
Abstract
The growing number of patients requiring liver transplantation for chronic liver disease cannot be currently met due to a shortage in donor tissue. As such, alternative tissue engineering approaches combining the use of acellular biological scaffolds and different cell populations (hepatic or progenitor) are being explored to augment the demand for functional organs. Our goal was to produce a clinically relevant sized scaffold from a sustainable source within 24 h, while preserving the extracellular matrix (ECM) to facilitate cell repopulation at a later stage. Whole porcine livers underwent perfusion decellularization via the hepatic artery and hepatic portal vein using a combination of saponin, sodium deoxycholate, and deionized water washes resulting in an acellular scaffold with an intact vasculature and preserved ECM. Molecular and immunohistochemical analysis (collagen I and IV and laminin) showed complete removal of any DNA material, together with excellent retention of glycosaminoglycans and collagen. Fourier-transform infrared spectroscopy (FTIR) analysis showed both absence of nuclear material and removal of any detergent residue, which was successfully achieved after additional ethanol gradient washes. Samples of the decellularized scaffold were assessed for cytotoxicity by seeding with porcine adipose-derived mesenchymal stem cells in vitro, these cells over a 10-day period showed attachment and proliferation. Perfusion of the vascular tree with contrast media followed by computed tomography (CT) imaging showed an intact vascular network. In vivo implantation of whole intact nonseeded livers, into a porcine model (as auxiliary graft) showed uniform perfusion macroscopically and histologically. Using this method, it is possible to create an acellular, clinically sized, liver scaffold with intact vasculature in less than 24 h.
Introduction
Liver disease presenting as fibrosis, cirrhosis, and end-stage liver failure is a worldwide health problem affecting 50 million people and is becoming an epidemic [1]. In Europe, 29 million people suffer from chronic liver disease resulting in 47,000 deaths per year [2], while in the United States, 5 million people live with chronic liver disease. By 2020, cirrhosis will be the 12th leading cause of death [3]. Notwithstanding the financial cost, surgery-related complications and the lifelong dependence on immunosuppressive drugs, the current standard of care for cirrhosis and end-stage liver failure is still a liver transplantation.
Worldwide, ∼20,000 liver transplants have been carried out, between 1968 and 2009, 13,984 transplants were performed in the UK alone [2]. Europe as a whole reported more than 5,500 liver transplants per year [4]. The number of transplants performed has steadily increased due to improved survival rates (1-year survival is ∼83% with all factors considered). However, the greatest limitation to providing this life-saving procedure is a shortage in donor tissue, and it is becoming increasingly clear that this demand cannot be met by tissue/organ donation alone [5].
To meet this demand, alternative solutions are required, and innovative attempts to address this shortage have focused on cell therapy approaches such as hematopoietic and mesenchymal stem cell and human primary hepatocyte transplantations. Although the cell therapy approach has shown promising results in both preclinical and clinical settings, limitations to wide-scale usage include access to liver tissue, from which to harvest good quality cells and associated donor-site morbidity [6]. In parallel, considerable work has been reported on the development of bespoke tissue-engineered livers using an acellular matrix configured to resemble liver tissue and populated with cells [7]. To date, the most frequently reported matrix has been obtained using existing liver tissue made acellular using a process of decellularization. Alternative matrices using three-dimensional (3D) printing approach [8] or polymeric scaffolds have also been reported, but it is currently difficult to create a liver matrix with the exact microarchitectural structure of the liver encompassing the vascular network, the biliary tree, and the appropriate molecular cues with which to attract and retain cells to its surface. Biologically derived tissue matrices have a significant advantage over synthetic or 3D printed hydrogels [9], since the starting material not only resembles the organ being “engineered” but also the extracellular matrix (ECM) that has the ability to retain on its surface its molecular fingerprint.
To date, different approaches have been developed for creating acellular liver matrices from different species, for example, rodents [10,11], porcine [12 –14], sheep [15], and human [16,17] using various combinations of chemicals, enzymes, and physical forces. Detergents and enzymes are known to have a negative impact on the ECM of biological tissue; this damage can be mitigated by limiting the duration and exposure [18]. In addition, efficient delivery of detergents and enzymes using existing vascular circulatory systems in larger species also helps to reduce the time and potentially provide a more uniform distribution to the deeper cellular compartment [13,15].
Several groups have studied the pig liver as a potential source for decellularization and repopulation in view of the availability of pig liver and the widespread use of porcine tissues in current clinical practice. Previous studies reporting the production of acellular porcine livers using a decellularization approach have used freeze/thaw cycles followed by Triton X-100 alone, in combination with sodium dodecyl sulfate (SDS), or a combination of SDS and ethylenediaminetetraacetic acid (EDTA) [19]. The time taken to produce the acellular matrix varied between 11 days and under 24 h (this was achieved using oscillating pressure conditions [20]). All reported protocols have resulted in acellular liver tissue matrices with significant reduction in DNA content when compared to normal liver tissue. In addition, the majority of the decellularized tissue was capable of supporting cells, either when engrafted or in vitro, indicating preserved ECM. Retention of the proteins within the ECM is vital for providing a platform for subsequent seeding of the organ. The ECM plays a number of roles with respect to cellular activity [21], both positive (ie, cellular differentiation, neovascularization, hepatocyte growth, and proliferation) and negative (ie, fibrosis and cancer progression) [22]. Consequently, the ECM can no longer be viewed as an inert or barren landscape but one which plays an active and dynamic role in the biological function of the liver as a whole. As such, the process by which liver tissue is rendered acellular is crucial, and damage to the ECM should be kept to a minimum.
Different cell types have been used for recellularization of decellularized liver matrices, including iPSC-induced hepatocytes [7], human primary hepatocytes [23], and mesenchymal stromal cells. The seeding of the latter onto a liver scaffold supported hepatic differentiation and the ability to restore liver function in a severe liver failure model [11]. As such, the use of mesenchymal stromal cells as a potential cell source may be more favorable as they do not carry the potential tumorigenic risk associated with iPSCs.
This study reports the development of a vascular perfusion-based decellularization protocol capable of producing a whole acellular porcine liver in less than 24 h using sodium deoxycholate (SOC) as opposed to the more commonly used sodium dodecyl sulfate (SDS). The resultant scaffold was assessed for vascular integrity by engrafting the entire nonseeded liver scaffold into an in vivo porcine model to determine its ability to withstand physiological perfusion pressures. In addition, the presence of detergent residue as a potential cytotoxic residue was assessed by evaluating the decellularized scaffolds ability to support porcine adipose-derived mesenchymal stem cells in vitro, since there are concerns that detergents remaining on the ECM matrix may be detrimental to the viability and long-term differentiation potential of seeded cells [10].
Materials and Methods
Regulatory guidelines
This study was carried out following Ethical permission from NPIMR Ethics Review Committee for animal experimentation and in accordance with the UK Animal (Scientific Procedures) Act 1986, which conforms to the European Convention for the protection of Vertebrate Animals Used for Experimental and Other Scientific Purposes (Strasbourg, Council of Europe).
All chemicals and reagents were purchased from Sigma-Aldrich Ltd. (Dorset, UK) unless otherwise stated.
