
Abstract
Fetal bovine serum (FBS) is widely used to culture mesenchymal stem cells (MSCs) in the laboratory; however, FBS has been linked to adverse immune-mediated reactions prompting the search for alternative cell culture medium. Platelet lysate (PL) as an FBS substitute has been shown to promote MSCs growth without compromising their functionality. Fibrinogen contained in PL has been shown to negatively impact the immune modulating properties of MSCs; therefore, we sought to deplete fibrinogen from PL and compare proliferation, viability, and immunomodulatory capacities of MSCs in FBS or PL without fibrinogen. We depleted fibrinogen from equine platelet lysate (ePL) and measured platelet-derived growth factor-beta (PDGF-β), transforming growth factor-beta (TGF-β) and tumor necrosis factor-alpha (TNF-α) through ELISA. First, we determined the ability of 10% ePL or fibrinogen-depleted lysate (fdePL) compared with 10% FBS to suppress monocyte activation by measuring TNF-α from culture supernatants. We then evaluated proliferation, viability, and immunomodulatory characteristics of bone marrow-derived MSCs (BM-MSCs) cultured in FBS or ePL with or without fibrinogen. Growth factor concentrations decreased in ePL after fibrinogen depletion. Lipopolysaccharide (LPS)-stimulated monocytes exposed to ePL and fdePL produced less TNF-α than LPS-stimulated monocytes in 10% FBS. BM-MSCs cultured in fdePL exhibited lower proliferation rates, but similar viability compared with BM-MSCs in ePL. BM-MSCs in fdePL did not effectively suppress TNF-α expression from LPS-stimulated monocytes compared with BM-MSCs in FBS. Depleting fibrinogen results in a lysate that suppresses TNF-α expression from LPS-stimulated monocytes, but that does not support proliferation and immune-modulatory capacity of BM-MSCs as effectively as nondepleted lysate.
Introduction
Mesenchymal stem cells (MSCs) have been extensively studied with promising results for the treatment of musculoskeletal, inflammatory, and immune-mediated diseases [1,2]. More recently the focus of MSC-based therapy has shifted from promoting tissue regeneration to relying on their ability to orchestrate healing responses through an immunomodulatory effect [1,3]. Regardless of their mode of action, MSC function is likely affected by laboratory manipulation and exposure to culture medium such as fetal bovine serum (FBS), which is widely used as a source of growth factors and nutrients [4,5]. Unfortunately, FBS contains bovine serum antigens [6], high endotoxin concentrations [7], and may be a source of microbial contaminants, such as bacteria, fungi, viruses, and prions [8,9]. Furthermore, FBS is a xenogenic culture supplement and immune reactions have been recorded in animal and human patients after the administration of MSCs cultured in FBS-supplemented medium [9]. As a result of the mentioned concerns, the need exists to investigate equally efficient growth medium supplements homologous to the species from which MSCs are obtained to minimize medium-dependent immune responses [9 –11].
Platelet lysate (PL) is a promising homologous alternative to FBS as a cell culture supplement [5,12]. Our laboratory has shown that equine platelet lysate (ePL) can be manufactured according to established protocols from platelet concentrates obtained through platelet apheresis [13]. Moreover, ePL suppresses the activation of lipopolysaccharide (LPS)-stimulated monocytes and offers an alternative to FBS as a serum-free medium supplement for the culture of equine bone marrow-derived MSCs (BM-MSCs) without compromising their functional characteristics [14,15]. Based on our results and on the existence of conflicting data regarding the ability of MSCs to exhibit immunomodulation when exposed to PL [16 –19], we designed a series of experiments aimed at further refining the use of PL as a culture medium supplement. During the process of platelet apheresis, it is common practice to add the platelet suspension in 10% of the anticoagulant acid citrate dextrose to avoid clotting. The resulting PL is rich in clotting factors such as fibrinogen, which may have a deleterious effect on the function of cells exposed to a fibrinogen-rich medium. Fibrinogen is a soluble 340-kDa acute phase protein known to mediate inflammatory processes [20] and has been shown to bind to MSCs and result in an increased production of proinflammatory cytokines such as monocyte chemoattractant protein-1 (MCP-1), interleukin (IL)-8, and IL-6, polarizing MSCs toward the proinflammatory phenotype once the Toll-like receptor 4 (TLR-4) complex is activated [21 –24]. Specifically, a study published by Copland et al. demonstrated that the presence of fibrinogen in PL can affect the immunomodulatory ability of MSCs by compromising their ability to upregulate indoleamine dioxygenase (IDO) and suppress T cell proliferation [21]. Moreover, fibrinogen can bind and activate a wide range of immune cells including leukocytes, monocytes, dendritic cells, macrophages, neutrophils, and some B cells [20]. In equine medicine, fibrinogen plasma concentrations can increase up to 10 times soon after a stimulus and increased concentrations of fibrinogen during inflammatory conditions have been linked with poor prognosis [25]. The role of fibrinogen on equine MSCs development and functionality has not been investigated.
MSCs regulate immune responses through many biological pathways [26] and promote tissue repair through the secretion of paracrine immunomodulatory factors or through direct cell-to-cell contact [27]. For example, MSCs can effectively inhibit the proliferation of stimulated lymphocytes [28 –31]. There is also a growing interest in developing MSC-derived acellular products for cell therapies, in which trophic and paracrine factors are retained, but no cells are present, thus minimizing issues of allogenicity [32]. An example of an acellular preparation is stem cell-derived supernatant, which is referred to the medium in which MCSs are proliferated that contains cell-derived secreted factors known as the secretome. This is easy to manufacture, sterilize, and store, offering a convenient off-the-shelf preparation [32,33]. Studies have previously shown that MSC supernatant derived from activated MSC cultures exhibits immunomodulatory abilities mainly through the release of trophic factors [31,34,35] and affects tissue repair [36]. In veterinary medicine, MSC supernatant generated from equine amniotic membrane-derived MSCs suppressed the proliferation of peripheral blood mononuclear cells (PBMCs) in vitro [37]. To our knowledge, there are no studies that have evaluated the effects of the culture medium on the secrotome produced by equine MSCs.
