells (hPSCs) into hindbrain-specific oligodendrocytes has not been well studied, in contrast to the large number of studies into the forebrain and spinal cord [32 –34].
The cerebellum, a major structure of the hindbrain, has crucial roles in controlling coordination, balance, motor skill, and posture. In rodent development, the majority (∼94%) of cerebellar oligodendrocytes originate from ventral rhombomere 1 (En1+ area) [35,36]. Although the origin of human cerebellar oligodendrocytes is largely unknown, it is very likely that human oligodendrocytes with hindbrain identity originate from the same segment (ventral rhombomere 1) as in the rodent [3].
Recently, Tailor et al. established several anterior hindbrain (caudal to the midbrain–hindbrain boundary, corresponding to rhombomere 1) neuroepithelial stem (hbNES) cell lines from human embryos (week 5–7) [37]. Although these cells exhibited high neurogenic potency, these findings prompted us to investigate whether hbNES cells could be generated from hPSCs and whether gliogenic progenitor cells could be generated with anterior hindbrain identity which might recapitulate the origin of human cerebellar oligodendrocytes.
In this study, we investigated whether expandable EN1+/GBX2+ glial-restricted progenitor-like cells (GPLCs) with anterior hindbrain identity can be differentiated from hPSCs through stepwise developmental stages (primitive, rosette, intermediate, and glial restricted) and determined how the generated GPLCs acquire gliogenic properties using RNA-seq. We successfully established expandable GPLCs that could differentiate into both GFAP+ astrocytes and MBP+ oligodendrocytes in vitro and in vivo (shiverer mice). Furthermore, RNA-seq revealed that regional identity was maintained from NSCs to GPLCs and that the machinery related to mRNA transport, protein translation, and targeting was upregulated as the cells shifted to the gliogenic phase.
Thus, our approach for generating anterior hindbrain-specific GPLCs will facilitate clinical studies of various demyelinating diseases and provide a useful tool for the development of disease models of hindbrain development.
Materials and Methods
Cell culture and conditions
H9-hESCs (WiCell Research Institute, Madison, WI) and BJ-iPSCs (previously established [38]) were maintained in E8 medium on dishes coated with Matrigel (BD Biosciences Clontech, Palo Alto, CA). For differentiation into pNSCs, dissociated iPSCs and embryonic stem cells (ESCs) were seeded at 1 × 105 cells/well in Matrigel-coated six-well plates, and medium was replaced with LSC medium containing 10 ng/mL LIF (Leukemia Inhibitory Factor, EMD Millipore, Burlington, MA), 2 μM SB431542 (Tocris, Bristol, United Kingdom), and 1 μM Chir99021 (Tocris). Basal medium was used to make LSC, FEP, neural, and glial differentiation medium, consisting of DMEM/F12 supplemented with 543 μg/mL sodium bicarbonate (Sigma-Aldrich, St. Louis, MO), 1× B-27 Supplement without vitamin A (Thermo Fisher Scientific, Waltham, MA), 1× N-2 Supplement (Thermo Fisher Scientific), 1% penicillin–streptomycin, 1%
In vitro differentiation into neurons, astrocytes, and oligodendrocytes
For neuronal differentiation, rosette-type NSCs were seeded at low density on poly-L-ornithine (PLO)/laminin-coated plates and cultured in neural differentiation medium consisting of Basal medium supplemented with 10 ng/mL brain-derived neurotrophic factor (BDNF) (Peprotech) and 10 ng/mL glial cell derived neurotrophic factor (GDNF) (Peprotech). For glial differentiation, rosette-type NSCs were seeded on PLO/laminin-coated plates and cultured in glial differentiation medium consisting of Basal medium supplemented with 10 ng/mL NT-3 (Peprotech), 10 μM forskolin (Tocris), 60 ng/mL 3,3′,5-triiodo-L-thyronine (T3; Sigma-Aldrich), 20 μg/mL ascorbic acid (Peprotech), and 25 μg/mL insulin (Sigma-Aldrich). To promote glial specification, neurospheres were suspended in PDGF medium consisting of Basal medium supplemented with 20 ng/mL PDGF-AA (Peprotech), 10 ng/mL IGF-1 (Peprotech), 10 ng/mL NT-3, 10 μM forskolin, 60 ng/mL T3, and 10 μg/mL insulin (Sigma-Aldrich) for 2 or 3 weeks.
RT-PCR and qPCR analyses
For RT-PCR and qPCR, cDNA was amplified in triplicate. Negative controls included a reverse transcription–negative blank of each sample and a no-template blank. Gene expression levels were normalized against the corresponding level of GAPDH, which was used as an internal control. Primers used for RT-PCR and qPCR are listed in Supplementary Table S1.
Immunostaining
Cells were fixed for 10 min at room temperature in 4% paraformaldehyde, rinsed thrice with phosphate-buffered saline (PBS), and then permeabilized for 15 min at room temperature in PBS containing 0.2% Triton X-100. The fixed permeabilized cells were blocked with 5% normal donkey serum and 0.01% Triton X-100 for 1 h and then incubated with the appropriate primary antibodies overnight at 4°C. To label A2B5 and O4, nonpermeabilized cells were incubated with the appropriate antibody. Following the primary antibody exposure, the cells were washed thrice with PBS and incubated with the appropriate fluor-conjugated secondary antibodies at a dilution of 1:500 for 1 h at room temperature. Nuclei were counterstained with 1 μg/mL 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich) for 5 min, and the samples were rinsed thrice with PBS. Images were obtained on an Olympus 1 × 81 inverted fluorescence microscope.
