
Abstract
Although treatment strategies for pediatric leukemia have improved overall survival rates in the recent past, relapse rates in certain subgroups such as infant leukemia remain unacceptably high. Despite undergoing extensive chemotherapy designed to target the rapidly proliferating leukemia cells, many of these children experience relapse. In refractory leukemia, the existence of cell populations with stemness characteristics, termed leukemia stem cells (LSCs), which remain quiescent and subsequently replenish the blast population, has been described. A significant body of evidence exists, derived largely from xenograft models of adult acute myeloid leukemia, to support the idea that LSCs may play a fundamental role in refractory disease. In addition, clinical studies have also linked LSCs with increased minimal residual disease, higher relapse rate, and decreased survival rates in these patients. Recently, a number of reports have addressed effective ways to utilize new-generation genomic sequencing and transcriptomic analyses to identify targeted therapeutic agents aimed at LSCs, while sparing normal hematopoietic stem cells. These data underscore the value of timely translation of knowledge from adult studies to the unique molecular and physiological characteristics seen in pediatric leukemia. We aim to summarize this article in the rapidly expanding field of stem cell biology in hematopoietic malignancies, focusing particularly on relevant preclinical models and novel targeted therapeutics, and their applicability to childhood leukemia.
Introduction
While the response rate in pediatric leukemia has increased significantly in the past two decades, relapsed disease remains a critical determinant of the overall outcome in these children. In fact, recent data indicate that relapsed acute lymphoblastic leukemia (ALL) by itself has become the fourth most common cancer in children [1,2]. Furthermore, among the patients diagnosed with leukemia, certain molecularly and clinically defined subgroups experience significantly worse outcomes. These include subtypes such as acute myeloid leukemia (AML), T-cell ALL, and infant leukemia (IL), and those with high-risk molecular aberrations such as Bcr-Abl fusions. IL, which affect children <1 year of age, have one of the lowest event-free survival rates, with an exceptionally high relapse rate [2]. The high rate of relapse following initial response suggests the inability of current treatments to completely eradicate certain components of the blast population.
In the recent past, the identity, biological evolution, and molecularly definable growth regulatory mechanisms of such cell populations have been investigated in adult leukemia. Findings from these laboratory and preclinical studies have generated significant interest among pediatric oncologists as they suggest potential approaches in novel therapeutics development for refractory leukemia in children. In the treatment of IL, studies have found that intensification of treatment reduces early relapse, but carries increased risk of toxicity, particularly during induction, without improving long-term outcomes. However, a recent report from the Children's Oncology Group showed that de-intensification of induction therapy and better supportive care result in considerably less induction mortality without affecting complete remission rates [3].
Pediatric Leukemia
Normally, the white blood cell components are generated from lymphoid and myeloid progenitors and lineages originating from hematopoietic stem cells (HSCs) [4]. Through distinct stages of immunoglobulin gene recombination events and changes in surface antigen expression, lymphoid progenitor cells differentiate into B or T cells [5,6]. Similarly, myeloid progenitor cells generate erythrocytes, platelets, monocytes, and eosinophils [5]. In most cases, leukemia is characterized by the transformation of a progenitor cell arrested at an early stage of differentiation, leading to excessive production of leukemic blasts [6,7].
ALL represents more than a quarter of all childhood cancers and about 80% of all childhood leukemia [8]. It is characterized by an excessive production of immature lymphoblasts and is classified as precursor B cell (B-ALL), precursor T cell, and B cell leukemia. Recently, Pui et al. proposed a modified classification system using molecular diagnostic methods involving the genetic and epigenetic alterations affecting cellular growth regulatory pathways [9].
AML, which accounts for ∼20% of all pediatric leukemia, originates from myeloid precursors and is characterized by an increase in progenitor cells [5]. At least one genetic alteration is found in over 90% of pediatric AML cases, and more than 200 distinct genetic abnormalities have been described, including mutations, translocations, and deletions [4,5]. Furthermore, abnormalities in pathways that regulate growth, proliferation, and apoptosis have also been described, including Fms-like tyrosine kinase (FLT3), c-Kit, Ras, nucleophosmin (NPM1), and Wilms' tumor-1 (WT1) [5,10]. Interestingly, despite this diversity, all such subtypes of AML cells show activities in shared pathways with potential to promote leukemogenesis [5].
Relapsed leukemia shows significantly more aggressive growth properties than the cells taken at diagnosis [11]. This is reflected in findings from molecular comparison studies using paired initial and relapsed leukemia cells. It has been shown that relapsed leukemia cells increase transcription of genes regulating growth and survival, including those affecting DNA repair, cell cycle progression, and apoptosis inhibition [11].