After manufacturing and characterizing fibrinogen-depleted lysate (fdePL), we hypothesized that fdePL would suppress the production of tumor necrosis factor-alpha (TNF-α) from LPS-stimulated monocytes more effectively than ePL. Because we ultimately propose to use fdePL to culture MSCs, we hypothesized that BM-MSCs cultured in fdePL or the supernatant generated from fdePL BM-MSC cultures would suppress TNF-α production from LPS-stimulated monocytes better than BM-MSCs grown in ePL or FBS or their respective supernatant media.
Materials and Methods
Depletion of fibrinogen from ePL
Horses involved in this study were housed in the University of Georgia research facility for monitoring and acclimation. All of the procedures required to conduct this study were approved by the Institutional Animal Care and Use Committee of the University of Georgia, study protocol No. A2018 01-013-Y1-A2. ePL pooled from five female healthy mixed breed university owned horses with a mean age of 13 ± 6 years was obtained as previously described [13]. The mean platelet count was 350 ± 106 × 103/μL. Pooled ePL was thawed at 37°C and mixed with 20 mM of CaCl2 dissolved in Plasma-Lyte in a glass container to create a coagulum that was observed to form within 15 min of incubation at 37°C and 5% CO2. The clotted preparation was stored at 4°C overnight and subsequently centrifuged at 3,485 g for 30 min. The coagulum separated from the fibrinogen-free supernatant was collected and 2 IU/mL of heparin was added and stored at −80°C.
Characterization of fdePL
The concentration of fibrinogen before and after depletion was measured with VetScan VS pro that measured the concentration of fibrinogen through a clotting rate assay and has been validated for use in horses (Abaxis, Union City, CA) [38]. The concentrations of platelet-derived growth factor-beta (PDGF-β), transforming growth factor-beta (TGF-β), and TNF-α were determined in ePL and fdePL through ELISA (R&D Systems) [13,14].
Effects of fdePL and ePL on LPS-stimulated monocytes
Blood from two female and one male adult healthy horses ranging from 3 to 20 years old was obtained for the isolation of equine PBMCs. One or two 60 mL syringes containing 1.5 mL of 100 μM EDTA were filled with blood from the left jugular vein from each horse. Equine PBMCs were isolated according to established protocols [14]. In brief, leukocyte-rich plasma was expressed and diluted with calcium and magnesium-free phosphate-buffered saline (PBS) (Corning Cellgro®, Mediatech, Inc., Manassas, VA). The mononuclear cells were isolated by single step density gradient centrifugation over leukocyte separation medium (Corning Cellgro®, Inc., Manassas, VA), collected and counted on a hymatocytometer. The cells were suspended in RPMI-1640 with
Monocytes were stimulated with 50 ng/mL Escherichia coli LPS 0111:B4 and incubated with RPMI-1640 containing either 10% DHS, FBS, ePL, or fdePL [14]. Equine monocytes incubated in RPMI-1640 supplemented with 10% DHS, FBS, ePL, or fdePL without LPS served as controls groups. After 6 h of incubation, cell culture supernatants were collected and assayed for the production of TNF-α [14].
Culture of BM-MSCs with fdePL medium and generation of MSC supernatant
Bone marrow was obtained from three horses different from those from which lysate or blood for monocyte isolation was obtained. These were healthy female mix-breed horses between 3 to 20 years old and marrow was processed and BM-MSCs were expanded and cultured in the presence of standard medium containing 10% FBS as previously described. Upon reaching 80%–90% confluency, cells were harvested with 0.05% trypsin–EDTA (Gibco, Invitrogen, Auckland, AZ), counted and cryopreserved until use. Experimental cell lines were established by plating BM-MSCs (P2; n = 3) at a density of 6,000 cells/cm2 in 150 mm culture dishes with MSC basal medium supplemented with either 10% FBS or 10% ePL or 10% fdePL [15,26,40]. Cells were incubated at 37°C with 5% CO2 and medium was replaced every 2 days. Upon reaching 80% confluence, cells were passaged, replated, or cryopreserved for future use in either FBS, ePL, or fdePL medium containing 10% DMSO.
We prepared MSC supernatant by culturing three different BM-MSC lines for 24 h at 6,000 cells/cm2 in medium supplemented with 10% FBS, 10% ePL or 10% fdePL until cells were 80% confluent. After 24 h, the medium from each BM-MSC line was pooled, while keeping the medium groups separate, centrifuged at 557 g for 10 min, and filtered through 0.22 μm Millex GP PES syringe filters [41]. This produced the stock of acellular MSC supernatant needed to conduct the subsequent experiments.
Cell growth kinetics: population doublings and doubling time
BM-MSCs (P2; n = 3) were plated in triplicates at a density of 6,000 cells/cm2 in six-well culture plates (Corning™ Costar™, Thermo Scientific, Hampton, NH) with 10% FBS or 10% ePL or 10% fdePL and passaged under standard cell culture conditions until P5. At every passage (P2–P5), BM-MSCs in each medium formulation were harvested through digestion with 0.05% trypsin and counted through an automatic cell counter (Bio Rad Laboratories, Hercules, CA). Population doublings (PD) was calculated using the following formula: PD = ln (N f/N i)/ln(2), where N f is the final number of cells and N i is the initial number of cells. All counts were performed in triplicates [42].
Cell viability
Cell viability was assessed with Live/Dead flow cytometry [15]. BM-MSCs from all samples in each medium formulation were harvested at P5 and centrifuged at 200 g for 4 min at room temperature. Cells were washed three times with PBS (+/+) and PBS (−/−) followed each time by a centrifugation cycle and were counted. One million BM-MSCs cultured in FBS or ePL or fdePL, resuspended in 1 mL of PBS, were stained with 4 μM ethidium homodimer (Biotium, Fremont, CA) and 2 μM Calcein Blue AM (Thermo Fisher Scientific, Waltman, MA). Single staining with either ethidium homodimer or Calcein Blue AM alone was used as control. BM-MSCs fixed with 4% paraformaldehyde for 20 min on ice, washed with PBS, and stained with both ethidium homodimer and Calcein Blue AM served as negative control groups. Samples were analyzed by flow cytometry and 50,000 events were collected per sample. Data were analyzed by Flow Jo software (NIH).