Organ retrieval
Female Large white/Landrace crossbred pigs (n = 10) ∼35–40 kg underwent liver harvest under terminal anesthesia. On the day of surgery, each pig was premedicated with ketamine (5 mg/kg Zoetis)/xylazine (1 mg/kg) intramuscularly (Bayer UK both obtained from The National Veterinary service, Stroke on Trent UK). General anesthesia was induced with isoflurane over oxygen and nitrous oxide. After intubation, anesthesia was maintained with isoflurane. A midline incision was made from the sternum to pubis and a lateral incision from the midline under the bottom left hand rib. The skin was reflected back to provide access to the bowel, which was placed in a sterile bowel bag containing a moist swab for ease of access. The liver was then mobilized from the surrounding tissue until only the hepatic portal vein (HPV) and hepatic artery (HA) remained as a vascular connection. The liver was freed from the diaphragm. Each animal then received 20 KIU of heparin. Both the HPV and HA were cannulated, the thoracic aorta was clamped and the inferior and superior vena cava incised and perfusion was initiated through both the HPV and HA simultaneously.
Scaffold production
While still in situ, the liver was decellularized by vascular perfusion using a Watson Marlow peristaltic perfusion pump at an rpm of 30. Soltran and heparin 20 KIU (2 L) were simultaneously perfused through HPV and HA followed by 0.25% saponin in warm saline (2 L) and then in warm deionized water (2 L). An additional soltran flush (2 L) was repeated followed by a series of washes with deionized water (2 L), 0.2% SOC in warm saline (2 L), and finally deionized water (2 L). At this stage, the liver was pale in color and was removed from the animal and placed into an enclosed container and the decellularization process continued on the bench using the same perfusion system (40 rpm for the remaining protocol). The protocol was continued with a saline and heparin 5 KIU flush (1 L) followed by 0.2% SOC in phosphate-buffered saline (PBS) (5 L) and saline heparin 5 KIU (1 L). The liver was then flushed with 0.2% Tween in deionized water (15 L) and 0.2% SOC in PBS (5 L). This cycle was repeated three times and then followed by 0.1% SOC (2 L). The final set of washes consisted of water only (15 L). The entire protocol was completed within 24 h. Following decellularization, the livers were either flushed with increasing concentration of ethanol (20%, 40%, 50%, and 70%), with each concentration cycle lasting 30 min or stored in PBS in preparation for the Fourier-transform infrared spectroscopy (FTIR) analysis.
Sterilization and residue assessment
To assess sterility, swabs were collected from the surface of the decellularized liver as well as deep to the surface through an incision and plated onto nutrient agar plates for bacterial and Sabouraud dextrose for fungal presences. Plates were incubated at 37°C for 48 h. The plates were examined at 12 h, 24 h, and 48 h for any fungal or bacterial growth.
To detect any detergent residue within the decellularized tissue, FTIR analysis was undertaken in control tissue (nondecellularized n = 5) and decellularized tissue washed in ethanol (n = 5) or PBS (n = 5). For FTIR point measurements, the measurement site was ∼1 × 1 mm and 30 scans were coadded for each point. Scans were performed between 550 and 4,000 cm−1 at a resolution of 4 cm−1, and 20 points were scanned for each sample (10 on each side of the sample). Each spectrum was subjected to an automatic baseline correction, normalized to the highest, most stable, and reproduced peak and the spectra average determined (Omnic Spectra Software Nicolet iS50 Edition; ThermoFisher Scientific, UK).
Scaffold characterization
Histology: Following decellularization, up to five representative full-thickness samples were taken from each lobe of the liver, fixed in 10% neutral buffered formal saline (NBF) and subsequently processed for routine wax embedding. Five micron paraffin sections were cut and stained with hematoxylin and eosin (H&E), Picro-Sirius red combined with Miller Elastin Stain (PSR-ME) and a reticulin stain.
Immunohistochemistry
The following antibodies were used to assess the basement membrane on paraffin sections: collagen I (Abcam ab6308 mouse mAb, antigen retrieval: citrate buffer 90°C for 20 min, incubation 1:200 for 1 h), collagen IV (Abcam ab6586 rabbit pAb, antigen retrieval: trypsin digestion at 37°C for 30 min, incubation 1:100 for 2 h, and Laminin (Abcam ab11575, rabbit pAb, antigen retrieval: trypsin digestion at 37°C for 30 min, Incubation 1:100 for 2 h). All washes were performed using PBS, and the secondary antibody was either Impress anti-mouse or Impress anti-rabbit immunoglobulin IgG peroxidase kit, (Vector Laboratories, Peterborough, UK) for 30 min at room temperature (RT), the chromogenic substrate diaminobenzidine (Impact peroxidase substrate; Vector Laboratories) was applied for 3 min at RT. On completion, sections were counterstained using Harris' hematoxylin.
DNA quantification and gel electrophoresis
The GenElute mammalian genomic DNA Miniprep Kit (Sigma-Aldrich) was used for DNA extraction and quantification following the manufacturer's instructions. Total DNA was quantified in all samples by measuring the absorbance using the NanoDrop Spectrophotometer (NanoDrop ND1000; Thermo Scientific, Wilmington), and from this, the absolute amount of DNA per milligram of tissue was calculated. The size, quality, and purity of the extracted DNA were determined by agarose gel electrophoresis. A 1.2% agarose (Agarose Type I, low EEO; Sigma-Aldrich) gel with 1 × Tris-borate-ethylenediaminetetraacetic acid (TBE—Bio Reagent, 10 × ; Sigma-Aldrich) running buffer was run at 4 to 5 V/cm between the electrodes. Equal volumes of DNA (5 μL) and 1 μL of loading buffer (5 × DNA loading buffer; Yorkshire Bioscience Ltd., York, UK) were loaded into each well. Visualization was achieved by staining with 1% ethidium bromide, and DNA was measured via ultraviolet transillumination against a 1-kb DNA ladder (Q-Step 4 quantitative DNA ladder; Yorkshire Bioscience Ltd.).
Analysis of ECM components
Glycosaminoglycan quantification
To quantify the glycosaminoglycan (GAG) content for both control and decellularized liver samples, the Blyscan GAG Assay Kit (Biocolor, Carrickfergus, Northern Ireland) was used. In brief, 50 mg of finely minced wet tissue was placed in a microcentrifuge tube and incubated with 1 mL of papain digestion buffer at 65°C for 18 h. Aliquots of each sample were mixed with 1,9 dimethylmethylene blue (DMMB) dye and reagents from the GAG assay kit. Two hundred microliters of each sample was added in triplicate into a 96-well plate. The absorbance was measured using a plate reader (VersaMax, Molecular Devices LLC) at 656 nm and the absolute GAG content was calculated per milligram of tissue.
Collagen extraction
Collagen was extracted from the control and decellularized samples according to published protocol [24]. In brief, samples were digested using a solution of guanidine hydrochloride buffer (4 M) (Sigma-Aldrich) with 12 μL of protease inhibitors (Sigma-Aldrich) left for 18 h at 4°C with agitation. Samples were then centrifuged for 10 min at 12,000 g to separate tissue debris. Supernatant was transferred to a microcentrifuge tube and nuclear material was digested by using RNAse enzyme (Sigma-Aldrich; activity of 140 EU/mL) for 2 min at RT, followed by DNAse I (Sigma-Aldrich; activity of 100 EU/mL). Samples were agitated without vortexing and incubated at 37°C for 1 h, and then dialyzed against distilled water overnight at 4°C. After dialysis, the supernatant was transferred to an Amicon ultra filter 2 (Ultra-4 3 kDa Ultracel-PL memb; Merck Millipore, Billerica, MA) to remove the hydrolyzed DNA and salt, and then centrifuged for 30 min at 4,000 g. For protein isolation, the centrifuged dialysate was added to 50 μL of Tris base (pH 9.5) with ice-cold phenol-chloroform-isoamyl alcohol (PCIA; Sigma-Aldrich) and allowed to stand for 24 h at 4°C, followed by centrifugation for 10 min at 12,000 g. The upper layer (containing DNA/RNA) and the lower organic phase (containing solvents) were individually retrieved by pipetting. The interface containing proteins was carefully removed and transferred to another microcentrifuge tube. Protein was precipitated using ice-cold ethanol and centrifuged for 10 min at 15,000 g at 4°C. The resultant pellet was allowed to dry and solubilized with 100 μL of ultrapure acidic water (pH = 3 with acetic acid).