Infant Leukemia
Leukemia diagnosed in children <1 year of age is classified as IL. This subgroup represents about 5%–10% of leukemia in children and contains both ALL and AML. For reasons that are yet to be determined, the incidence of IL appears to be increasing [12]. IL often contains distinct biological properties and clinical presentation, including high white blood cell count (>100,000/μL), hepatosplenomegaly, and leukemic infiltration of the central nervous system. In addition, rearrangement of the mixed-lineage leukemia (MLL) gene, FLT3 mutations, and other molecular abnormalities have been frequently found [9,13,14]. The most commonly used chemotherapy protocols for IL include high doses of cytarabine and methotrexate [5]. Event-free survival rates, especially for infants with MLL rearrangements, are significantly lower than the rates seen in older children, ranging from 30% to 40% [5,15]. The high rates of relapse observed in pediatric leukemia, especially AML and IL, suggest that current treatment regimens often fail in complete eradication of the malignant cells.
Leukemia Stem Cells and Leukemia
Leukemia stem cells and AML
The increased incidence of relapse observed in high-risk leukemia patients implies the existence of a subset of rare, persistent cells that are not always affected by current treatments. There is general agreement that in leukemia, such cells contain stem cell-like properties, and have therefore been termed leukemia stem cells (LSCs). A significant body of evidence indicates that LSCs contain biological properties distinct from the more mature leukemic cell population [16 –23]. In many ways, LSCs resemble normal HSCs, including their ability to remain quiescent, self-renew, and generate substantial numbers of differentiated leukemic progeny. These attributes provide LSCs the ability to maintain a low, but critical number of cancer stem cells to effectively sustain the disease [24]. These properties also help to describe how LSCs elude aggressive chemotherapy and remain quiescent, eventually repopulating during relapse. Many of the physiological mechanisms shared with HSCs may allow them to survive in hematopoietic niches, which also makes targeting these cells a challenging task [25]. To achieve sustained blood cell production, HSCs must have both self-renewal capacity and the ability to differentiate and form progeny.
Several studies have aimed to characterize the essential properties of LSCs [26 –28], and some important differences from HSCs have been noted. Although similar to normal HSCs with a surface marker profile including CD34+, CD38−, HLA−, and DR−, LSCs may express surface antigens that can differentiate these rare cells from the larger HSC population. These include CD90, CD117, and CD123, among others [21 –23]. The ability to distinguish LSCs from normal HSCs will prove to be exceedingly important in designing therapeutics that are able to selectively target LSCs without simultaneously causing nonspecific toxicities. The ability to engraft and propagate leukemia in immune-deficient mice has been developed in an effort to distinguish LSCs from both normal HSCs and more mature leukemic cells [29].
In the past, immunocompromised NOD/SCID mouse repopulating assays have been used to show that AML stem cells are present in the CD34+CD38− hematopoietic cell fraction [30,31]. In a recent adult AML study, the level of CD34+CD38− blast cells at diagnosis correlated with lower treatment response and shorter disease-free survival [32]. CD34+CD38+ cells, on the other hand, have not, using standard experimental methods, shown long-term repopulating capacity, indicating that this fraction may represent more mature differentiated leukemic cells [33,34]. Initial studies detected leukemia-initiating cells (LICs) exclusively in the CD34+CD38− fraction [18,19,35,36]; however, some studies have suggested that both CD34− and CD38+ cells can initiate and maintain leukemia [37 –41]. For example, LSCs of patients with NPM-1 mutations have been found in the CD34− compartment [39,42]. Presently, however, a complete agreement with respect to the exact phenotype of LSCs has not been achieved. Previous studies have described a number of distinct surface markers for the isolation and characterization of these cells, including CD25, CD32, CD34, CD38, CD44, CD47, CD90, CD96, CD117, CD123, CD133, TIM3, and CLL1 [18,24,43]. Taken together, these findings underscore the immunophenotypic heterogeneity present in LSCs and the complexity associated with the isolation of LSCs correctly, entirely based on a uniform set of limited cell surface markers.
In addition to cell surface antigens, abnormal expression of the enzyme aldehyde dehydrogenase (ALDH) has been proposed as a characteristic of LSCs. It was found that ALDH status may enable the identification of a CD34+CD38− population that effectively generated AML in transplanted animals, but it was also highly predictive of relapse in the donor patient population [29]. A subpopulation of CD34+CD38− AML cells with intermediate-level ALDH (ALDHint) has been shown to efficiently engraft immunocompromised mice to produce AML. It was suggested that minimal residual disease (MRD) was highly enriched for the CD34+CD38− ALDHint population, as this abnormal ALDH expression correlated with subsequent clinical relapse [29]. Interestingly, in this particular study, of the patients who achieved clinical remission, all with the MRD pattern of ALDHint cells subsequently relapsed, while all without this MRD pattern maintained remission. Overall, the current data suggest the capacity of ALDH expression to predict MRD is of high clinical relevance and it provides a way to recognize patients who have achieved true remission from those with high risk for relapse.