Effects of coculture of BM-MSCs or BM-MSC supernatant on LPS-stimulated monocytes
Equine PBMCs were isolated from healthy mixed-breed horses as described previously [14,43]. Isolated PBMCs were resuspended in medium consisting of RPMI-1640 supplemented with 10% DHS and plated in 150 mm plates for 2 h at 37°C and 5% CO2. After 2 h of incubation, adherent PBMCs, now mainly monocytes [39], were harvested, counted, and used in subsequent experiments.
The ability of BM-MSCs cultured in either 10% FBS, 10% ePL, or 10% fdePL, to suppress monocyte activation, was determined using two different experimental protocols. First, we used a stimulation assay in which BM-MSCs and monocytes were in a transwell culture system, but without direct cell-to-cell contact as previously described [44]. In this experiment, equine BM-MSCs (n = 3) at P6 were placed at the bottom of 12-well transwell plates at 100,000 cells/well and allowed to adhere for 12 h in the presence of their corresponding media. The following day, the medium was aspirated and replaced with 1.5 mL of standard monocyte medium (RPMI +10% DHS). In total, 0.5 mL containing 400,000 equine monocytes (ratio 1:4; n = 3) was added to each insert of a transwell plate with pore size of inserts 0.4 μm (Corning, NY), stimulated with 50 ng/mL of E. coli 0111:B4 LPS (List Biologicals, Inc.) and incubated at 37°C and 5% CO2. Eighteen hours later, cell culture supernatants were collected and assayed for the production of the proinflammatory cytokine TNF-α as an indicator of inflammatory responses [14]. We chose this time point because previous experiments conducted in our laboratory gave us confidence that MSCs would be able to modulate monocyte activation 18 h after coincubation [15]. Also, prolonged incubation time might be required for the MSCs to release the factors that could impact monocyte activation.
In a second set of experiments, monocytes were stimulated with 50 ng/mL E. coli LPS 0111:B4 and at the same time 10% or 50% of BM-MSC supernatant generated from BM-MSCs (n = 3) cultured in either FBS, ePL, or fdePL was added to the culture. We decided to test two different concentrations of our medium to determine a possible concentration-dependent effect, but also because a 100% medium substitution would have resulted in the absence of standard monocyte medium that is required to provide factors, such as Lipid A, responsible for LPS-driven monocytes activation [14]. In addition to FBS BM-MSC supernatant, we incorporated in our design regular FBS, without MSC exposure, as an additional control group. In contrast to the previous experiment, we chose to collect cell culture supernatants to measure TNF-α through ELISA 6 h (not 18 h) after stimulated monocytes had been incubated in the different media preparations. This was done in part because the MSC supernatant already contains secreted factors, including growth factors, chemokines, and cytokines that are directly implicated in the ability of MSCs to modulate immune responses [31,34,35]. We, therefore, hypothesized that MSC supernatant would require less time to modulate immune responses compared with direct incubation of MSCs and immune cells. Moreover, choosing this time point would not affect the potential of MSC supernatant to suppress immune activation in a highly inflammatory microenvironment since the release of proinflammatory cytokines from monocytes stimulated with LPS peaks at 6 h and remains stable until 18 h [45].
Statistics
Normality of the data was evaluated by visual examination of histograms of the residuals, normal plots of residuals, and by using Shapiro–Wilks test. Equality of variances was assessed by using Levene's test and plotting residuals against the fitted value. One way repeated measures ANOVA was used to detect significant differences for viability, monocytes inflammatory responses, and immunomodulatory abilities of BM-MSCs and BM-MSC supernatant in different media formulations. A linear mixed model was used to assess the effect of medium on BM-MSCs proliferation at different time points. Multiple pairwise comparisons, if necessary, were obtained by using the Tukey–Kramer test or Sidak test. A statistical analysis could not be performed for the growth factor, cytokine, and fibrinogen concentration after removal of fibrinogen since ePL was pooled from three donors resulting in only one biological replicate. The level of significance was set at P < 0.05. Results are reported as mean ± SD. For all analyses, commercially available statistical packages were used (Stata version 13.1; StataCorp LP, College Station, TX® or GraphPad Prism 7.0 c, La Jolla, CA).
Results
Depletion of fibrinogen from ePL and fdePL characterization
The addition of 20 mM CaCL2 to ePL resulted in clot formation within 15 min. The recovery rate after fibrinogen depletion ranged between 65% and 75% and the concentration of fibrinogen was reduced to undetectable concentration compared with 250 mg/dL in pooled ePL (Fig. 1). PDGF, TGF-β, and TNF-α concentrations declined after fibrinogen depletion. Specifically, the concentration of PDGF-β dropped from 5,049.46 to 2,999 pg/mL. There was also a decrease of the concentration of TGF-β after depletion from 6,237 to 5,356.1 pg/mL. Finally, the concentration of TNF-α was reduced from 2,216.9 to 1,037.6 pg/mL (Fig. 2).