For immunostaining of brains, samples were embedded in O.C.T. compound (Tissue-Tek, Sakura Finetek U.S.A., Inc., Torrance, CA) and cryosectioned at a thickness of 10 μm. Sections were blocked with 2% normal donkey serum and 0.2% Triton X-100 in PBS for 1 h, incubated overnight at 4°C with the indicated primary antibodies, and then incubated at room temperature for 1 h with the appropriate Alexa Fluor 488– or 594–conjugated secondary antibodies (Thermo Fisher Scientific). Nuclei were counterstained with 1 μg/mL DAPI (Sigma-Aldrich) for 5 min. The samples were rinsed thrice with PBS and observed under an Olympus confocal laser scanning microscope. Antibodies used for immunocytochemistry are listed in Supplementary Tables S2 and S3.
Flow cytometry
To analyze expression of A2B5, PDGFRα, and O4, cells were harvested with Accutase, rinsed thrice with cold PBS, blocked with 5% normal donkey serum for 15 min, stained with the appropriate primary antibodies for 30 min, incubated with fluor-conjugated secondary antibodies for 30 min, and then fixed with 0.5% paraformaldehyde (PFA). To label EN1, cells were permeabilized with 0.1% Triton X-100 during the blocking process. All flow cytometry analyses were performed on a FACSVerse flow cytometer (BD Biosciences, San Jose, CA).
Transplantation into shiverer mice
O4+ 200,000 H9-hESCs (n = 6) or iPSC-derived glial-restricted progenitor-like cells (GPLCs; n = 6) were transplanted into the corpus callosum (coordinates: AP = +0.9 mm, ML = 0 mm, DV = −2.8 mm) of 4-week-old homozygous shiverer mice (The Jackson Laboratory, Bar Harbor, ME). O4-positive cells were purified by Magnetic-Activated Cell Sorting (MACS; Miltenyi Biotec). Before transplantation, mice were randomly selected and examined in a blinded manner. Cells were delivered through a Hamilton syringe (Hamilton, Reno, NV) on a stereotaxic frame instrument (David Kopf Instruments, Tujunga, CA). All animals, including the PBS-treated group, received 5 mg/mL cyclosporine daily starting the day before transplantation. Eight weeks after transplantation, mice were anesthetized and perfusion-fixed with 4% paraformaldehyde (for IHC), 2% paraformaldehyde, and 2.5% glutaraldehyde (for transmission electron microscopy [TEM]). All animal experiments were performed in accordance with procedures approved by the Institutional Animal Care and Use Committee of Korea University.
TEM: sample preparation and analysis
Shiverer mice were anesthetized and perfused with normal saline, followed by 2% paraformaldehyde/2.5% glutaraldehyde in 0.1 M phosphate buffer (PB, pH 7.4). Brains were removed and stored overnight at 4°C in the same fixative. Small blocks of sectioned brains using brain matrix were rinsed thrice with PB for 10 min and then postfixed in 1% osmium tetroxide in 0.1 M PB for 90 min. Tissues were dehydrated through a graded ethanol series and embedded in epoxy resin mixture. Ultrathin sections (70 nm) were cut using an ultramicrotome (UC7; Leica Microsystems, Wetzlar, Germany) and mounted on 200-mesh grids contrasted with uranyl acetate and lead citrate. Transmission electron microscopic images (15,000–60,000 × ) were randomly acquired on a transmission electron microscope (H-7650, Hitachi, Tokyo, Japan) at an acceleration voltage of 80 kV.
RNA-seq data analysis
Total RNA extraction from iPSCs, ESCs, NSCs, IPC, GPLC, and BJ fibroblasts was performed using TRIzol reagent (Thermo Fisher Scientific), and genomic DNA was removed by DNase I. RNA quality was checked on an Agilent 2100 Bioanalyzer (RNA Integrity Number [RIN] > 8). cDNA libraries were constructed using the TruSeq RNA library Prep Kit (Illumina, San Diego, CA) by Theragen Etex Bio Institute (Korea), and libraries were sequenced on an Illumina HiSeq 2500 system. Gene expression levels were measured with Cufflinks v2.1.1 using the latest release of the Ensembl gene annotation database. To improve the accuracy of measurement, the multi-read-correction and frag-bias-correct options were applied; all other options were set to default values. Differentially expressed genes (DEGs) were identified using Cuffdiff with a false discovery rate (FDR) <0.05 and further screened by two- or fourfold changes of Fragments Per Kilobase of transcript per Million mapped reads (FPKMs) in at least one sample of the group. RNA-seq data are available from the NCBI GEO database (accession number GSE117664).
Statistical analysis
Data were analyzed by unpaired two-tailed Student's t test or one-way ANOVA. Data are shown as mean ± standard deviation (SD) for three to six replicates. P < 0.05 was considered statistically significant. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Results
Generation of rosette-type NSCs from pNSCs
To generate GPCs with anterior hindbrain identity, we modified the recently established culture systems based on developmental processes (Fig. 1A) [20,23,25,33]. Recently, Lu et al. established a differentiation method to induce rhombomeric segments 2–3 (HOXA2+/EN1−) by modulating WNT activation in a dose-dependent manner (1.4 μM CHIR99021) [20].