Detection of LSCs in ALL
Although the existence of an LSC population in AML is generally acknowledged, the data for ALL are less well defined [44,45]. However, emerging experimental evidence indicates that LSCs are also likely to be a key feature of high-risk ALL and chemotherapy resistance. In the most common subtype of childhood ALL, ALL/t (12;21), malignant cells have been found exclusively in the CD19+ compartment, reflecting a more mature cell population [46,47]. Studies have shown the more immature CD34+CD19− compartment to be involved in both infant t(4;11) ALL and in Philadelphia chromosome-positive ALL, both of which constitute high-risk populations [46,47]. Recently, Aoki et al. investigated the cell surface and molecular characteristics of LICs from children with MLL+ leukemia [48]. Their findings indicated variability in these cells, defined by the distinct fusion partners of the MLL gene. For instance, in MLL-AF4, the LIC fraction included CD34+CD38+CD19+ and CD34−CD19+ populations, and in MLL-ENL, they were found either in the CD34+ or CD34− population. However, in MLL-AF9 cells, leukemia initiation function was observed exclusively in the CD34−CD19+ fraction. Interestingly, their studies have shown that, within the MLL nonrearranged population, a fraction enriched for normal HSC characteristics can lead to repopulation of the normal hematopoiesis compartment in transplanted animals.
LICs from ALL share a number of functional properties with AML LSCs, but a number of important differences exist. For example, there are no uniform surface marker panels to characterize ALL leukemia-initiating populations in xenograft studies [49]. Hence, it has been postulated that the relapse in ALL may involve a stochastic process compared to the hierarchical stem cell model proposed for AML. Molecular analyses have shown the presence of multiple subclones in ALL patient samples, suggesting a functional diversity in tumor-initiating capabilities and their potential to be affected by the selective pressure exerted by therapeutic agents [50].
Clonal diversity is thought to be a key factor in ultimate treatment response as resistant clones may arise from previously present clones or from newly acquired molecular alteration during treatment [51]. A number of recent studies have evaluated the mutational landscape of relapsed and refractory pediatric ALL. For example, next-generation sequencing studies of 240 pediatric leukemia specimens and matched remission specimens showed significant genetic heterogeneity and low occurrence of persisting somatic mutations in remission [52]. Overall, the mutational landscapes at diagnosis and relapse reveal that clones observable at relapse were present as minor populations initially, but are not commonly derived from the dominant clone detected at diagnosis.
Features of clonal evolution can also be studied by comparative analyses of HSCs and LSCs in individual leukemia patients. In ALL, researchers were able to separate and investigate HSCs from leukemic cells. For example, Wang et al. were able to isolate normal HSCs from malignant leukemic cells in a subgroup of adult B-ALL patients [53]. Their studies showed that CD34+CD38−CD19−ALDH+ cells are enriched for HSC potential. Treatment resistance may also be generated by the genetic alterations during clonal evolution of critical cell populations, allowing the benefit of the protective microenvironment of the bone marrow (BM) through adhesion or related processes. For example, alterations in the IKZF1 gene leading to the generation of drug resistance in ALL have been described [54].
LSCs as a prognostic indicator
A number of recent studies have attempted to identify putative biomarkers in LSCs, which can further inform about prognosis. Intermediate ALDH expression in the LSCs has been described as an indicator of MRD and increased likelihood of relapse [29]. In addition, gene expression analyses of subpopulations with validated stem cell engraftment potential have shown LSC gene expression signatures that are highly predictive of poor survival in patients. For example, a report from van Rhenen et al. evaluating AML cells taken at the time of diagnosis from 92 AML patients has demonstrated that a high proportion of CD34+CD38− cells with engraftment potential in SCID/NOD mice correlated with a high MRD and poor patient survival [55]. In a recent study, Long et al. evaluated 112 pediatric ALL patients and found that an increased CD34+CD38− population inversely related to the outcome [56].
In pediatric AML, an increased CD34+, CD38−, and CD45−/low population has been shown to correlate with reduced treatment response [57]. These findings implicate the activity of CD34+CD38− cells at diagnosis as an adverse prognostic factor predicting resistance to chemotherapy, higher MRD, and more likely relapse. Another study has shown that patients with cells carrying LSC or HSC gene expression signatures experience the worst disease outcome [58]. Gene expression studies of the LICs in MLL-rearranged leukemia have found, in addition to patient-specific gene signatures, distinct expression patterns in certain genes and clusters of genes depending on the given MLL fusion partner and individual phenotype [59]. Furthermore, studies by Shlush et al. have shown that preleukemic HSCs present at diagnosis can develop normal hematopoiesis, but carry a competitive repopulation advantage over nonleukemic HSCs [60]. It has been shown that preleukemic stem cells are highly enriched in cells with mutations in genes such as DNMT3A and IDH2. These cells are resistant to chemotherapy and persist in the BM, providing a potential supply of cells for leukemic relapse. Collectively, these data point toward the information present in gene expression signatures of LSCs to identify potential prognostic indicators in leukemia patients.