Collagen quantification
Collagen was quantified using Sircol Collagen Assay Kit (Biocolor). In brief, 90 μL of each sample was subjected to Sirius red dye binding, followed by washing with ice-cold acid-salt reagent to remove unbound dye. Samples were centrifuged, and dye bound to collagen was released from the pellet with alkali reagent (1 N NaOH). Extracts were placed in a 96-well plate in triplicates and spectrophotometric readings were taken at 555 nm on a microplate reader (VersaMax, Molecular Devices LLC). Absolute values were attained with a standard curve composed of type I bovine skin collagen solution (0.5 mg/mL) in the range of 5–100 μg per 0.1 mL. Total collagen was normalized per milligram of tissue.
In vitro cytocompatibility assessment using adipose-derived mesenchymal stem cells
Cells isolation and culture
Adipose tissue was collected from Large White/Landrace crossbred pigs. Under anesthesia and using aseptic techniques, [the abdomen was cleaned using 10% Iodinated iodine followed by sterile saline (Baxter 0.9% NaCl)] a 3 cm-transverse cutaneous incision was made. Approximately 5 g of adipose tissue was harvested from beneath the dermis, cut into small pieces, and prepared for digestion. Collagenase 0.1% in PBS was added to the tissue and allowed to digest for up to 2 h at 37°C with agitation (50 rpm). The stromal vascular fraction (SVF) was separated from the remaining fibrous material and the floating adipocytes separated by centrifugation at 300 g for 10 min. The sedimented SVF cells were filtered through a 100 μm pore filter followed by an incubation step in an erythrocyte lysing buffer (160 mM NH4Cl) for 10 min. For initial cell culture and expansion of the adipose derived mesenchymal stem cells (AS-MSC), low-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 Ug/mL streptomycin was used. Cultures were washed with PBS 24 h after plating to remove unattached cells and then fed with fresh medium. Cultures were maintained at 37°C with 5% CO2, and medium was changed three times per week. Cells were grown to confluence after the initial plating (P = 0), typically within 7 days. Once confluent 80%, the adherent cells were released with 0.5% trypsin–EDTA and then either plated at 25 × 103 per cm2 or used for experimental analysis. All cells used for analysis were early passage (passages 2 to 4).
In vitro cytocompatibility assessment
Before the in vitro assay, liver scaffolds were prepared as described above. In brief, decellularized samples were cut into 1 × 1 × 0.2 cm samples and individual pieces placed into a 24-well plate and incubated in growth medium (DMEM with 10% fetal calf serum [FCS] and 1% antibiotics and antimycotic [AA]) overnight at 37°C, including 5% CO2. AD-MSC were released from the culture flasks using a nonenzymatic solution, counted, and seeded onto each decellularized liver sample at 2.5 × 105 cells per cm2 in triplicate. Cells were left undisturbed to adhere for 2 h and then fresh growth medium was added into each well. The medium was changed every other day. After 1, 2, 4, 7, and 10 days of culture, the liver scaffolds were collected, gently washed with PBS, fixed in NBF 10% for 24 h, and processed for histological analysis.
In vivo assessment
Vascular integrity by contrast perfusion and computed tomography imaging
To assess the integrity of the vasculature following decellularization, a single decellularized liver was perfused with contrast media (ex vivo) (Omnipaque GE HealthCare, UK) initially through the HA and then through the HPV and imaged (GE Healthcare Innova 4100) as an assessment for leakages and or blockages.
In vivo implantation
Two intact decellularized livers were implanted into two separate pigs under terminal anesthesia (ie, nonrecovery) following left-sided nephrectomy to evaluate (1) whether the decellularized liver would be reperfused under physiological conditions (2) the technical aspects of vascular reconstruction, and (3) uniformity and pattern of the perfusion. This procedure was undertaken without clamping or removal of the host's native liver.
Procedure 1
The decellularized liver was primed with 5,000 IU of heparin in 1 L of RT saline (Baxter) through the HP and HPV. A left-sided nephrectomy was performed in the host animal and HA of the decellularized liver connected to the recipient left renal artery, the decellularized inferior vena cava to the host left renal vein and the decellularized HPV to the host HPV. The decellularized superior vena cava was tied close. The liver was allowed to perfuse under constant monitoring for ∼90 min.
Procedure 2
The decellularized liver was primed using 2 × 5,000 IU of heparin in 500 mL of RT saline. Implantation of the decellularized liver was via direct anastomosis as described as above. The decellularized inferior vena cava was sutured to the renal vein, the decellularized HPV to the host portal vein and the decellularized HA to the host renal artery. The liver was allowed to perfuse under constant monitoring for ∼70 min.
For each animal, vital signs, including blood pressure, heart rate, and oxygen saturation were monitored. Following termination, the implanted liver was removed and samples were taken from each lobe and prepared for histological evaluation as previously described.
Results
Scaffold characterization
Following decellularization, the entire liver tissue mass turned to a pale golden color; the gallbladder became white, (indicating sufficient perfusion through the HPV and HPA), as well as all the blood vessels (HPV and HPA) associated with the liver. Histological assessment of H&E-stained sections showed an intact hepatic lobular structure together with all the main features of the portal triad (bile ducts HPV and artery) could be easily identified. All structures showed complete absence of any nuclear material or cellular structures. The absence of cellular and nuclear material was also confirmed by DNA molecular analysis, which showed a 70% reduction in DNA content of the decellularized tissue compared to native liver tissue (control 634.4 ± 14.6 ng/mg vs. de-cell 3.36 ± 7.66 ng/mg) and no visible DNA bands were observed by gel electrophoresis (Fig. 1).