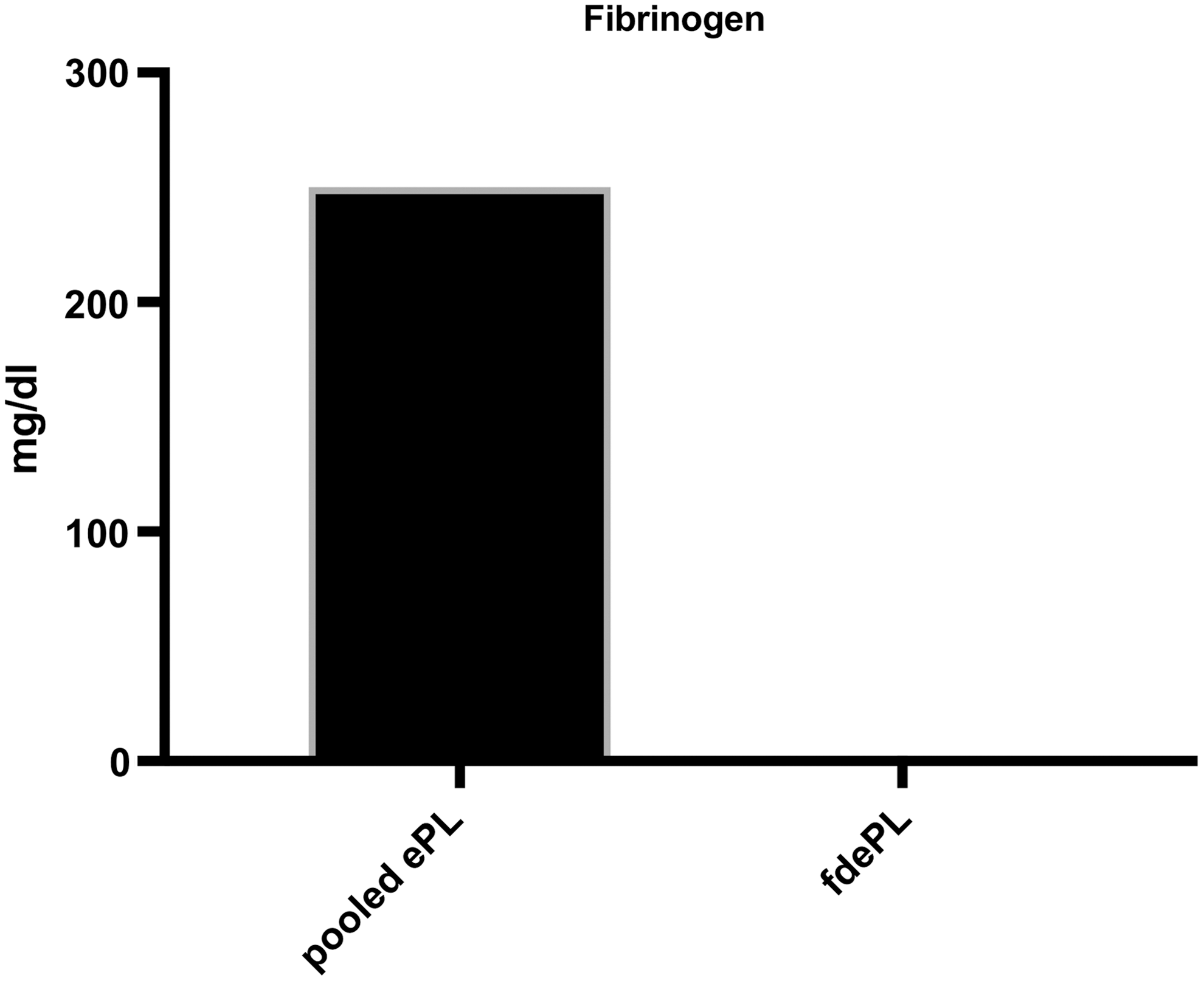
Fibrinogen concentration in pooled ePL before and after fibrinogen depletion. ePL, equine platelet lysate; fdePL, fibrinogen-depleted lysate.

Comparison of the concentrations of growth factors and cytokines, including PDGF-β, TGF-β, and TNF-α in pooled ePL and fdePL. PDGF-β, platelet-derived growth factor-beta; TGF-β, transforming growth factor-beta; TNF-α, tumor necrosis factor-alpha.
Effects of ePL and fdePL on LPS-stimulated monocytes
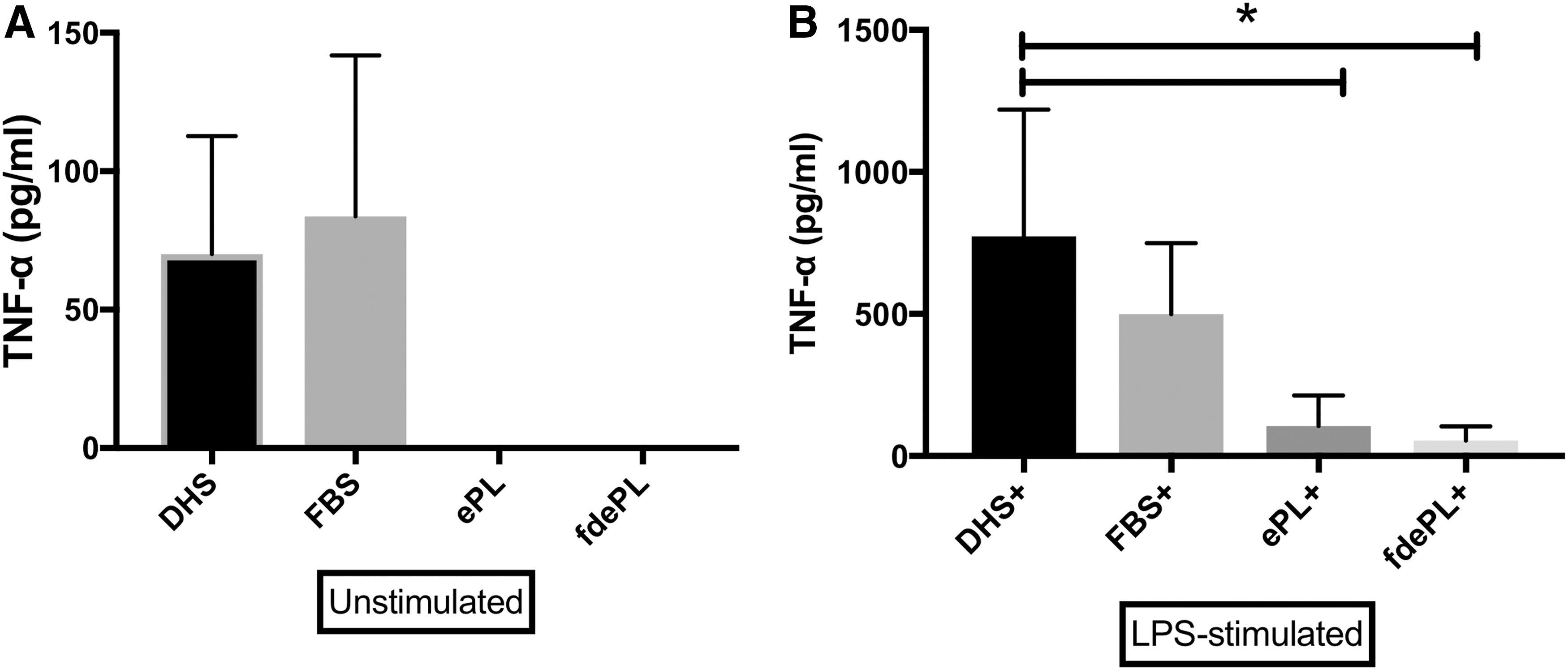
The addition of 10% ePL or fdePL to unstimulated equine monocytes resulted in undetectable concentrations of TNF-α. When FBS was added to unstimulated monocytes, TNF-α was detected at a concentration of 83.68 ± 58.19 pg/mL (Fig. 3A). The addition of DHS (positive control) to LPS-stimulated monocytes leads to a statistically greater increase in TNF-α (773 ± 446.2 pg/mL) than those without the addition of LPS, indicating proper stimulation of monocytes. TNF-α concentrations did not statistically differ when FBS (499.3 ± 250.61 pg/mL) was added to LPS-stimulated monocytes compared with the positive control group. However, monocytes cultured in ePL or fdePL produced significantly lower concentrations of TNF-α (106.04 ± 106.8 and 54.4 ± 50.5 pg/mL, respectively) than the positive control (773 ± 446.2 pg/mL) (Fig. 3B). No differences were recorded between ePL and fdePL.