Targeting LSCs
For the most part, current chemotherapy protocols aim to target rapidly dividing cells. The high rate of relapse identified in pediatric leukemia may be a consequence of the inability of current chemotherapy regimens to address the survival of LSCs. Although research specific to pediatric LSCs is limited, several strategies have been described to preferentially target LSCs, while sparing normal stem cells.
Targeting the microenvironment encompassing HSC interactions
A report by Colmone et al. has suggested that leukemia cells alter the BM microenvironment and generate malignant stromal niches that block the engraftment and growth of healthy hematopoietic progenitor cells [61]. These microenvironmental niches have been thought to provide a safe haven for LSCs to sustain their survival while self-renewal capacity. Several recent studies have also attempted to better understand the components of the nonleukemic stem cell niche within the BM. One study on activated osteoblast-specific parathyroid hormone (PTH) receptors in mice showed increased stem cell production, in addition to increased production of the Notch1 ligand, Jagged1 [62]. This led to the suggestion that Notch1 activation may be one of the mechanisms that enhances stem cell growth within the BM niche. Another mouse model reported that bone morphogenetic protein receptor type IA knockout increased both osteoblasts and stem cells [63]. Osteoblast production of angiopoietin-1, which activates Tie-2 on stem cells, has been shown to promote tight adhesion of stem cells to their niche, resulting in quiescence and increased survival [64]. Extracellular matrix components have also been shown to play a role in stem cell regulation, as the absence of osteopontin resulted in increased stem cells, with an accompanying increase in both Jagged1 and angiopoietin-1 levels [65]. The role of osteopontin as a limiting factor in stem cell production was replicated in another study that demonstrated osteopontin interacted with different stem cell surface molecules [CD44, B1 integrins, and with very late antigens (VLA)-4 and VLA-5] to inhibit stem cell proliferation [66].
Furthermore, studies have sought to establish if LSCs share the same niche as normal stem cells to determine if modifying the normal stem cell niche will similarly affect LSCs. Another study using specific fluorescence labeling found that LSCs and normal HSCs localize to the same periendosteal region of the marrow [67]. When addressing the niche-stimulating effects in both normal and leukemic cells by introducing PTH to the marrow, they found differing sensitivities to the niche-derived signals between LSCs and normal HSCs. Signals induced by PTH were beneficial for the growth and engraftment of normal HSCs, but the LSCs did not appear to be stimulated by those signals in the same way. It is conceivable that PTH-induced signals allowed the normal HSCs to out-compete the LSCs for growth opportunities. This suggests that, although they may occupy the same location in the marrow under normal conditions, it may be possible to alter the niche to make it less accommodating to malignant stem cells. Support for this idea comes from the studies that have shown a decrease in the development of leukemia in rats treated with PTH [67].
The effective signals received from the BM microenvironment include various cytokines such as stromal cell-derived factor (SDF)1, insulin growth factor, fibroblast growth factor, interleukin 6, and vascular endothelial growth factor, in addition to various adhesion molecules [68]. For example, one potentially important aspect of the homing and localizing of LSCs in a protective BM niche is the interaction between the CXCR4 with the stromal-derived cytokine SDF1a (CXCL12). Overexpression of CXCR4 has been observed in leukemia cells and was shown to correlate with poor clinical outcome [69]. Furthermore, working with adult AML specimens, Rombouts et al. have found that increased CXCR4 expression in the CD34+ subpopulation conferred an increased chance of relapse and substantially reduced survival rate [70]. It was also found that CXCR-4 expression was significantly increased in Flt3 internal tandem duplication (ITD) leukemia cells compared to wild-type controls. It is relevant to note that FLT3/ITD is the most commonly found mutation with a prevalence of 10%–17% of pediatric AML [71]. A study investigating leukemia precursor cells found that FLT3/ITD was present in all CD34+/CD33+, but in only 19/24 CD34+/CD33− cells from AML patient samples and in whom CD34+/CD33− precursors harbored the FLT3/ITD had worse clinical outcome [72]. This indicated that FLT3/ITD involvement in the CD34+/CD33− leukemia precursors is heterogeneous and may correspond to resistance disease. Recently, Ma et al. have demonstrated the utility of combining all-trans retinoic acid with FLT3 tyrosine kinase inhibitors to eliminate FLT3/ITD+ LSCs [73].