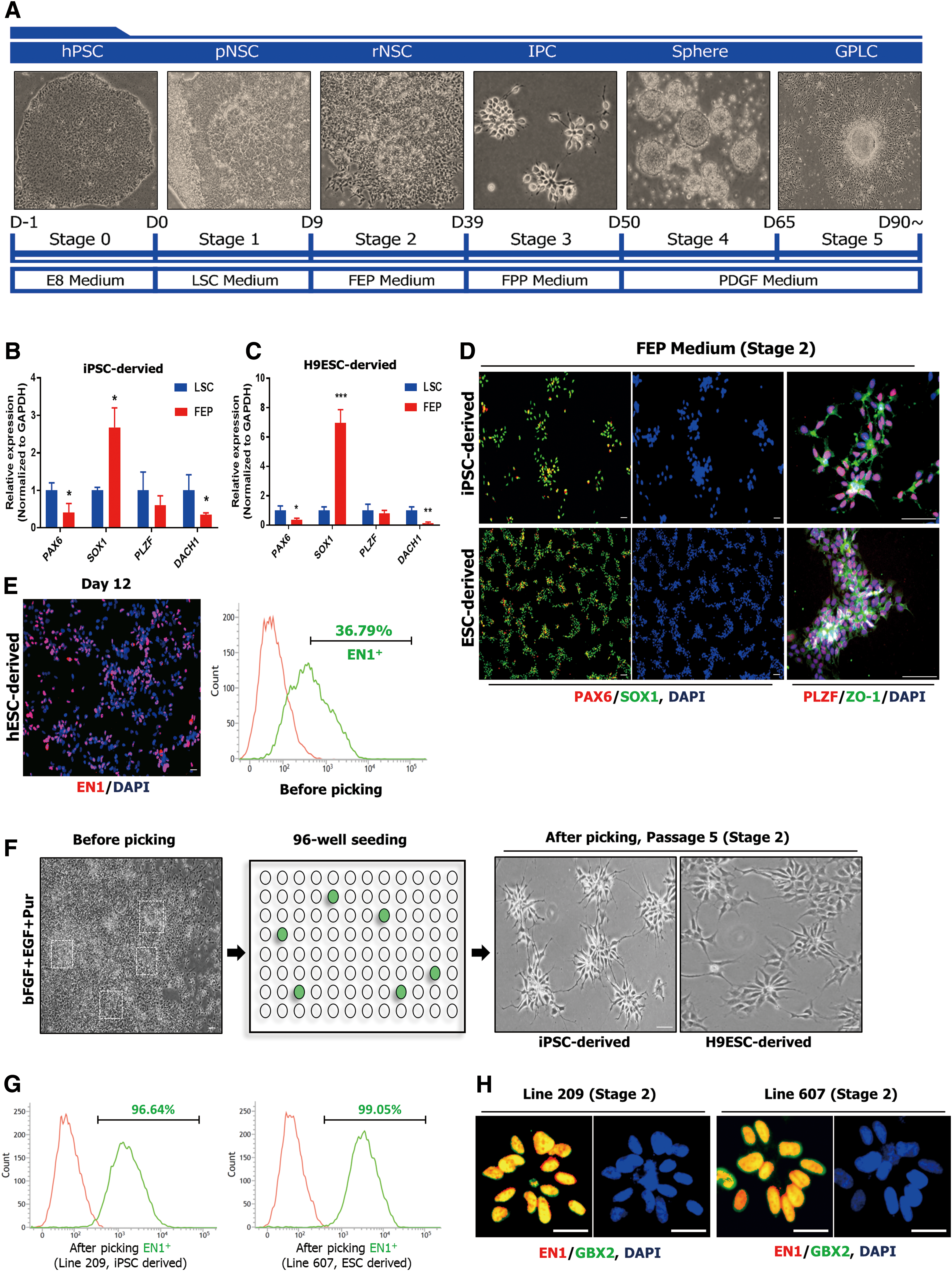
Generation of homogeneous EN1- and GBX2-positive neural stem cells.
To induce near rhombomere 1 identity (no expression of any HOX gene) [39,40], a feature of cerebellar oligodendrocytes during rodent development [35], we used 1.0 μM CHIR99021 in combination with LIF and SB431542, an inhibitor of activin-nodal signaling which have been used to promote neuroectodermal differentiation (LSC culture medium) during initial periods of differentiation.
Next, we subcultured neuroepithelial colonies at low density onto Matrigel-coated dishes and replaced the medium with FEP medium to induce ventralization [25,32]. After 8 days, radial-shaped colonies appeared (Fig. 1A, F) and we determined whether FEP medium could induce the expression pattern of hbNES cells [37]. In contrast to LSC medium, in FEP medium the yield of SOX1 increased by 2.6-fold (Fig. 1B) and 6.9-fold (Fig. 1C), respectively, but expression of PAX6 and DACH1 decreased (Fig. 1B, C). Consistent with the upregulation of SOX1 expression, immunocytochemistry confirmed that virtually all (>99%) hESC- and hiPSC-derived cells were positive for SOX1 and exhibited typical rosette-specific structures, as observed previously for hbNES cells [37] (Fig. 1D).
However, we could not increase the proportion of EN1+ cells in this culture (less than 60%) (Fig. 1E). To isolate EN1+ populations, each colony was manually picked, attached to a 96-well plate, propagated until passage 5 (Fig. 1F), and then characterized by flow cytometry.
During the screening period (until passage 5), cells exhibiting highly homogenous populations with apical–basal patterns and high proliferation rates were selected and propagated (Stage 2). Line 607, derived from H9-hESCs, contained a nearly homogeneous (>99%) EN1-positive population, as did Line 209, which was derived from hiPSCs (Fig. 1G).
To assess regional specification into anterior hindbrain, we examined expression of GBX2, another marker of anterior hindbrain. Immunocytochemical analysis revealed that Lines 607 and 209 strongly coexpressed GBX2 and EN1 (Fig. 1H). Accordingly, we designated these EN1- and GBX2-positive rosette-type NSC lines (Lines 607 and 209) as H9-rNSCs and iPSC-rNSCs, respectively.