Cell growth kinetics and viability of BM-MSCs in fdePL
Figure 4 shows the growth kinetics of BM-MSCs cultured in different media supplements. BM-MSCs grown in FBS or ePL exhibited comparable PD from passages 2 through 5. However, MSCs grown in fdePL exhibited a significant reduction in their PD compared with those cultured in ePL (P < 0.05). No significant differences were detected between BM-MSCs cultured in fdePL or FBS.
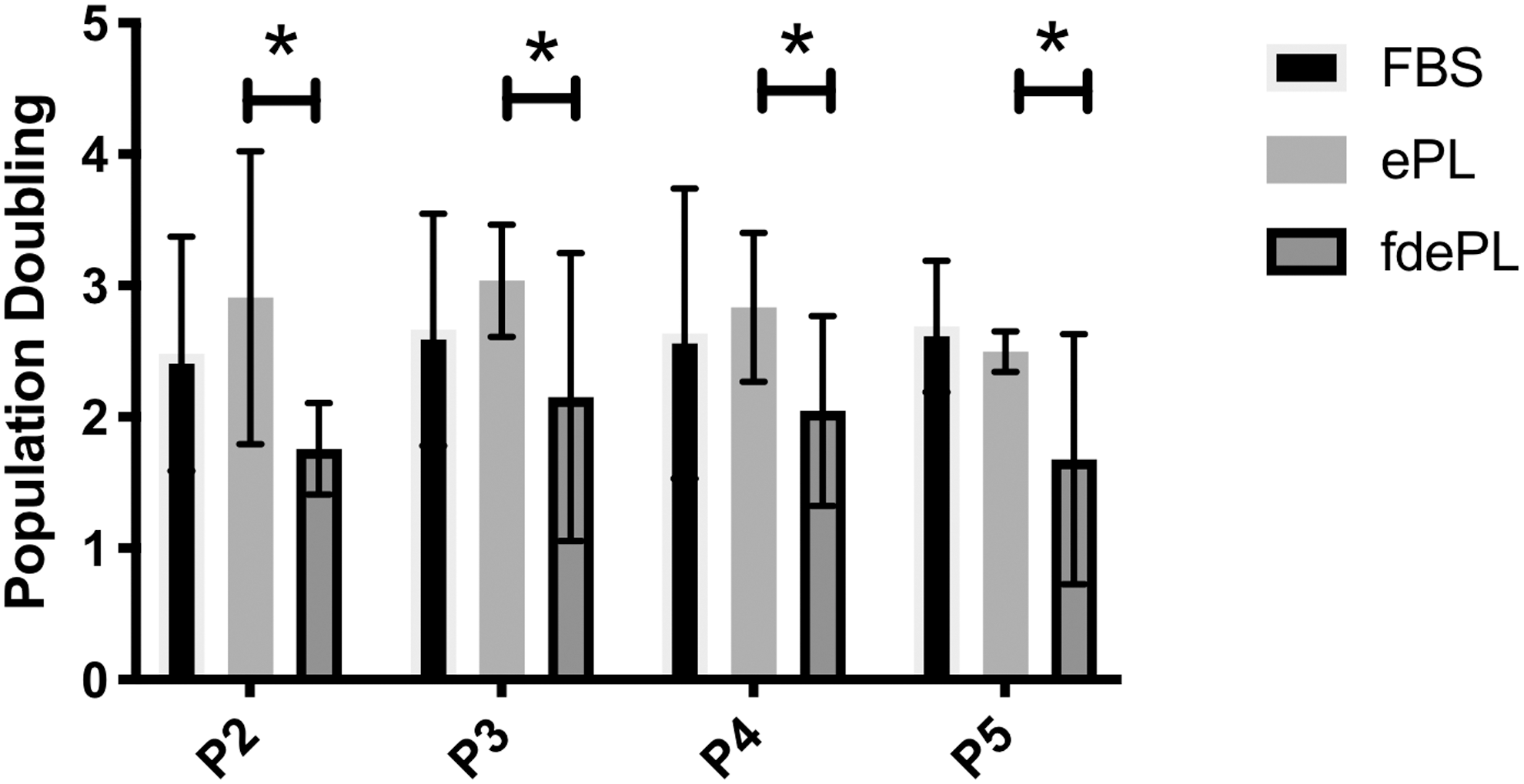
Population doublings of BM-MSCs cultured in medium containing 10% FBS, ePL, or fdePL from passage 2 to passage 5. Data presented as mean ± standard deviation; n = 3. *Significant difference: P < 0.05. MSCs, mesenchymal stem cells.