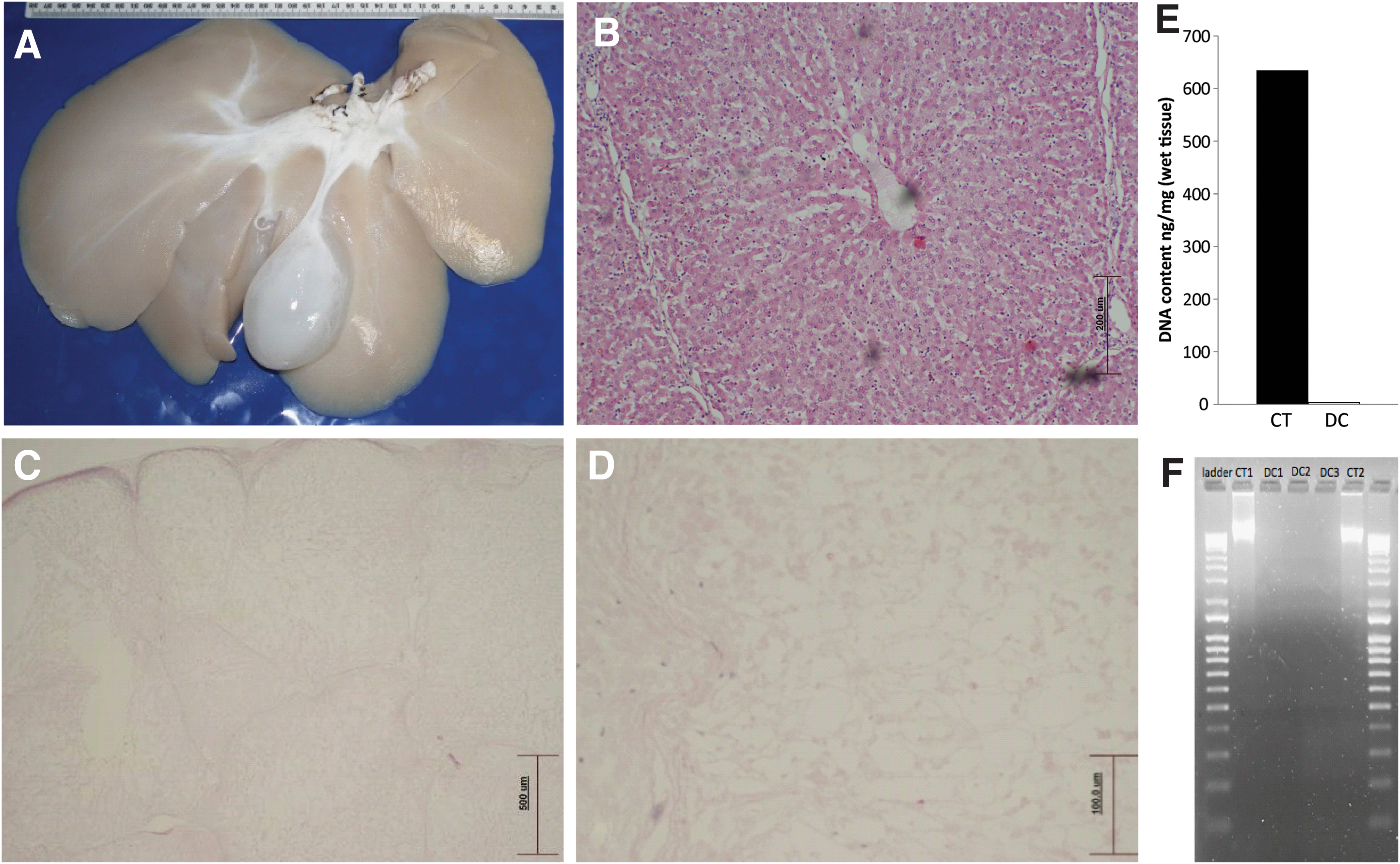
Scaffold characterization showing the liver postdecellularization
Following decellularization, microbiology confirmed sterility of the tissue for both bacterial and fungal contamination over a period of 10 days (data not shown).
FTIR spectra for the three groups, control (ie, nondecellularized), decellularized (washed in PBS), and decellularized (ethanol wash) are shown in Fig. 2. Regions of the spectra associated with proteins, lipids and/or polysaccharides, and nucleic acids were identified and key peaks numbered 1–10. No differences were observed between groups for peak 1 (3,100–3,600 cm−1) relating to proteins, water, and alcohols, peak 4 (1,600–1,700 cm−1) identifying amide I, and peak 5 (1,500–1,600 cm−1) associated with amide II bands of protein in the three groups suggesting preserved ECM integrity. However, peak 2 (2,800–3,000 cm−1, -CH stretching) and peak 6 (1,300–1,500 cm−1, -CH bending) associated with lipids and or polysaccharides showed significantly stronger transmittance in the decellularized-PBS wash tissue than in both the control and decellularized ethanol samples showing the same transmittance. This difference may be attributed to residual deoxycholine retained from the decellularization process within the decellularized PBS-washed samples. Strong peaks observed at 2,800–3,000 cm−1 and 1,700–1,725 cm−1 are characteristic of deoxycholate and lipids. The intensity of peak 3 (1,700 cm−1, associated with Carbonyl and lipid groups) was reduced in both decellularized samples, but substantially more in the decellularized PBS wash; an ethanol wash did not restore the levels to that seen in control tissue. This suggests potential ECM alteration by the decellularization process as characterized by increased carbonyl groups in the liver ECM. Crucially, however, peaks (800–1,200 cm−1) associated with deoxyribose stretching and symmetric and asymmetric PO2 − groups was stronger in the control sample (peak 9, 1,026 cm−1 and peak 10, 1,078 cm−1) than in both the decellularized samples, where it had shifted to weaker peak at 1,065 cm−1, with the decellularized PBS-ethanol sample being lower of the two. It appears that a final wash with ethanol removed residual detergent and lipids while also removing nucleic materials without any detrimental impact on the ECM as demonstrated by the restoration of the FTIR spectral profile after ethanol wash to closely match that of the control (nondecellularized).
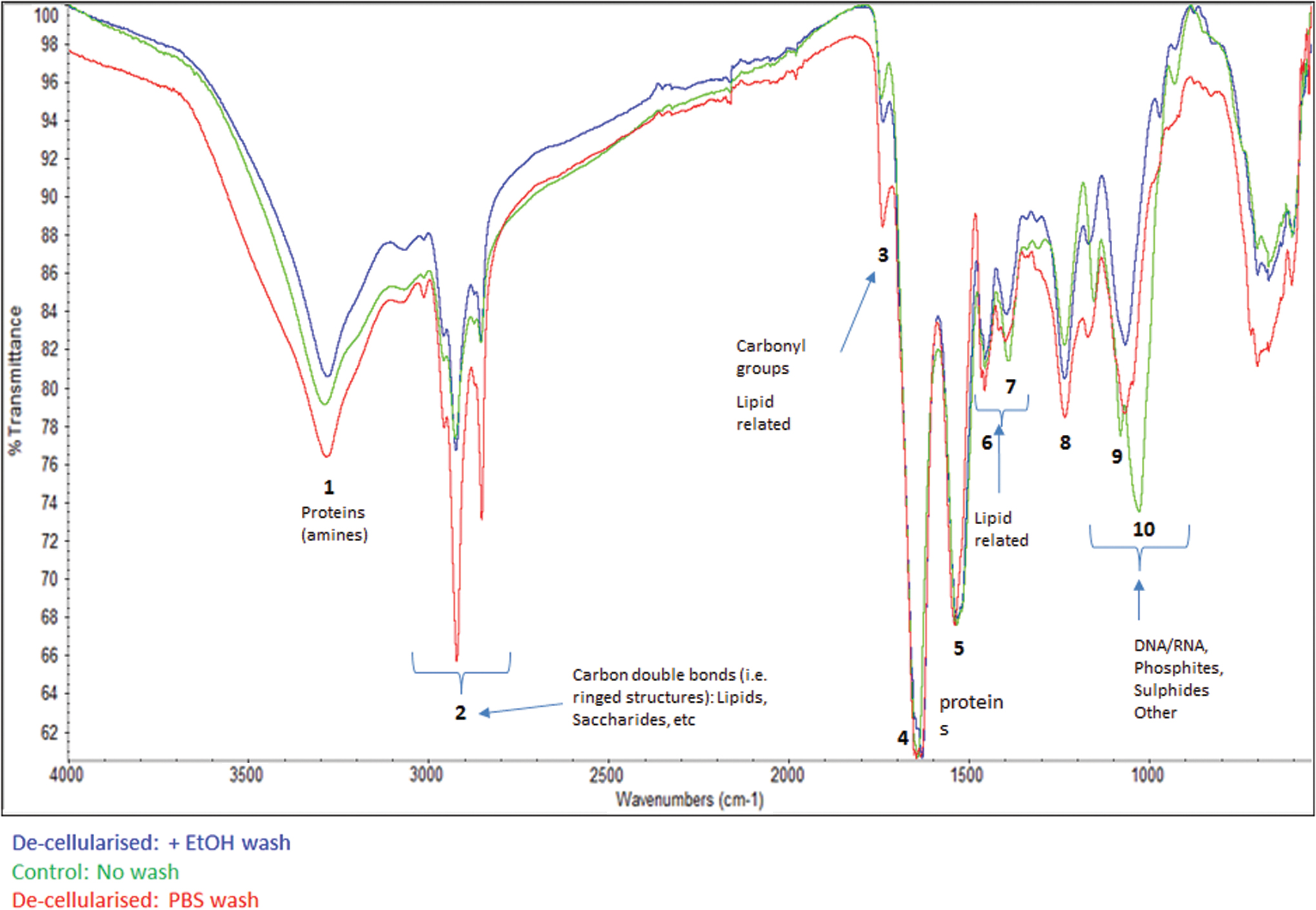
The FTIR profile is presented for control samples (green), decellularized samples washed in PBS (red) and decellularized samples washed in ethanol (blue). Different peaks have been annotated to represent different molecular structural components. FTIR, Fourier-transform infrared spectroscopy; PBS, phosphate-buffered saline. Color images are available online.