The leukemia niche contains areas of hypoxia in which malignant cells have been shown to thrive. In this milieu, the overexpression of hypoxia-inducible transcription factor-1 alpha (HIF-1α) has been noted [74]. It appears that the hypoxic BM microenvironment generates a fertile stem cell niche through HIF-1α, which may provide support for HSC migration and survival through the FLT3/HIF-1α/SDF-1/CXCR4 axis. This possibility is supported by the observation that antibodies directed against CXCR4/SDF1 block AML engraftment in SCID/NOD mice [75]. These findings indicate a critical role for FLT3-mediated pathways in the homing and survival of LSCs and present a potential target for intervention. Early-phase clinical trials have begun in adult leukemia patients testing anti-CXCR4 [24], but advancement in pediatric patients awaits additional positive preclinical studies.
Targeting HSC survival, growth, and self-renewal mechanisms
Crucial evidence for the presence of mechanisms that inhibit apoptosis in LSCs comes from studies carried out in chronic myelogenous leukemia (CML) where BCR-ABL+, CD34+ leukemic progenitor cells have shown increased resistance to imatinib-induced apoptosis [76]. Furthermore, ABT-737, a Bcl-2 inhibitor, was able to generate a proapoptotic phenotype and promoted apoptosis in quiescent CD34+ cells from primary CML samples [77]. In AML, the CD34+CD38−CD123+ stem cell population was shown to be more sensitive to ABT-737 than the conventional antileukemia chemotherapeutic agent cytarabine, whereas normal HSCs were unaffected by ABT-737 [78].
To downregulate apoptosis, LSCs may also utilize alterations in a diverse group of growth regulatory pathways to increase their survival. These include nuclear factor-kappa B (NF-κB) and PI3K/AKT/mTOR [79,80]. Constitutive upregulation of PI3k/AKT/mTOR signaling has been found in both AML blasts and their stem cell precursors, where it enhanced survival and chemoresistance through NF-κB [81]. Inhibition of NF-κB, which has shown constitutive activity in primary AML specimens, is one proposed strategy to target LSC survival [82]. Although active in many other malignancies [83 –85], NF-κB was found to be active in the quiescent LSCs, suggesting a potential role as a therapeutic target. Proteasome inhibitors are a class of drugs that have shown promising effectiveness in inhibiting NF-κB and inducing LSC apoptosis [79]. Interestingly, anthracyclines, commonly used in AML induction therapy, have been shown to upregulate NF-κB [86,87]. When used in combination, the proteasome inhibitor MG-132 and the anthracycline idarubicin induced a rapid, strong apoptotic response in the LSC population with minimal effect on the normal HSCs in primary AML specimens [88].
Two other agents, Parthenolide (PTL) and TDZ8, have also been shown to specifically target LSCs, while sparing normal stem cells by altering NF-κB signaling. PTL is an active compound in the plant Tanacetum parthenium, known as feverfew or bachelor's buttons, and a potent NF-κB inhibitor, which for centuries has been used as a herbal remedy. PTL has been shown to preferentially target both LSCs and AML progenitor cells in NOD/SCID xenograft mouse models [88], but problems have been encountered in its pharmacologic development due to lack of water solubility. 4-Benzyl-2-methyl-1,2,4-thiadiazolidine-3,5-dione (TDZD-8), initially studied for the treatment of neurological diseases, also acts as an NF-κB inhibitor. Studies have shown the ability of TDZD-8 to selectively induce death in primary AML progenitor cells [89]. Further studies using CD34+CD38− AML progenitor cells showed that TDZD-8 is more effective than PTL in reducing survival of these cells. In addition, TDZD-8 selectively inhibited engraftment of AML progenitor cells into NOD/SCID mouse models without significantly affecting the engraftment of normal HSCs.
In addition to alterations in apoptosis and survival mechanisms, pathways that regulate self-renewal capacity in LSCs have also been investigated. Signaling molecules and pathways such as HOX, Notch, hedgehog, and Wnt/beta catenin have been implicated with HSC self-renewal and may play a similar role in the LSC [90,91]. There are suggestions that LSCs may depend on signaling pathways such as Wnt/beta catenin for self-renewal [92] and both myeloid and lymphoid leukemias have shown dysregulation of HOX genes [93,94]. Additional research is needed to characterize the exact role of these signaling molecules in facilitating self-renewal processes in LSCs and to identify effective therapeutics targeting these processes.
A number of cell surface markers identified on leukemic cells with stem-like characteristics have also been explored as targets for therapeutics. A report by Cheng et al. has shown that in AML, CD33, and CD44 antigen expression is significantly higher in LSCs compared to normal HSCs [95], implying the potential of targeting these molecules in future therapeutics for pediatric AML.
Investigations have also focused on targeting LSCs based on their potential unique metabolic vulnerabilities. Recent studies by Jones et al. evaluated the metabolic differences between AML cells and corresponding LSCs isolated from de novo leukemia specimens from adult patients, and found a critical dependence of LSCs on oxidative phosphorylation (OXPHOS) and amino acid metabolism for their survival [96]. However, similar cells isolated from relapsed specimens were not found to be dependent on amino acid metabolism as they were able to compensate by enhanced fatty acid metabolism. In addition, this study has also shown metabolism-based mechanisms by which agents such as venetoclax and azacitidine may be used to target LSCs in AML. These data provide the basis for future focused investigations to identify analogous processes that can be developed for leukemia treatment.