The viability of BM-MSCs cultured in FBS, ePL, or fdePL was comparable among the groups as shown in Fig. 5. Specifically, BM-MSCs cultured in FBS were 61.83% ± 10.42% viable, cultured in ePL were 64.6% ± 7.67% viable, and cultured in fdePL were 68.07% ± 14% viable with no statistically significant differences (P > 0.05).
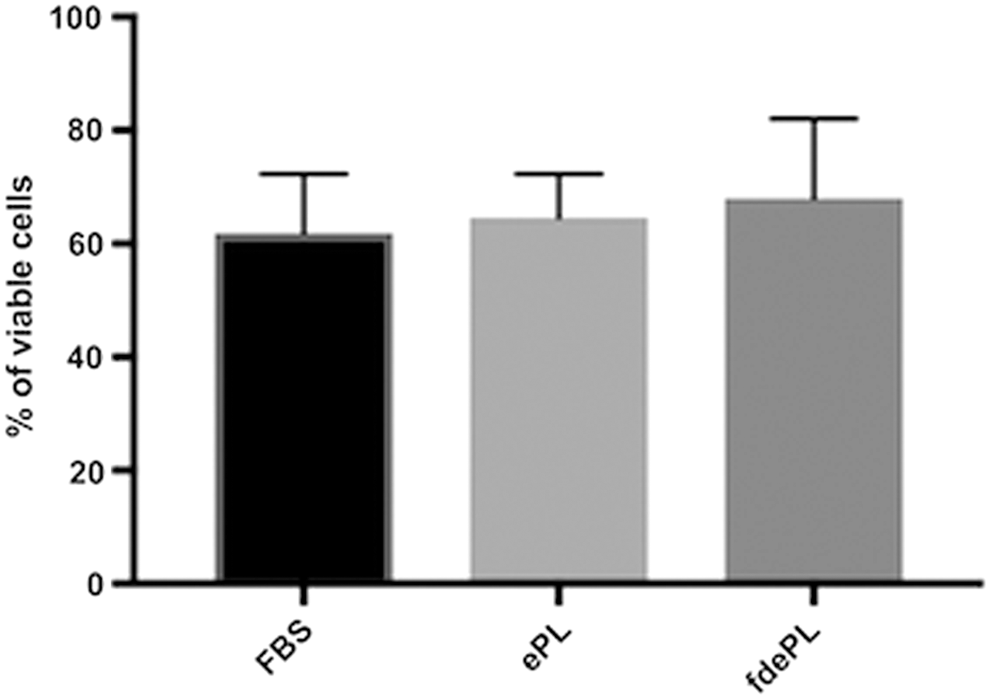
Percentage of viable BM-MSCs cultured in medium containing 10% FBS, ePL, or fdePL at passage 5. Data presented as mean ± standard deviation; n = 3.
Effects of coculture of MSCs or MSC supernatant on LPS-stimulated monocytes
First, BM-MSCs were developed in the different media conditions and were then cocultured with LPS-stimulated monocytes using a transwell system. BM-MSCs in FBS and BM-MSCs in ePL significantly decreased levels of TNF-α (1,744.38 ± 658.21, 2,321.15 ± 977.79 pg/mL, respectively) produced by LPS-stimulated monocytes compared with LPS-stimulated monocytes alone (3,958.33 ± 943.07 pg/mL). Compared with FBS and ePL, BM-MSCs in fdePL medium did not significantly suppress the production of TNF-α (3,101.9 ± 1,576.88 pg/mL) (Fig. 6).
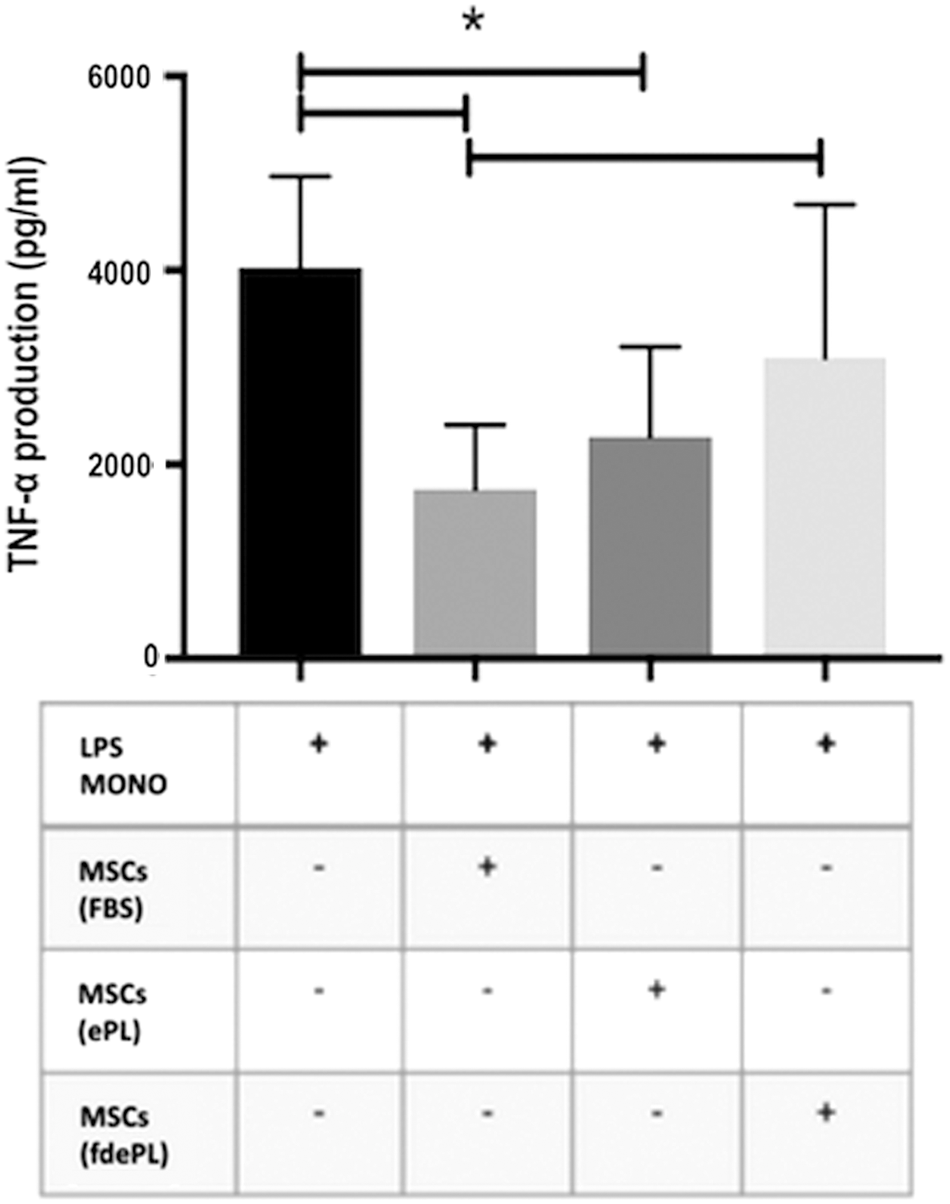
TNF-α production in 16 h in LPS-stimulated monocytes (n = 3) in the presence of BM-MSCs previously exposed to medium containing 10% FBS, ePL, or fdePL. Data presented as mean ± standard deviation; n = 3. *Significant difference: P < 0.05.