As the functionality of the ECM of the decellularized liver will be vital for cell repopulation, when implanted at a later stage, detailed analysis was undertaken. PSR-ME-stained sections clearly showed intact boundaries of the hepatic lobes (ie, interlobular septa); while at a higher magnification, intact elastin within each blood vessel wall could also be seen. When viewed under polarized light, the collagen structure showed excellent birefringence with the collagen appearing as bright orange/red indicating potential for functionality (Fig. 3A–C). Reticulin fibers were also well preserved, illustrating preservation of the fine fibrillary structure of the stroma within the connective tissue, this collectively demonstrated retention of key ECM components (Fig. 3D). Detailed IHC staining of the sections showed the appropriate location and presence of laminin (outlining the basement membrane of the blood vessels Fig. 3E, F), collagen 1 (liver capsule and portal stroma Fig. 3G, H) and collagen IV (basal lamina of blood vessels and bile duct, Fig. 3I, J) within the stroma of the decellularized tissue. Both the molecular collagen (control: 141.4 ± 18 μg/mg vs. decellularized: 104 ± 22.44 μg/mg; 26% decrease P < 0.05) and GAG data (control: 0.12 μgs GAG/mg ±0.03 vs. decellularized: 0.08 μgs GAG/mg ±0.007, P < 0.05 representing a 28% decrease in the decellularized tissue) were in accordance with the morphology data (Fig. 3K, L).
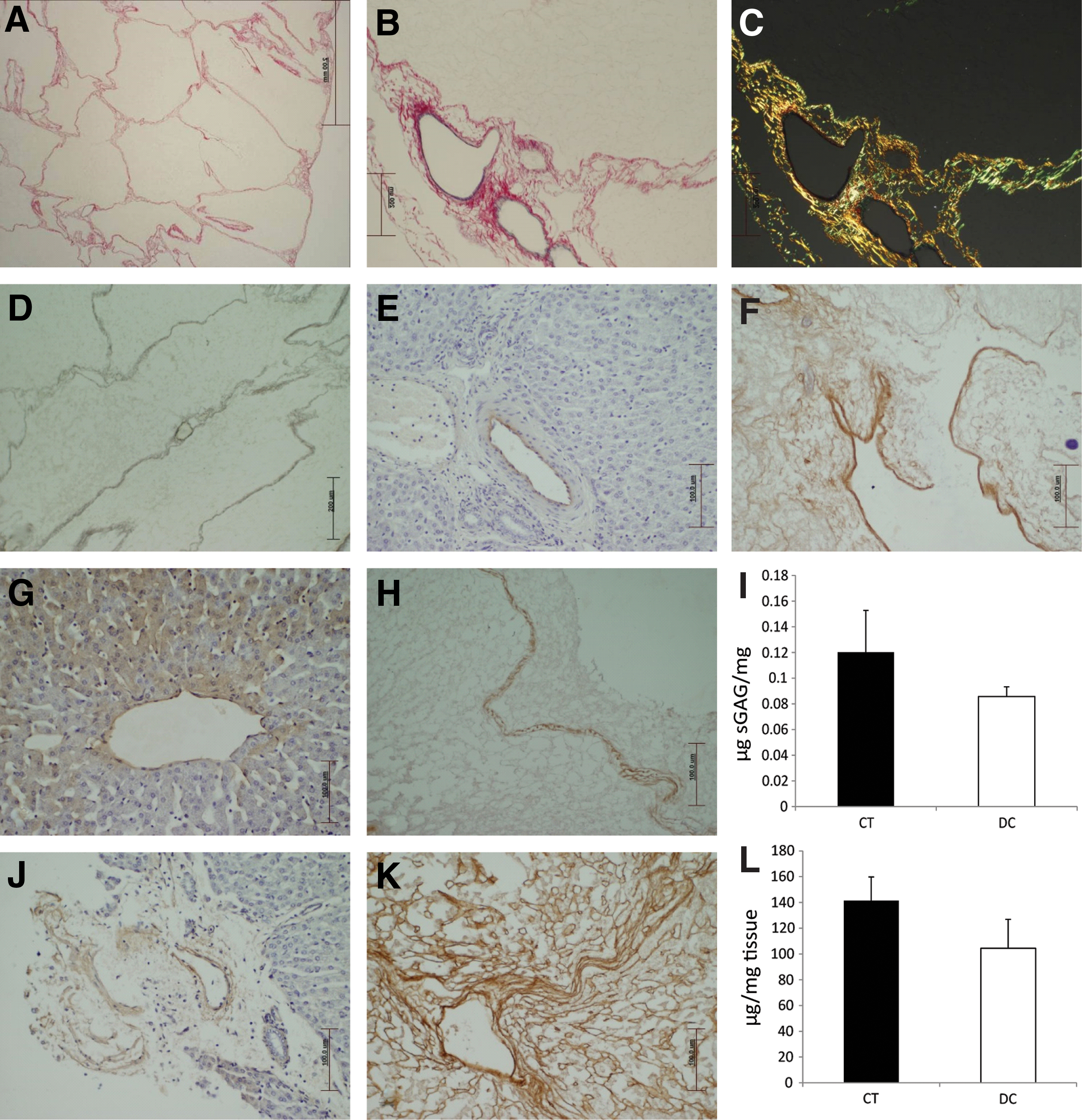
ECM characterization of the decellularized scaffold. Decellularized tissue was initially stained with PSR-ME to show the fine collagen fibers outlining each liver lobule
In vitro analysis
Samples of sterile decellularized liver matrix were used to confirm cytocompatibility of the tissue using porcine AD-MSC. AD-MSC were seeded onto the parenchymal (Fig. 4A–E) and serosal (Fig. 4F–J) surface individually over a 10-day period. Cells readily attached to both surfaces and showed good morphology with little cell death accompanied by cellular migration into the tissue. Twenty-four hours postseeding, cells were easily identifiable on both surfaces, by day 2, both surfaces showed cells with elongated morphology. By day 4, cells seeded onto the cut parenchymal surface had multiplied and were seen to migrate into the tissue while those on the serosal surface were beginning to form a multilayered feature consisting of elongated viable cells. By day 10, both surfaces showed cells in various stages of differentiated morphology based on appearance.

Shows the in vitro cellular biocompatibility of the seeded scaffold for both the parenchymal and serosal surface over the 10-day period. For each seeded surface, viable cells can be clearly seen changing in morphology with increasing time. Color images are available online.