EN1- and GBX2-positive NSCs maintain anterior–ventral hindbrain specific spatial properties
We next asked whether these selected NSCs could maintain rosette identity in FEP culture conditions. Indeed, at passage 10, we observed that PAX6 expression was markedly downregulated and that glial markers BLBP, SOX9, and OLIG2 were highly expressed (Fig. 2A, B). Expression of PLZF was also downregulated, but typical rosette structure and expression of ZO-1 were maintained (Fig. 2A, B). Further characterization revealed that iPSC-rNSCs proliferated stably beyond passage 50 and maintained a stable karyotype despite undergoing numerous cellular divisions (Fig. 2C, D).
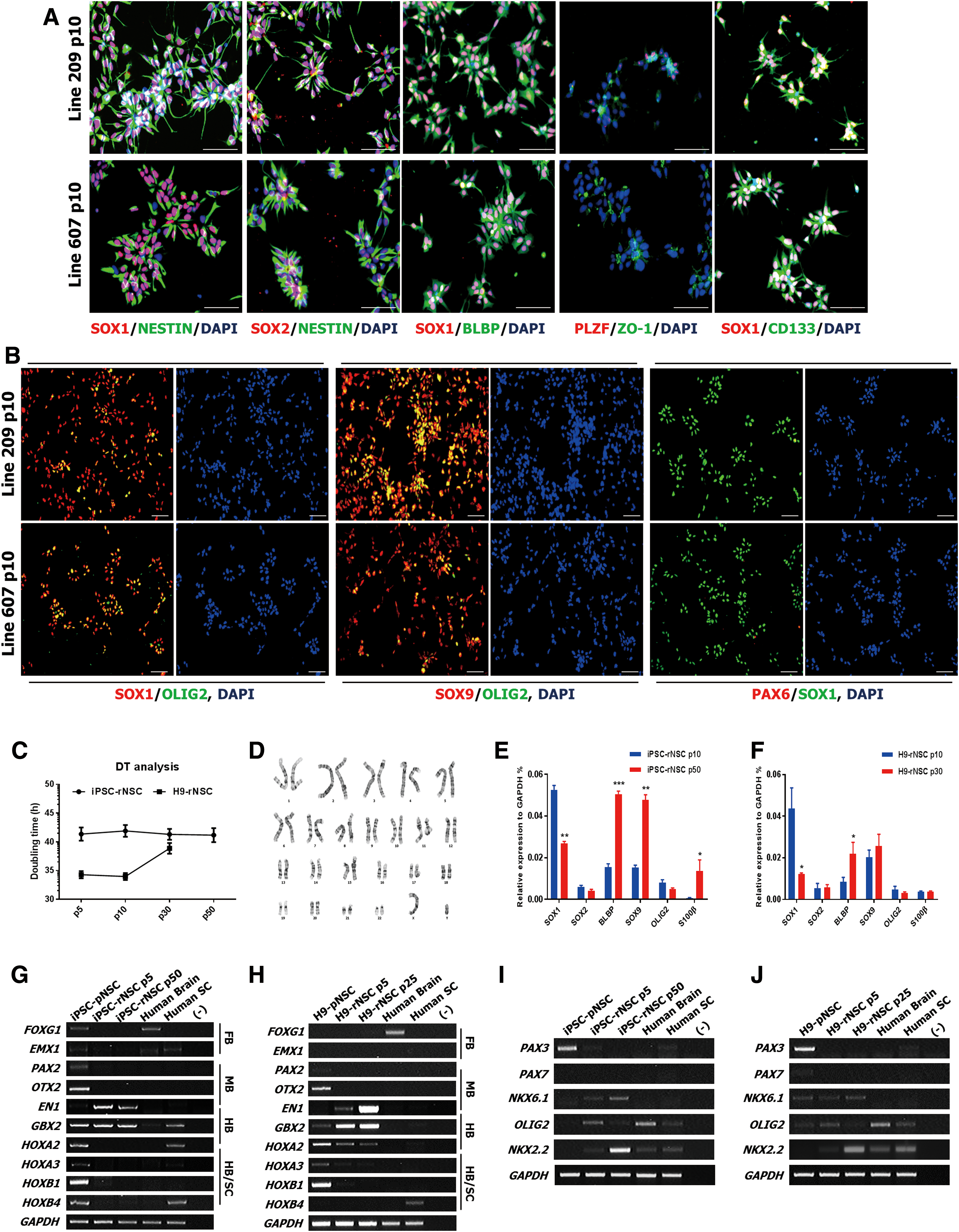
EN1-positive neural stem cells can maintain anterior–ventral hindbrain restricted spatial properties.
Interestingly, real-time PCR data showed that glial markers BLBP and SOX9 were gradually upregulated over the course of passage in both iPSC-rNSCs (Fig. 2E) and H9-rNSCs (Fig. 2F), whereas expression of SOX1 was reduced, providing evidence that continuous FEP culture conditions induce a developmental shift from rosette stage to glial stage, mimicking in vivo development (Supplementary Fig. S1A–C) [25,41].
CNS development is associated with differentiation of rosette-type NSCs into glial cells through the caudal–rostral and dorsal–ventral axes [42 –44]. Hence, we investigated whether rNSCs could adopt another regional identity following passage. RT-PCR analysis of various regional markers revealed that pNSCs cultured in LSC medium heterogeneously expressed markers of regions, including the midbrain, hindbrain, and spinal cord (Fig. 2G, H). Interestingly, iPSC-rNSCs mainly expressed EN1 and GBX2 and maintained regional identity by passage 50 (Fig. 2G). These results were consistent with those obtained in the H9-ESC line (Fig. 2H and Supplementary Fig. S1D), but H9-rNSCs also expressed some HOXA2, a marker of rhombomeres 2 and 3 [20].
In the D-V axis, representative dorsal markers PAX3 and PAX7 were not detected, whereas typical ventral markers NKX6.1, OLIG2, and NKX2.2 were expressed in both iPSC-rNSCs and H9-rNSCs (Fig. 2I, J and Supplementary Fig. S1E). Notably, expression of NKX2.2 was upregulated following passage, providing evidence that continuous FEP culture conditions induce more ventral patterning.