Second, we measured TNF-α concentrations from LPS-stimulated monocytes exposed to supernatants prepared by culturing BM-MSCs in FBS, ePL, or fdePL (Fig. 7). Even though statistically significant differences were not seen, LPS-stimulated monocytes exposed to 10% or 50% FBS BM-MSC supernatant expressed higher TNF-α concentrations (1,062.22 ± 422.83 and 1,088.23 ± 397.88 pg/mL, respectively) than monocytes without BM-MSC supernatant (792.84 ± 238.36 pg/mL). Although failing to reach significance, LPS-stimulated monocytes cultured in 10% or 50% ePL BM-MSC supernatant appeared to produce less TNF-α (respectively, 350.72 ± 110.5 to 447 ± 429.2 pg/mL) than those exposed to FBS BM-MSC supernatant. This reduction in TNF-α was not proportional to the percentage of ePL MSC supernatant used. Finally, LPS-stimulated monocytes in 50% fdePL BM-MSC supernatant produced less TNF-α (236.68 ± 50.01 pg/mL) than monocytes in 10% or 50% FBS BM-MSC supernatant. Exposure to 10% fdePL BM-MSC supernatant did not achieve a significant reduction in TNF-α production (708.1 ± 131.45 pg/mL).
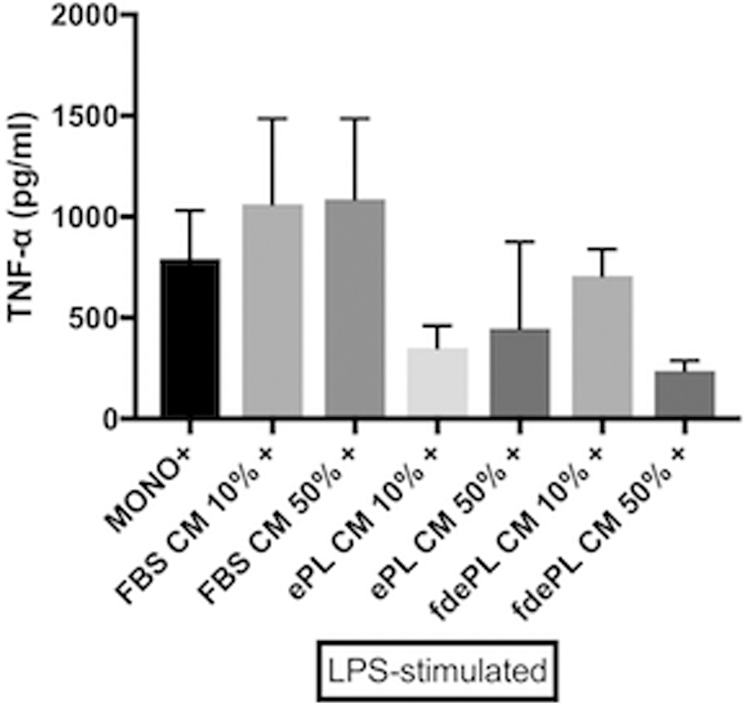
TNF-α production at 6 h in LPS-stimulated monocytes (n = 3) in the presence of BM-MSC supernatant produced from cultures with medium containing 10% or 50% FBS, ePL, or fdePL. Data presented as mean ± standard deviation; n = 3.
Discussion
In this study, we continued in our efforts to optimize the use of PL as a homologous cell culture medium supplement that could suitably replace FBS. Having previously shown that ePL is an effective alternative to FBS for cell culture, we wanted to deplete fibrinogen from lysate to further refine this product and assess its effect on MSC functionality. In our previously published work, we found that MSCs cultured in ePL retained their ability to suppress monocyte activation in a manner similar to those cultured in FBS [15]. In an attempt to improve on this and because evidence would suggest that fibrinogen affects the immunomodulatory ability of MSCs, we decided to study the characteristics of ePL without fibrinogen [21].
The process of depletion was straightforward and resulted in elimination of fibrinogen from our product. Our protocol was based on the addition of CaCl2 that initiated gelation of ePL, resulting in fibrinogen being trapped in the resulting clot. This meant that some of the initial lysate could not be completely retrieved, leading to a recovery rate of ∼70% of the initial lysate.
We initially presumed that the removal of fibrinogen would alter the lysate, but at the same time eliminate fibrinogen as one potential source of monocyte activation and TNF-α release. Our effort was prompted by evidence that fibrinogen can activate TLR-4 evoking the release of MCP-1, IL-6, and IL-8 [20,46,47].