Liver perfusion analysis
To determine whether an intact vasculature had been retained following exposure to the detergents as part of the decellularization process, a single liver was perfused with contrast media and computed tomography (CT) imaged initially through the HPV (Fig. 5A, B) and then the HA (Fig. 5C, D). In both cases the venous and arterial vasculature was uniformly perfused to the peripheral edges of the liver, including all the smaller capillary vessels. No leakage was seen indicating that the blood vessels were intact throughout the liver.
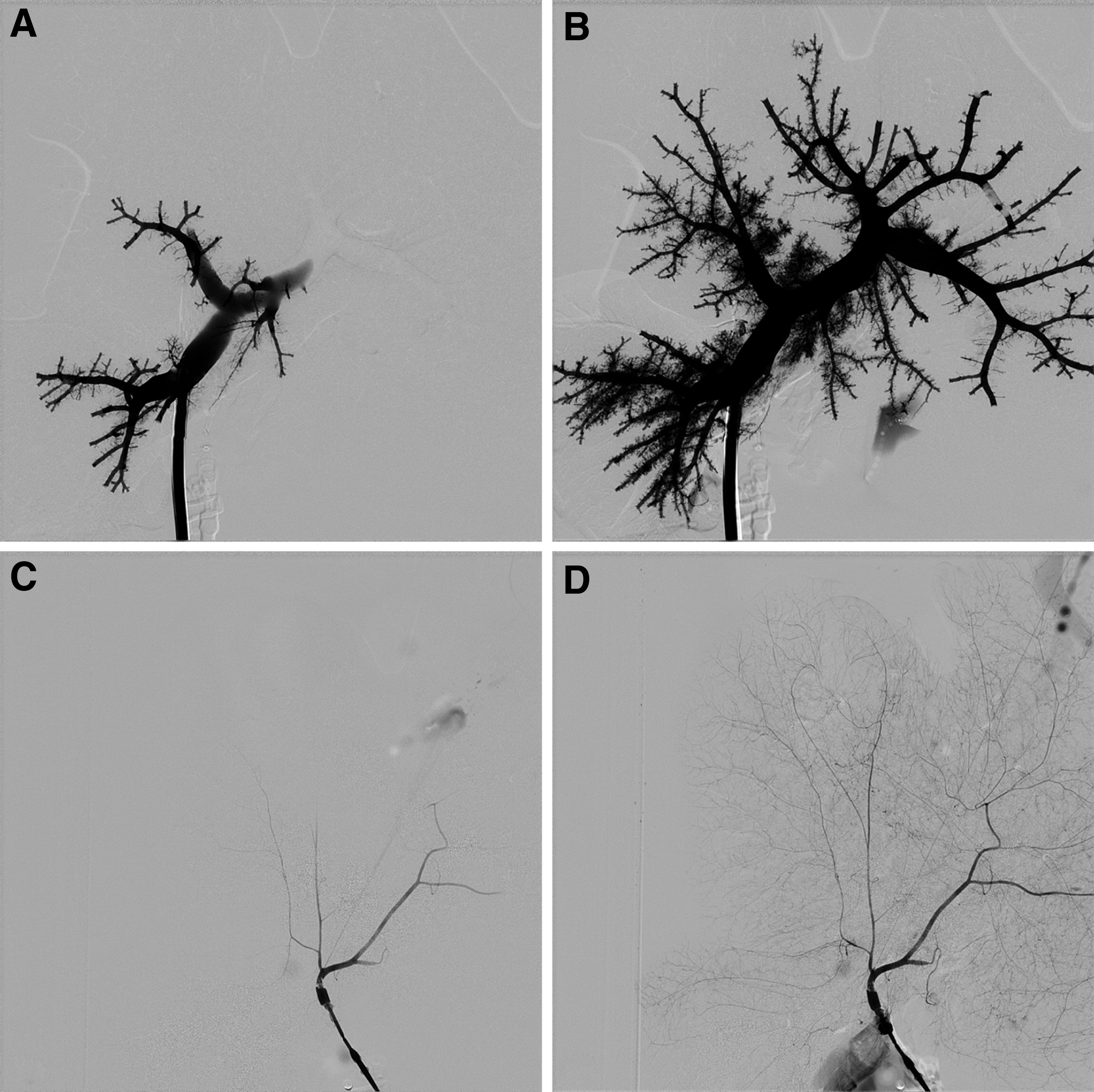
In vitro perfusion and CT imaging was undertaken to show intact retention of the both the venous
In vivo analysis
Following, priming decellularized liver scaffolds were implanted into two animals in nonrecovery procedures (Fig. 6A, B). Perfusion of hosts blood into the liver scaffold occurred slowly, but over time resulted in the entire liver scaffold taking on the characteristic color and consistency of a normal liver. However, darker patches of potentially pooled or coagulating blood became apparent macroscopically. The first animal was monitored for 90 min, but was terminated due to circulatory failure, accompanied by a loss of blood pressure. For the second animal, the liver scaffold was primed with a lower volume of saline (500 mL vs. 1 L) This animal was monitored for 70 min and terminated due to marked elevation in heart rate (>240 bpm). Although each animal was given Hartmann fluid throughout the procedure, no additional blood was administered, the increased heart rate was probably due to large volume of blood being shunted into the implanted liver scaffold.