LSCs have also been shown to maintain altered DNA repair mechanisms that facilitate survival under cytotoxic conditions. Therefore, effective inhibition of these pathways has been explored in novel treatment approaches [97]. A recent investigation has shown that DNA-dependent protein kinase (DNA-PK)-deficient quiescent leukemia cells are susceptible to PARP1 inhibitors, as a single agent or in combination with chemotherapy, and are subjected to dual cellular synthetic lethality—potentially leading to the eradication of LSCs [98].
Targeting aberrant epigenetic mechanisms
DNA methylation changes occur frequently in cancer and have been considered to play a critical role in the generation of aggressive and refractory disease. Normally, epigenetic mechanisms regulate transcriptional programs during embryogenesis and the maintenance and development of normal stem cells, contributing to differentiation and self-renewal processes. Similarly, such activities can also help to generate a survival advantage and self-renewal capacity in LSCs. Recently, attempts have been made to generate DNA methylation profiles in difficult to cure pediatric malignancies, including childhood B-cell ALL. An investigation by Sandoval et al. identified distinct DNA methylome profiles in pediatric B-ALL patients with relapsed samples, showing significant association between distinct DNA methylation signatures and disease-free survival [99]. With respect to cancer stem cells, research has been focused largely on understanding the epigenetic alterations that promote these cells to gain essential stemness characteristics. For example, it has been shown that inhibiting Dnmt1 DNA methyltransferase expression impeded the development of leukemia [100]. Epigenetic modifications can also provide survival advantage through the facilitation of beneficial interactions with the microenvironment. For instance, hypoxia has been shown to induce the expression of the histone methyltransferase MLL1 in tumor stem cells than non-stem tumor cells [101]. Downregulation of MLL1 also led to the reduction in self-renewal capacity and tumorigenicity of the stem cells.
Recent research has focused on identifying agents to block the deleterious methyltransferase activity of the MLL complex in IL cells. For instance, a report by Cao et al. described targeted inhibition of the MLL1-WDR5 complex by a novel small-molecule inhibitor MM-401 that resulted in cell growth inhibition and apoptosis, specifically in leukemia cells [102]. Similarly, a number of inhibitors against the histone methyltransferase DOT1L have shown selective antileukemic activity in preclinical models and have entered into early-phase clinical trials [103]. Evaluating the activity of these agents against LSCs will provide critical information regarding their use in eradicating refractory disease in future clinical trials.
Genomic Analysis of Growth Regulatory Pathways
Advances in the interrogation of the cancer genome, achieved primarily through large institutional initiatives, have produced new knowledge about the genomic and epigenomic events that initiate and sustain malignant transformation. This information has made available a potential new approach for the development of novel therapeutics. These research efforts have been facilitated by discovery tools such as massively parallel nucleic acid sequencing, leading to the identification of critical mutations, copy number variations, and promoter methylation patterns. This technology is beginning to be used effectively to categorize defining molecular alterations in subpopulations of many different malignancies and to identify novel therapeutic avenues. For example, data from The Cancer Genome Atlas and the International Cancer Genome Consortium initiatives provided evidence for tumor subtypes identified by transcriptional signatures. One very important accomplishment of these initiatives is the realization that the response to specific molecular alterations and pathways can be influenced by other co-occurring aberrations in the cancer cell. For instance, in breast cancer cells positive for HER2, the activity of trastuzumab appears to be influenced by the presence of concurrent PIK3CA mutations [104]. Consequently, co-treatment with a PI3K inhibitor leads to highly effective combination therapy and restored cytotoxicity against trastuzumab-resistant tumor cells. However, the advancement of pathway-targeted drug discovery approaches can present complexities, as many of the low-frequency aberrations have not been functionally evaluated. One limitation of many of the currently available algorithms is the redundancy of the pathways analyzed. However, it has been shown that redundant pathway elements can be computationally removed to produce a “super pathway,” of which the activity differs between two comparative cell populations [105].
Previously, Hassane et al. have investigated the use of gene expression signatures to identify agents with activity against AML stem cells [106]. Based on the information that PTL, the active compound from the feverfew plant, was able to kill AML LSCs, the investigators determined the global transcriptional response to PTL using microarray analysis. This information was then used to identify other compounds that produced a similar transcriptional signature. Importantly, such an approach has shown the utility of using similar gene expression profiles to discover novel antileukemic stem cell therapeutic agents. Gene expression profiles of LICs from ALL with MLL rearrangement have been generated, which have identified a number of cell surface markers that are specific for LICs and not present in HSCs [48]. These include CD9, CD32, and CD24. It is anticipated that targeting these molecules will elicit LIC-specific inhibitory effects, while sparing treatment-related toxicity in the closely related normal HSC population. Recently, Duployez et al. examined the previously described highly prognostic adult LSC12 gene expression signature and demonstrated its prognostic relevance in pediatric AML [107].