Fibrinogen depletion resulted in an overall decrease in the concentration of PDGF-β, TGF-β, and TNF-α in ePL, but it appears that even without fibrinogen, ePL could suppress TNF-α production from LPS-stimulated monocytes with no appreciable differences between depleted and nondepleted lysate. Because of the lack of a fibrinogen effect, it may be possible that fibrinogen does not activate inflammatory pathways in equine monocytes to the extent that has been described in people [20]. It is also plausible that the chemokines and other factors yet to be characterized in lysate achieve the desired monocyte suppressant effect regardless of the presence or absence of fibrinogen.
fdePL had a negative effect on BM-MSC proliferation compared with ePL. The lower PD rates of BM-MSCs cultured in fdePL could likely be attributed to a decrease in the concentration of PDGF-β, TGF-β, and other growth factors important for cell proliferation. Interestingly, however, BM-MSC viability was unaffected regardless of the use of FBS, ePL, or fdePL, suggesting that depleting fibrinogen does not influence cell viability. It should be pointed out that in this study, we conducted extensive washes of our cells before measuring viability [15]. This is a common practice when cells are prepared for clinical application to remove potential FBS-derived xeno-contaminants from MSC preparations. We maintained this practice when conducting this study; however, our recent evidence has changed our laboratory protocols so that when MSCs are developed in lysate, we no longer perform extensive cell washing cycles, thus significantly improving the number of viable cells retrieved from culture [15]. MSCs are clinically attractive because of their ability to modulate immune responses after delivery within a site of injury [26]. Although we hypothesized that BM-MSCs grown in fdePL would exhibit improved immunomodulatory capacity, our data indicate that this was not the case. BM-MSCs cultured in fdePL were unable to suppress TNF-α production from LPS-stimulated monocytes as effectively as those grown in ePL or FBS. It has been previously shown that activation/priming of MSCs with TNF-α and IFN-γ significantly enhances their in vitro immunosuppressive abilities as reflected by a suppression of T cell proliferation [48]. Because we showed a decrease in the concentrations of TNF-α in lysate after removal of fibrinogen, it is possible that MSCs in fdePL medium were not exposed to sufficient TNF-α priming as compared with those in ePL, leading to a less efficient monocyte suppression of TNF-α production. It is also possible that the depletion process eliminated or decreased the concentrations of factors that allow MSCs to exert their monocyte suppressive effect.
It is worth pointing out that we only assessed the ability of BM-MSCs to control monocyte activation and did not evaluate responses in other cell types, such as T cells or neutrophils. This is important because others have shown that fibrinogen has an important effect on the induction of IDO production from MSCs which is reportedly a key cytokine in the MSC-driven modulation of T cell proliferation [21]. It is, however, known that equine MSCs do not modulate immune responses through IDO. In contrast, it has been reported that the immunomodulatory capacities of the equine MSCs are attributed to the production of PGE2, IL-6, TGF-β, and NO [30]. It would be interesting in a future study to evaluate whether different media preparations affect the expression levels of these factors. Moreover, since the effect of fibrinogen on equine MSCs has not been investigated, future studies should explore how equine MSCs cultured in fdePL influence the biological properties of other types of immune cells.
In addition to our experiments conducted in transwell systems, we wanted to test the effect of medium developed from BM-MSCs, but without the presence of cells. We designed these experiments to test 10% and 50% BM-MSC supernatant to determine a possible dose-dependent effect because we assumed that increased concentrations of BM-MSC-derived supernatant will affect suppression of monocyte activation differently. We were unable to justify using higher concentrations than 50% because the standard monocyte medium could not be completely replaced as it provides essential components such as Lipid A, necessary for the effective stimulation of monocytes with endotoxin [14]. As shown in Fig. 7, it appeared that supernatant obtained from BM-MSCs grown in 10% ePL and 50% fdePL was trending to suppress monocyte activation better than medium from 10% FBS BM-MSC culture medium, but we were unable to prove significance. Future studies should include a larger sample size. The fact that we were not able to identify statistically significant differences between the groups can be attributed to the small size of biological replicates.
There is merit in investigating “stem cell-free” products as potential therapeutics in wound healing and other injuries [49]. Rather than using cell-based preparations, studies are suggesting that acellular products may be more suitable for therapy because of ease of preparation and storage, reduced immune reactions in the recipient, and an overall decrease in the teratogenic potential when compared with traditional cell-based products [50,51].
Substituting FBS with ePL may have a substantial effect on the profile of factors secreted by MSCs in culture, and this aspect of MSC biology should be further investigated. In fact, reducing the concentrations of serum in culture medium improves the angiogenic potential of MSCs, but reduces their proliferation rates since the medium contains lower amounts of growth factors [52,53]. Moreover, there is enough evidence suggesting that the secretion of soluble factors important for MSCs immunomodulatory features might be influenced by cytokines and factors accumulated or secreted in the microenvironement [54,55]. We need to further investigate growth factors, paracrine factors, and cytokines present in ePL and fdePL because they are likely to affect the secretion of immunomodulatory factors by MSCs impacting the composition of the MSC secretome.
In this study, we showed that fibrinogen can be effectively removed from ePL and that fdePL alone can suppress the release of TNF-α from LPS-stimulated monocytes. When we evaluated the effect of fdePL on BM-MSCs cultures, we found no change in viability, but lower proliferation rates, and, importantly, a reduced immunomodulatory capacity compared with BM-MSCs cultured in ePL or FBS. When we evaluated medium obtained from BM-MSCs cultured in lysate, a statistically significant difference was not detected and further conclusions cannot be extrapolated from the data reported. Overall, contrary to our hypotheses and in contrast to the available literature on people, we were unable to demonstrate a clear advantage of pursuing fibrinogen depletion from ePL.
Footnotes
Acknowledgments
The authors thank Annie Bullington for her technical support during the platelet apheresis procedure and Anna Chocallo and Hannah Kemelmakher for their help in performing laboratory assays.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was funded by Morris Animal Foundation (grant number D17EQ-021).