Together, the results show that we were successful in establishing iPSC-rNSC and H9-rNSCs possessing an anterior–ventral hindbrain identity similar to that of ventral rhombomere 1.
H9-rNSCs and iPSC-rNSCs have high neurogenic potential despite expressing glial genes
To assess neuronal differentiation potential, we cultured iPSC-rNSCs and H9-rNSCs in neuronal differentiation medium containing BDNF and GDNF instead of FGF2 and EGF. After 2 weeks of growth factor withdrawal, most cells were positive for Tuj1 (>90%), but GFAP and O4 were barely detectable. Among Tuj1-positive neurons, more than 70% were GABAergic, whereas fewer than 5% were dopaminergic or serotonergic (Fig. 3A).

rNSCs have high neurogenic potential.
Unexpectedly, despite the broad expression of OLIG2, which recapitulates the pMN domain of the spinal cord, we could not detect any HB9/ChAT-positive motor neurons (Supplementary Fig. S2A). This may be related to oligodendrocyte specification of the spinal cord, in which FGF2 acts to inhibit motor neuron differentiation [32].
Moreover, we found that, as passage progressed, differentiation into serotoninergic neurons was barely detectable (Supplementary Fig. S2B). After 4 weeks in culture conditions that promoted spontaneous differentiation, which excluded all growth factors (including BDNF and GDNF), most cells differentiated into Tuj1+ neurons (>80%). Some GFAP+ astrocytes were also detected (<2%) (Fig. 3B), but we could not detect O4+ oligodendrocytes. These results demonstrate that both types of rNSCs had high neurogenic potential, as observed previously for hbNES cells [37], despite expressing SOX9, OLIG2, and NKX2.2 at later passages.
PDGF, but not EGF, induces more gliogenic potential
To induce gliogenic phase (Stage 3), we replaced the FEP medium with FPP medium (Fig. 3C) and found that expression of NKX2.2 was highly upregulated as a result (Fig. 3D). In contrast to expression of NKX2.2 within OLIG2-positive cells under FEP culture conditions, the NKX2.2-positive populations gradually increased (>80%) under FPP medium (Supplementary Fig. S2C, D). Moreover, under these conditions, the typical rosette structure was weakly maintained, and some glial-like cells appeared after 4 weeks of differentiation (Fig. 3E).