Targeting LSCs in Therapeutic Applications
A number of recent reports have focused on the development of LSC-targeted therapeutic approaches based on the critical role for these cells in the initiation, development, and disease relapse. To a large extent, these investigations aim to identify and target the key molecular determinants, mechanisms, and pathways that regulate the growth and survival of LSCs. Recently, Laverdière et al. have used a bioinformatics-based approach to probe gene expression signatures with preclinical drug screening to identify specific targets for therapies [108]. It was been suggested that the examination of equivalent genes and pathways in normal HSCs will help to minimize or eliminate treatment-related toxicities in LSC-directed therapies. Potential therapeutic approaches specifically targeting LSCs, while saving normal HSCs in CML patients, have been reviewed [109]. The two stem cell populations share common functional characteristics such as self-renewal, multipotency, and the ability to remain quiescent. Specific targeting of survival pathways that are critical for LSCs, but nonessential in HSCs, would help to design future treatments. For example, Chen et al. have reported that arachidonate 5-lipoxygenase gene (Alox5), which is involved in the synthesis of leukotrienes from arachidonic acid, is required for the induction of CML by BCR-ABL and its deficiency interferes with LSC function without affecting normal HSCs [110]. Consequently, targeting Alox5 has been suggested as an approach to prevent the initiation of BCR-ABL-mediated CML.
Specific cell survival pathways and epigenetic mechanisms found in LSCs have also been considered targets for new therapeutics development [109]. Overexpression of specific cell survival proteins such as Bcl-2 has been demonstrated in AML LSCs but not in HSCs, providing a potential target for stem cell-based therapy [111]. Targeting epigenetic mechanisms may also prove effective in the identification of LSC-based therapies. For instance, in MLL-rearranged leukemias, an association between distinct epigenetic regulators and the capacity of MLL fusion proteins to transform HSCs into leukemic cells has been demonstrated [112]. Investigators have also focused on targeting metabolic activities of LSCs. It has been shown that LSCs are aberrantly dependent for energy metabolism and these biochemical processes can be targeted in therapeutic strategies [113]. For example, the suppression of OXPHOS may inhibit LSCs by disrupting their energy metabolism [114]. In addition, LSCs have also been found to express distinct immunological profiles in the BM niche with mechanisms to evade immune-mediated killing. Hence, a number of immunotherapeutic strategies to target LSCs to overcome resistance to conventional treatments and relapse have been investigated [115].
Conclusion
Despite the advances made in the treatment and supportive care of pediatric malignancies in the past two decades, the overall outcome for relapsed pediatric high-risk leukemia remains unacceptably poor. With current treatment regimens, more than half of infants diagnosed with leukemia experience relapse, often after a short period of remission. Earlier studies carried out in adults have provided initial evidence for the presence of subgroups of cells with stemness characteristics. Current experimental evidence implies that LSCs develop from normal HSCs or from hematopoietic progenitors following decisive mutational events, initiating abnormally regulating self-renewal capacity as well as the ability to generate blasts with differentiated phenotype. In addition, a multitude of investigations have described the interrelationship between the LSCs and normal HSCs, and hematopoietic progenitor cells, as well as the growth regulatory conditions and supportive role of the BM stromal niche in the survival and maintenance of LSCs (Fig. 1). This is largely due to the measurable progress that has been made on methods to isolate rare populations of leukemia cells and to functionally validate their stemness in xenograft models. Molecular analyses have also expanded our knowledge of distinct biomarkers found on LSC candidates compared to HSCs, providing ways to develop selective and tolerable therapies. In addition, advances have also been made to estimate the LSC burden and to correlate this with clinical outcomes. Admittedly, the application of these approaches in high-risk pediatric leukemia will be logistically challenging due to low incidence, but it can be achieved in collaborations involving cooperative groups and multi-institutional clinical trial consortia. In addition, a significant amount of information is also available on the decisive molecular alterations that commonly occur in currently difficult to cure pediatric leukemia. This provides a vantage point for molecular analyses to detect tumor-initiating mutations, leukemia-promoting events, and the biology of LSCs. Hence, supporting the potential for the identification of promising new targets and effective novel therapeutics aimed at eliminating this disease in children and adults.