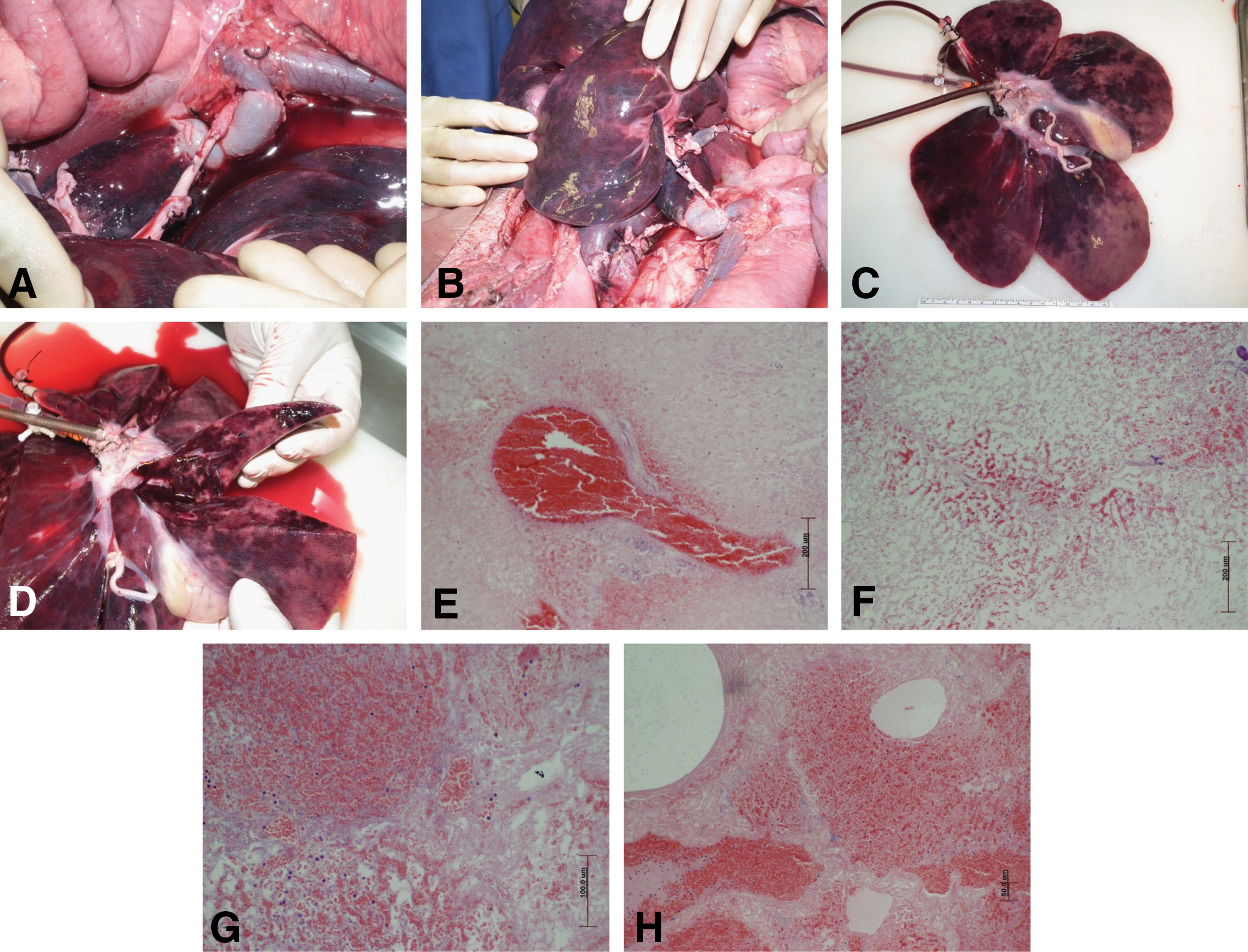
In vivo implantation was undertaken to demonstrate the ability of the scaffold to withstand physiological vascular perfusion pressures. On implantation, the scaffold was uniformly perfused with the host's blood
Following termination of each animal, the liver scaffold was removed and macroscopic inspection showed complete perfusion of the liver and some change in the color of the gall bladder (Fig. 6C, D). Each lobe was truncated and showed perfusion deep into the liver tissue, but accompanied by some dark patches. Analysis of H&E-stained sections not only showed engorged blood vessels but also occasional extravasation of red blood cells into the tissue matrix (Fig. 6E–H). Histologically, the tissue was not uniformly perfused with some areas showing accumulation of blood and other areas with little to no presence of red blood cells. Numerous nucleated white cells could be seen within the red blood cell population (Fig. 6G).
Discussion
This study was undertaken to develop a biologically derived liver scaffold applicable for human clinical application. Porcine livers were chosen as a starting material due to availability, clinical size, and ethical acceptability and to avoid any competition with human liver tissue destined for transplantation. Our approach demonstrated the production of a whole porcine acellular 3D scaffold suitable for future clinical translation in ∼18 h, which is less than the previously reported 24 h used for human livers [25]. Our scaffold had a preserved vascular circulatory system, was capable of supporting adipose-derived mesenchymal stem cell growth and migration, was capable of withstanding normal physiological perfusion pressures when implanted in vivo, and was perfused uniformly by the host's blood. The overall protocol is very effective with respect to time and cost and has the potential to be automated and translated to eventual clinical usage. An advantage in using porcine livers to create liver scaffolds for future human clinical implantation is the readily available supply of healthy tissue from food chain animals. This source of material limits and conflicts with the requirement for human livers which can be retrieved for transplantation. However, future studies would need to address any safety concerns around zoonosis; specifically, viral infections and issues surrounding potential galactose-alpha-1,3-galactose (alpha-gal) epitope.
Both the histological and molecular data demonstrated removal of all nuclear material leaving behind an intact ECM composed of collagen, elastin, and GAG's. Crucially to confirm that the entire liver had been decellularized, representative histological samples were taken from each liver lobe (∼5 per lobe) and analyzed individually. Unlike previously published studies [26] our scaffold did not become translucent as seen in other tissues from different species (eg, rodents), but remained a pale gold or beige color. This pale gold or beige appearance has been previously reported in both porcine and human decellularized livers and described as a possible consequence of lipofuscin accumulation which occurs with increasing age. Verstegen et al., noted the presence of brown deposits on unstained and H&E-stained histological sections from human liver decellularized tissue; they confirmed the presence of lipofuscin using Sudan Black B stain [17]. The pig livers used in this study were all from animals less than 50 kg in weight (∼12 weeks old), therefore, it is debatable whether the beige color of the scaffold at the end of the process is due to lipofuscin accumulation and is unlikely to be age related. In addition, no brown deposits were seen on H&E-stained sections either prior or postdecellularization. Our previous study on porcine esophagus scaffolds also showed a pale coloration of the scaffold tissue following complete decellularization, which was confirmed both molecularly and histologically [27].
Porcine livers were harvested using a similar approach to that used for harvesting human livers for transplantation. The liver's inherent vascular circulatory system (HPV and HA) was used to deliver decellularizing reagents into the tissue under peristaltic flow; the latter chosen to mimic normal liver physiology. Similar to previous studies using this route, the intricate vascular network remained intact, as did the internal elastic lamellae on small blood vessels. This is crucial as it may help to prevent blood clotting when reperfused. The absence of endothelium and exposure of collagen fibers from the basement membrane to circulating blood was thought to activate the clotting cascade and cause vascular thrombi noted in previously implanted decellularized tissues [28]. In addition, blood and cell culture media both exhibit different mechanical flow properties (non-Newtonian vs. Newtonian) and as such perfusion ex vivo may not replicate or mimic the in vivo scenario.
Soltran is an organ preservation solution and has been used clinically for many years. It was heparinized and used to flush the liver of red blood cells and any thrombi in preparation for cell lysis; this was accelerated by using saponin, which acts by causing the lipid bilayer to become more permeable to macromolecules. The use of SOC is a departure from the use of SDS or Triton x-100 as the main detergent. SDS as an ionic detergent causes proteins to denature, deoxycholic acid is naturally produced as bile acid by the body and the sodium salt of deoxycholic acid is known to solubilize cellular and membrane components by acting specifically on lipids. As a mild detergent it has previously been used to decellularize various tissues with varying outcomes depending upon tissue thickness [26]. Significantly, it has been used to decellularize 3 mm thick slices of porcine liver, but at a much higher concentration of 4% [29] compared to the 0.2% initially used followed by 0.1% used in this study. The sequential use of saponin followed by SOC was designed to increase the overall impact on cell breakdown, while keeping its impact on the ECM to a minimum.
The predominant component of our decellularization protocol was deionized water and 0.2% Tween 20, the latter acting as a nonionic nontoxic surfactant and was essential in removing the cell breakdown products from the scaffold. Previous uses of SDS at concentrations of 0.01%, 0.1%, and 1% when combined with Triton x-100 have all resulted in the production of acellular liver scaffold. However, residual SDS on the ECM has proven to be toxic to cells when scaffolds have been repopulated and ideally SDS should be avoided during the latter stages of any decellularization protocol [30]. Therefore, the final stage of our protocol was completed washes using water. This was followed by an ethanol rinse to assess whether this would remove any residual detergent bound to lipid breakdown products.