Notably, these cells were positive for GFAP and S100β (Fig. 3F), and these GFAP-positive astrocytes coexpressed EN1 (Fig. 3G). By contrast, when the cells were cultured in glial medium, we observed immature O4-positive oligodendrocytes (Fig. 3H) rather than MBP positive oligodendrocytes. These results indicated that PDGF induces more gliogenic properties than EGF but is not sufficient to generate mature oligodendrocytes. Hereafter, we designated the FPP-cultured intermediate progenitor cells (IPCs) derived from H9-hESCs and iPSCs as H9-IPCs and iPSC-IPCs, respectively.
Generation of GPLCs
We next asked whether the absence of mature MBP+ oligodendrocytes among rNSCs and IPCs was due to low expression of PDGFRα and SOX10, which are typical OPC markers [29]. To promote conversion into PDGFRα-positive cells, we generated neurospheres and cultured them in PDGF medium for 2 weeks (Stage 4) using a recently established sphere culture system [45], plated them onto PLO/laminin-coated dishes, and cultured thereafter in PDGF medium or glial medium (Fig. 4A). Flow cytometry revealed that distinct A2B5+ PDGFRα+ populations expanded after sphere culture (Fig. 4B), and expression of PDGFRα, CD9, and SOX10 markedly increased relative to the FEP culture condition, whereas OLIG2 and NKX2.2 were downregulated (Fig. 4C, G and Supplementary Fig. S3A).
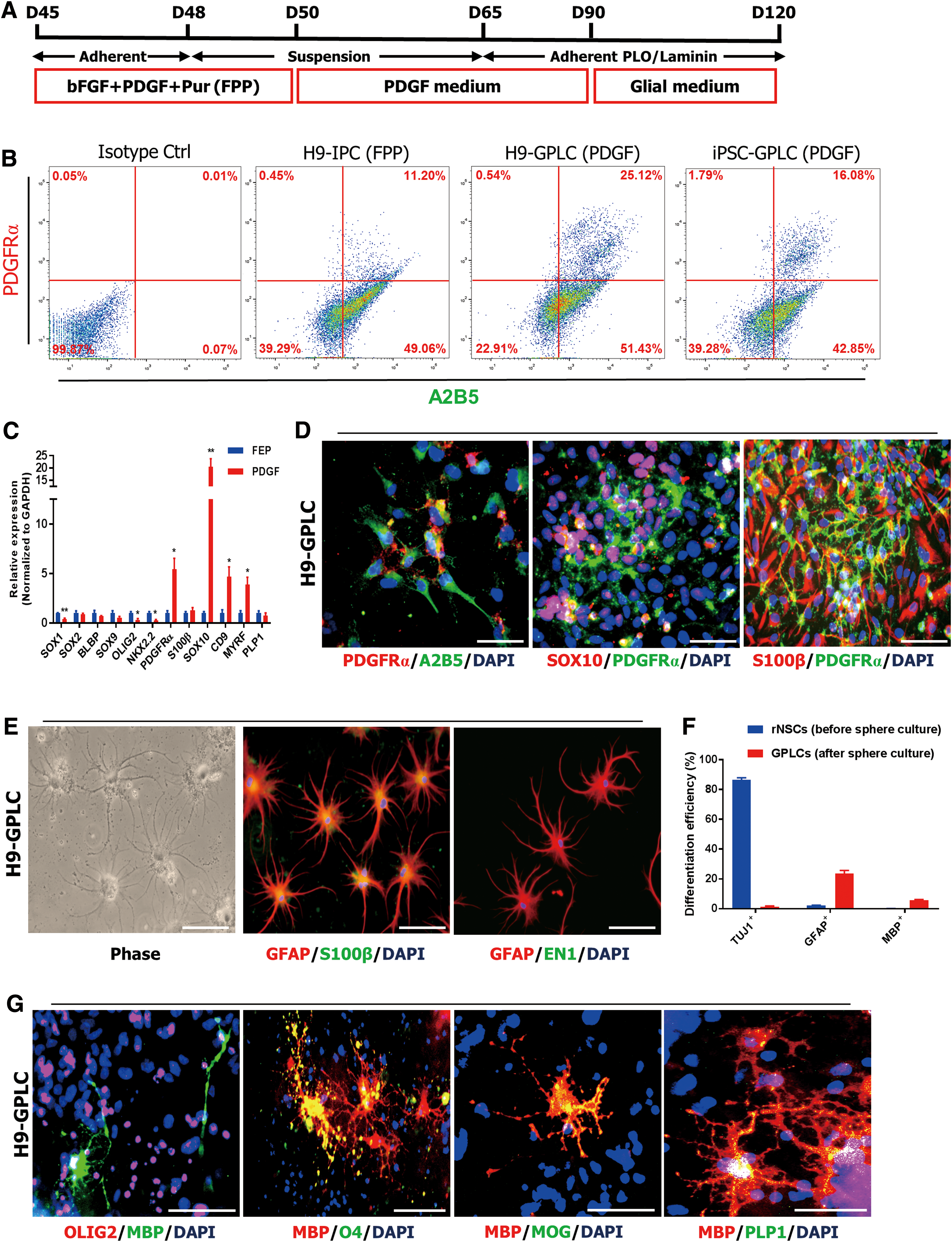
Generation of glial-restricted progenitor-like cells.
Immunocytochemistry confirmed that PDGFRα-positive cells coexpressed SOX10 and S100β (Fig. 4D and Supplementary Fig. S3B). The expression patterns of these sphere-derived progenitor cells (Stage 5) were similar to those of previously established GPCs in terms of upregulation of PDGFRα and CD9 [29] and expanded for at least five to eight passages; however, cell growth steadily decreased as the O4-positive population grew (Supplementary Fig. S3C, D). These results indicated that sphere-derived progenitor cells become more oligodendroglia as the number of passages increases.
After weeks of cultivation in glial medium, typical stellate astrocytes appeared that exhibited strong expression of GFAP and weak expression of S100β (Fig. 4E and Supplementary Fig. S3E). Moreover, sphere-derived progenitor cells mostly differentiated into GFAP-positive astrocytes, whereas rNSCs mostly differentiated into TUJ1-positive neurons (Fig. 4F and Supplementary Fig. S3F). We could not detect expression of EN1 at the protein level in these astrocytes (Fig. 4E). This may have been due to the reduced expression of EN1 and GBX2 during differentiation (Fig. 5B).
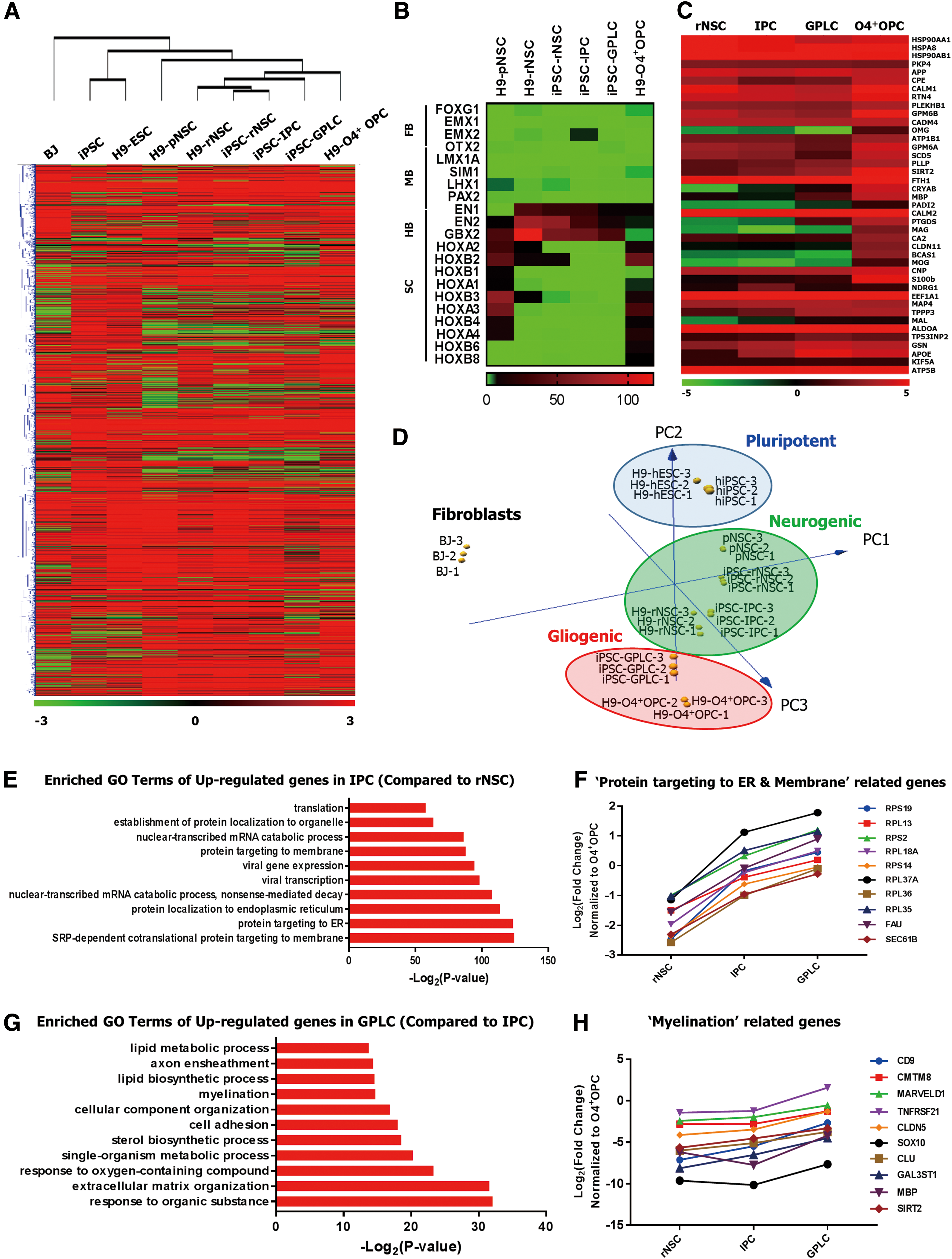
Global gene expression profiles of pNSCs, rNSCs, GPLCs, and O4+ OPCs.