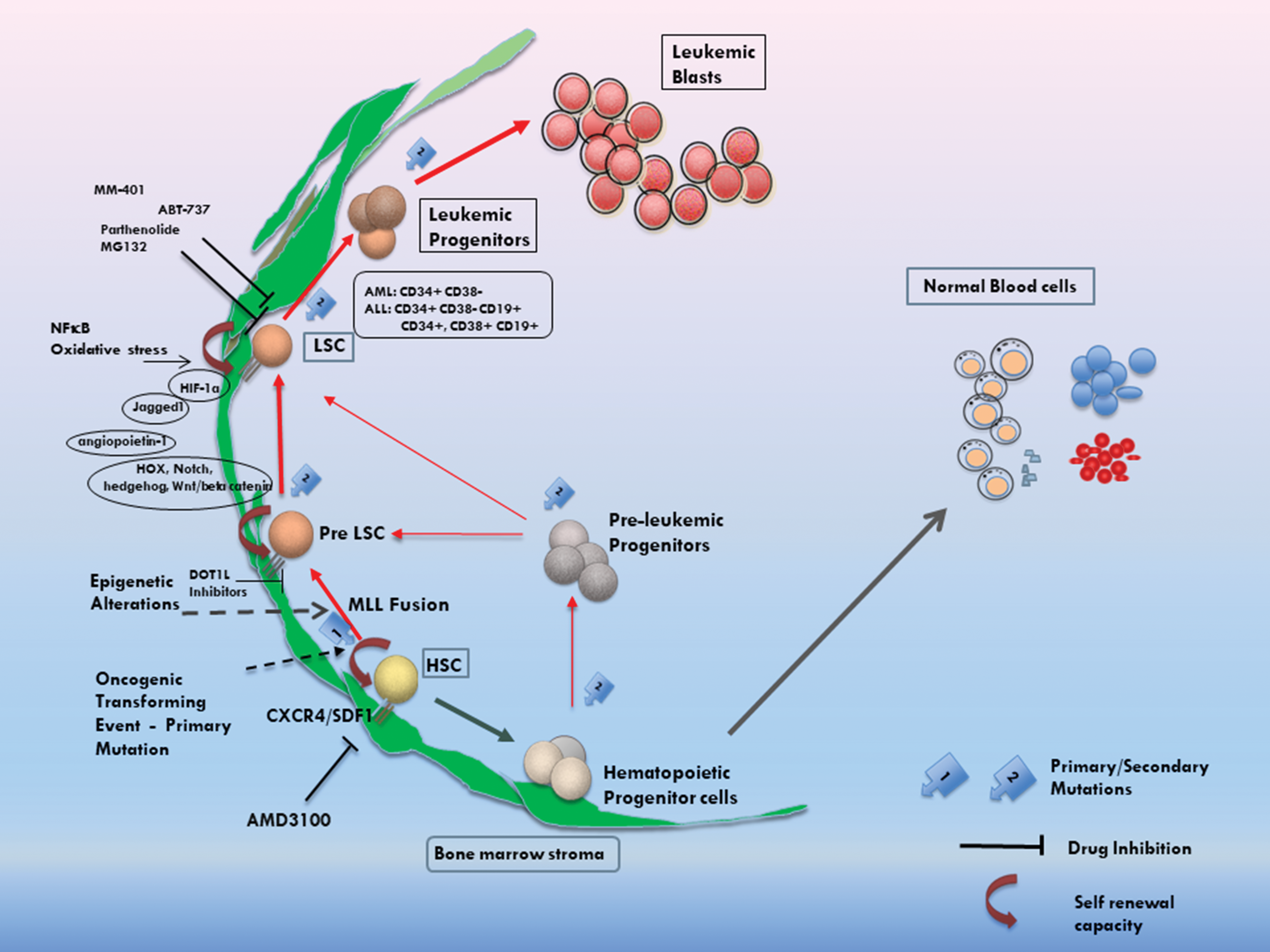
Schematic representation of key developmental pathways, known properties and potential targets for therapeutics of LSCs, and their relationship to HSCs and normal hematopoiesis. HSCs hold the potential to generate LSCs following an initial oncogenic genetic alteration, potentially represented by events such as MLL rearrangement. Such mutations lead to the generation of cells that can be enriched by cell sorting with previously identified cell surface antigens or by their absence. These cells also carry the capacity for self-renewal and the generation of differentiated leukemic blasts, supported possibly by additional mutations. A number of stromal-derived factors have also been identified, which regulate the survival, attachment, and differentiation properties of LSCs. Molecular and drug screen studies have detected a number of targets and small-molecule inhibitors to inhibit the growth of LSCs and hold promise for the development of future therapeutics for refractory leukemia by targeting the stem cell population. The cells depicted as Pre-LSCs and LSCs in this simplified schema may constitute significantly overlapping populations and/or functional characteristics. LSC, leukemia stem cell; HSC, hematopoietic stem cell; MLL, mixed-lineage leukemia. Color images are available online.
Footnotes
Acknowledgments
We acknowledge the contribution of Dr. Gaya Narendran in the preparation of the figure presented in this review and Ms. Charlotte Wilson for article support.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported, in part, by the Alberta Children's Hospital Foundation and the Kids Cancer Care Foundation (grant #10024168).