FTIR analysis is not routinely used for assessing the effect of decellularization on the ECM of biological tissue with only few studies reporting its use. However, FTIR can assess the composition of the ECM [31] and can produce standardized repeatable results. To date, it has been used to assess the ECM finger print in fibrocartilage bioscaffold for bone-tendon interface [32] and in the evaluation of decellularized human tracheal samples [33]. It has not to our knowledge been used to determine the presence of any residual detergent or enzymes remaining in biological tissue following decellularization. An additional advantage in using FTIR is its potential to be used as a quality control method for scaffolds destined for clinical application with respect to retained detergent or enzyme residue. Our data clearly demonstrated that additional treatment of the decellularized liver using ethanol-gradient washes helps to eliminate both detergent residue and sterilize the scaffold. It is possible that during the decellularization process, a point is reached where the ratio of detergent to lipid break down product favors the lipids, and as such, the detergent becomes bound up in the lipid forming a complex. This would then potentially remain behind as a residue and may cause cellular toxicity, as shown by previous in vitro studies, where the seeded cells did not attach to the matrix [30]. Additional wash steps using water alone (data not shown) were not sufficient to remove any remaining residue, and therefore, the use of an organic solvent such as ethanol, which is gentle on the ECM, was used to capture the lipid–detergent complex and remove it from the tissue. Overall, the FTIR data suggest that the final washes with ethanol removed any detergent residue, leaving behind a sterile matrix capable of supporting AD-MSC in vitro.
Alcohols have been reported to be effective at removing lipids, but this is accompanied by tissue dehydration, which leads to cell lysis and elimination of cellular materials [34,35]. However, alcohols have also been reported to precipitate some proteins [34 –36], with the potential to alter ECM ultrastructure, particularly structural proteins such as collagens, elastin, and growth factors. As such, the benefits of ethanol use observed in this study may not be applicable to other tissue types and should be used conservatively. However, it is worth noting that any residual chemical reagents and detergents retained in the ECM after decellularization can be cytotoxic to colonizing cells and potentially lead to graft failure in vivo. It has been suggested that the use of alcohols be limited to the elimination of phospholipids from tissue, since the preservation of phospholipids in the tissue could cause calcification [34,35].
Retention of a potentially functional ECM is important for future recellularization and implantation, and similar to previous studies our scaffold showed preservation of collagen (I and IV), laminin, and reticulin. Preservation of the basement membrane is important for recellularization since it controls cellular growth and differentiation during embryogenesis while directing cellular migration and epithelialization during tissue repair [37]. The major components of the basement membrane include collagen IV, laminin, perlecan, and nidogen, which have been retained in our decellularized scaffold. In addition, the internal elastic lamella of the basement membrane in smaller blood vessels was also preserved. This adds a level of confidence in using the vasculature as a route of delivery for recellularization at later date.
Although analysis of ECM in our scaffold demonstrated retention of the major ECM components, no attempt was made to determine whether key cytokines or growth factors normally associated with the ECM had been retained following the decellularization [38]. Along with the molecular integrity of the scaffold, the structural/mechanical integrity must also be assessed, since both are known to influence cell behavior (specifically hepatocytes) at least in in vitro cell culture [39]. An unstable or damaged liver ECM is unlikely to retain cellular phenotype and may deter cellular dedifferentiation. The presence of reticulin outlining the lobule boundaries indicates that a mostly complete ECM framework has been retained along with all the porous infra structure. This suggests that a significant degree of mechanical integrity has been retained; however, this can only be verified following large scale seeding and in vivo implantation.
To assess whether the decellularized scaffold was capable of supporting cells (any cells), it was seeded with porcine-derived AD-MSC's as a simple cytotoxicity study over a short period of time in vitro. Had the cells all died within the first few days, the decellularization process would have been considered unsuitable and would have required modification. Detergent residue left from the decellularization process can impact on cellular attachment, the FITR analysis was undertaken partially to address this issue of detergent residue. Since functionality of the cells was not relevant and nor were the cells likely to differentiate into liver specific cells, no further analysis was undertaken. We specifically did not set out to differentiate the AD-MSC's into liver specific or hepatic lineages in this study.
Previous studies have used numerous cell types to seed liver matrices, including iPSC, which showed significant expression of hepatic markers [40] following implantation and mesenchymal stromal cells. MSC's hold promise on various levels, when seeded onto a liver matrix, they have been shown to differentiate into hepatocytes and when transplanted to restore liver function in a lethal liver failure model [11]. Our scaffolds were seeded for 10 days with AD-MSC's, and although they did not express any markers of differentiation (hepatocyte nuclear factor 4 alpha or CK 19–data not shown), probably due to the short seeding period, the cells showed changes in cellular morphology and migration indicative of a degree of cellular–matrix cross talk. The use of primary porcine hepatocytes has also been reported as a potential cell source [41]; however, concern surrounding immune rejection and the possibility of xeno zoonosis have yet to be fully addressed. The advantage in using a xenogenic cell source is that there is no limitation to donor tissue availability for recellularization unlike the current clinical situation.
Characterization of our scaffold clearly demonstrates that it has the potential to support cells, while the intact vascular system points to a favorable route for the delivery of cells for recellularization. However, this observation is based on a limited number of animals (n = 2) and was used primarily to demonstrate that it is (1) surgically possible to suture the decellularized blood vessels to normal tissue and (2) whether the entire liver is capable of being uniformly perfused. Follow-up studies are required with a larger cohort of animals using seeded (hepatic progenitors) in a recovery model. A lack of an integral vascular supply and network to any tissue-engineered organ runs a significant risk of failure due to the inability to keep the cell supplied with sufficient nutrients and oxygen. Our ex vivo perfusion study using contrast media clearly showed that both the arterial and venous vascular system retained an intact basement membrane allowing for complete perfusion with no evidence of extravasation into the surrounding tissue. On implantation in vivo, the entire liver was perfused with blood demonstrating that it might be possible to deliver cells and nutrients into the thickest portion of the liver and maintain oxygen exchange within the tissue. Crucially, if the structural integrity of the ECM can be maintained following decellularization, molecular cues on the ECM may promote matrix-cell-crosstalk resulting in a more optimized recellularization process [42]. In a previous porcine small bowel decellularized scaffold, we demonstrated that by connecting the unseeded scaffold onto the renal artery and vein, it was possible to see host cells in the villi (located in the mucosa) of the small bowel tissue, following 24 h implantation [43]. This was possible because the scaffold had retained its overall structural integrity.
Previous studies of implanted decellularized liver scaffolds in preclinical models have highlighted a number of hurdles, including thrombosis and hemorrhage, which have prevented human clinical translation. A potential cause for the formation of blood clots in the reperfused liver is the interaction between blood and the blood vessel wall. In some cases, blood vessels within the decellularized livers lacked an intact endothelium and unless particular attention has been paid to the reagents used during decellularization, the internal elastic lamina may also be damaged. The consequence is that blood flowing through the vessels may be in direct contact with the collagen fibers and may activate the clotting cascade. Clinically, damage to the endothelium exposing the collagen within the basement membrane can result in intimal hyperplasia initiated by thrombus formation [44].
Conclusion
It is possible to create a clinically relevant sized acellular liver scaffold, which has the potential following additional investigation to address the clinical human liver transplantation shortage. A tissue engineering approach to creating a liver scaffold with both macro- and microarchitectural anatomy incorporating an intact vascular and biliary network is possible. The use of porcine livers as the starting material represents an appropriately sized tissue which can be readily obtained. In addition, the use of porcine livers will not compete with any human livers destined for transplantation. An integrated vascular network provides the optimum route for recellularization using any number of different cell types and supplies appropriate route for nutrient delivery. Our method is a low-cost approach and coupled with FTIR analysis for quality control and presents a potential pathway for creating tissue-engineered liver for future human application.
Footnotes
Acknowledgments
The authors acknowledge the Paul Foundation for its generous financial support. Additional thanks go to the husbandry and theater staff at NPIMR and Theoni Demcollari and Hemal Shah for technical assistance
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Funding for this article was supplied by the Paul Foundation.