However, these sphere-derived progenitor cells retained anterior hindbrain–specific regional identity relative to H9-O4+ OPCs derived from H9-hESCs using the recently developed efficient protocol (Fig. 5B and Supplementary Fig. S4) [45]. Furthermore, these cultures consisted of mature branched MBP+ oligodendrocytes, which coexpressed OLIG2, O4, myelin oligodendrocyte glycoprotein (MOG), and PLP1 (Fig. 4G and Supplementary Fig. S3G, I), and exhibited myelinating capacity with Tuj1-positive neurons (Supplementary Fig. S3H). The resultant progenitor cells obtained by sphere formation, which could differentiate into astrocytes and oligodendrocytes, are hereafter termed H9-GPLCs and iPSC-GPLCs (Glial restricted progenitor-like cells, GPLCs). Taken together, these results indicated that H9-GPLCs and iPSC-GPLCs have high gliogenic potential after sphere formation.
Transcriptome analysis of GPLCs
To clarify why rosette stage NSCs exhibited poor differentiation into mature oligodendrocytes and how GPLCs acquired gliogenic potential, we investigated dynamic changes of global gene expression in ESCs, iPSCs, rNSCs, IPCs, GPLCs, and H9-O4+ OPCs by RNA-seq.
Hierarchical clustering of 10,008 selected genes (FDR <0.05) revealed that ESCs and iPSCs clustered together, separately from NSCs (Fig. 5A). Moreover, iPSC derived progenitor cells became more similar to H9-O4+ OPCs as differentiation progressed (Fig. 5A). Furthermore, RNA-seq profiles of region-specific markers confirmed that established rNSCs, IPCs, and GPLCs prominently expressed EN1, EN2, and GBX2, which characterize anterior hindbrain identity, whereas H9-O4+ OPCs prominently expressed HOXA3, HOXB3, HOXA4, HOXB4, and HOXB6, which characterize spinal cord identity (Fig. 5B).
As expected, transcripts abundant in CNS myelin were highly enriched in GPLCs and H9-O4+ OPCs (Fig. 5C). In addition, principal component analysis (PCA) showed that iPSC-GPLCs co-clustered with H9-O4+ OPCs distinctly, while various rNSCs clustered closely together (Fig. 5D). Of note, we detected progression of differentiation from the pluripotent stage to the gliogenic stage (Fig. 5D). These results indicated that GPLCs were highly similar to H9-O4+ OPCs and had distinct regional identities similar to that of ventral rhombomere 1.
Next, to identify biological processes associated with genes differentially expressed between the three groups (rNSCs, IPCs, and GPLCs), we performed GO enrichment analysis on 534 DEGs upregulated in IPCs relative to rNSCs (≥2-fold) and 318 DEGs upregulated in GPLCs relative to IPCs (≥2-fold).
DEGs upregulated in IPCs were highly enriched in processes related to SRP-dependent cotranslational protein targeting to membrane, protein targeting to ER, and protein translation, all of which are also strongly upregulated in CNS myelin (Fig. 5E) [46]. Notably, expression of more than 50 genes involved in protein targeting to the ER and membrane steadily increased from the rNSC to GPLC stage (Fig. 5F). Furthermore, DEGs upregulated in GPLCs were enriched in processes related to extracellular matrix organization, myelination, and lipid biosynthesis (Fig. 5G). These myelination-related genes such as CD9, SIRT2, and MBP tended to be upregulated in GPLCs (Fig. 5H).
Together, these results indicate that the machinery related to mRNA transport, protein translation, and targeting is upregulated as cells shift to the gliogenic phase during sphere culture and that this upregulation leads to a reduction in the length of the differentiation period needed to acquire gliogenic potential.
GPLCs can differentiate into astrocytes and oligodendrocytes in vivo
Next, we investigated whether GPLCs could myelinate axons in congenitally hypomyelinated shiverer mice. For this purpose, we implanted 4-week-old homozygous shiverer (shi/shi) mice with 200,000 O4+ GPLCs (n = 12) or PBS control (n = 6). Eight weeks after engraftment, we sacrificed the mice and analyzed their brains to confirm the myelination competence of grafted GPLCs.
Notably, we observed MBP+ cells in corpus callosum of grafted brains, colocalized with human mitochondria, whereas control brains did not express MBP (Fig. 6A and Supplementary Fig. S5A, B). Moreover, the cells positive for human mitochondria also coexpressed MOG (Fig. 6B) and PLP1 (Fig. 6C), and a subset of them were also positive for GFAP (Fig. 6D). Furthermore, TEM of engrafted brain revealed compact myelin with major dense lines, providing evidence of myelin compaction, whereas untransplanted shiverer mice exhibited no such lines (Fig. 6E and Supplementary Fig. S5C).

Transplantation into congenitally hypomyelinated shiverer mice.
Taken together, these data indicate that grafted human GPLCs can differentiate into astrocytes and oligodendrocytes in vivo and remyelinate axons in demyelinated brains.
Discussion
In the spinal cord, oligodendrocytes are generated from the pMN domain in the ventral region of OLIG2 expressing populations [47 –49]. Using retinoic acid, the developmental process of the spinal cord can be easily mimicked during the differentiation of hPSCs [32,33,50]. Development of oligodendrocytes in forebrain has also been well studied, revealing that OPCs are generated from the NKX2.1-positive domain following two sequential rounds [50 –53].
Although numerous studies have explored cerebellar neurons and pathology, including cerebellar ataxia and MSA, cerebellar glial cell development has not been well characterized and it remains unclear where the various subtypes of glial cells come from [35,54]. Moreover, these glial cells are highly heterogeneous in terms of morphology, function, and anatomical location, despite the fact that they originate from the same segment [11,12,55].
Furthermore, there is no effective therapeutic agent for cerebellar disorders, including demyelinating diseases [56]; thus, the glial cell-based therapeutic approach has been considered a promising alternative [10,27]. To address the issue, Goldman's group developed a stepwise protocol for generating GPCs that could differentiate into astrocytes and oligodendrocytes both in vitro and in vivo when engrafted into congenitally hypomyelinated shiverer mice [28,29]. These cells exhibited high glial potential, but regional identity during the overall differentiation process remains poorly understood; however, the result of treatment with retinoic acid at Stage 3 suggests that these GPCs might have spinal cord identity [29].
In this context, we investigated how anterior hindbrain–specific GPCs are generated through stepwise developmental stages (primitive, rosette, intermediate, and glial restricted). To induce rhombomeric segments 1, we used 1.0 μM CHIR99021 because several HOX genes were upregulated and EN1 was downregulated when cells were treated with more than 1.0 μM CHIR99021 32.
However, under LSC culture conditions, EN1+ cells appeared with lower efficiency (less than 40%), but their density was enough to allow us to isolate them as colonies. To expand the EN1+ populations, we replaced the LSC culture medium with FEP medium. The characteristics of these isolated cells were similar to those of rosette-type NSCs, including hbNES cells [25,37], but their gene expression profiles differed slightly. During early differentiation, our established rNSCs lost expression of PAX6 subsequently and PLZF gradually, whereas expression of SOX1, OLIG2, and SOX9 gradually increased. These cells can be grown for 30–50 passages depending on the isolate, and gliogenic properties became predominant over neurogenic properties in late passage.
Notably, expression of OLIG2 decreased slightly, while expression of NKX2.2 increased, with increasing passage. However, it remains unclear whether the upregulation of NKX2.2 is accompanied by differentiation into oligodendrocytes or instead whether the spatial properties of rNSCs became more ventral toward the NKX2.2-positive domains. After upregulation of PDGFRα through IPC and sphere formation, we successfully generated EN1/GBX2-positive GPLCs. These GPLCs expressed CD9 and PDGFRα, which are typical markers of OPCs [28,29] and can differentiate into GFAP+ astrocytes and MBP+ oligodendrocytes in vitro and in vivo.
Indeed, RNA-seq analysis revealed that several genes associated with protein translation and targeting (eg, RPL37A, RPL35, and RPS2; encoding ribosomal proteins) increased gradually from NSCs to GPLCs. Interestingly, those ribosomal proteins are enriched in CNS-myelin and OPC-specific modules [46,57]. Based on these results, sphere culture led to shorter differentiation periods by upregulating the expression of those ribosomal proteins.
In this study, however, we could not determine the biological mechanism that which of the cells differentiated into astrocytes or oligodendrocytes among bi-potent GPLCs, although we selected homogeneous cell lines during initial differentiation periods by clonal analysis. Recently, Rosenberg et al. developed a useful tool, SPLit-seq, that can profile at the single-cell level in heterogeneous populations [58]. They showed that SPLit-seq facilitates exploration of gene expression in the developing brain and spinal cord. In the future, it should be possible to obtain valuable results using single-cell profiling tools, which would provide insight into the molecular mechanisms underlying glial development.
Although further preclinical investigations are clearly needed to demonstrate the therapeutic potential of GPLCs in demyelinating disorders (especially hindbrain), we demonstrated that GPLCs have functional myelinogenic potential in vitro and remyelinating capacity in myelin deficient shiverer mice (in vivo), suggesting that anterior-hindbrain specific GPLCs could be used to alleviate disease symptoms of myelin disorders and serve as a useful tool for studying disease progression in the hindbrain as an in vitro model.
In summary, we established region-specific NSCs and their differentiated glial progeny and investigated their characteristics. Our region-specific differentiation protocol will facilitate clinical research aimed at improving our knowledge of the development and progression of disease in various regions of the brain and spinal cord.
Footnotes
Acknowledgments
This work was supported by Korea University Grant, the Bio & Medical Technology Development Program of the National Research Foundation of Korea, funded by the Korea Ministry of Science, ICT, & Future Planning (MSIP) project no. NRF-2015M3A9B4071074 and NRF-2015M3A9B4071076, a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI18C2166), and the School of Life Sciences and Biotechnology for BK21 PLUS, Korea University.
Author Disclosure Statement
The authors declare no competing interests.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.