
Abstract
Restoring β-cell mass by the transplantation of pancreatic islets is an effective diabetes treatment, but it is limited by the shortage of donor organs. CD133-expressing pancreatic ductal epithelial cells (PDECs) have the ability to generate insulin-producing cells. The expansion of these cells is dependent on extrinsic niche factors, but few of those signals have been identified. In this study, CD133-expressing PDECs were purified by sorting from adult wild-type C57BL/6 mice and TGFβRIInull/null mice. Furthermore, using immunofluorescence and transplantation assays, we found that the inhibition of the transforming growth factor-β (TGF-β) pathway promoted the expansion of CD133-expressing PDECs for many generations and maintained the ability of CD133-expressing PDECs to generate insulin-producing cells. Moreover, western blot, qRT-PCR, and dual luciferase assays using TGF-β inhibitors were performed to identify the mechanisms by which TGF-β signaling regulates proliferation and differentiation. The results showed that the inhibition of TGF-β signaling enhanced Id2 binding to the promoter region of the cell proliferation repressor p16 and promoted the expansion of CD133-expressing PDECs, and the increased Id2 binding to NeuroD1 decreased the transcription of Pax6 to maintain CD133-expressing PDECs in the Pdx1-expression stage. Taken together, the effect of TGF-β antagonists on CD133-expressing PDECs reveals a novel paradigm of signaling that explains the balance between the expansion and differentiation of pancreatic duct epithelial progenitors.
Introduction
Diabetes mellitus is characterized by a progressive decrease in the functional mass of insulin-producing β-cells. Restoring β-cell mass by transplanting pancreatic islets is an effective diabetes treatment, but it may be limited by the shortage of donor organs. The islet β-cell is among the most differentiated mammalian cell type, whereas adult β-cells are largely quiescent. Therefore, multiple studies have been conducted to develop alternative sources of β-cells ex vivo [1 –4]. A promising approach for the generation of insulin-producing cells is inducing the differentiation of various types of stem/progenitor cells [5 –8].
During the development of the pancreas, the endocrine and exocrine cells are thought to develop from a common cell component associated with the pancreatic ductal epithelium [9,10]. It has been confirmed that CD133-expressing pancreatic ductal epithelial cells (PDECs) contain large amounts of pancreatic progenitor cells [11,12]. Manipulation of the ability of CD133-expressing PDECs to produce endocrine cells may open new avenues for the treatment of diabetes. However, the percentage of CD133-expressing PDECs in the pancreas is very low, and it is difficult to grow these cells for extended periods under in vitro conditions using conventional culture protocols [13,14]. This lack of an in vitro expansion protocol restricts further studies. Uncovering the molecular pathways that control CD133-expressing PDEC expansion is therefore critical to enable the potential application of stem cell therapy in diabetes.
The development of cell culture methods to produce PDECs, particularly from the adult tissue, has been slow. Before 1980, animal tissues were usually cultured from an explant obtained from a small dissected ductal fragment of autopsied organs [15]. In these culture systems, the ductal epithelial cells would grow out from the tissue explants and form cell monolayers. The development of techniques for the cultivation of isolated ductal epithelial cells eventually followed [16,17]. The systematic exploration of trophic growth factors and several extracellular matrices (agarose and collagen gel) has improved PDEC culture systems [18 –20]. More recently, PDECs were successfully cultured on Matrigel, which stimulates duct cell proliferation, cyst formation, and differentiation [14,21]. Despite these remarkable achievements, the prolonged expansion of adult PDECs is challenging. Furthermore, ductal epithelial cells are phenotypically unstable over time in culture systems [22 –24], suggesting that they require suboptimal culture conditions.
We recently reported the transcriptome profiling of pancreatic progenitor-like cells and found that these cells are enriched with transforming growth factor-β (TGF-β) signaling effectors [25]. The TGF-β superfamily signaling balances stem cell proliferation and differentiation and regulates the maintenance and dynamic behavior of normal reservoirs of stem cells [26,27]. Specific antagonists of TGF-β signaling have been previously used to improve the expansion and maintenance of some epithelial progenitor cells [28 –30]. However, the role of TGF-β signaling in the regulation of expansion and maintenance of CD133-expressing PDECs has not been explored.
In this study, we demonstrated that TGF-β signaling inhibition regulates the expansion and maintenance of CD133-expressing PDECs. We found that specific antagonists of TGF-β signaling promoted the proliferation of CD133-expressing PDECs and maintained their undifferentiation stage in vitro. The specific knockdown TGF-β type II receptor (TGFβRII) in pancreatic and duodenal homeobox 1 (Pdx1)-expressing cells using the Pdx1-cre/TGFβRIIflox/flox system caused hyperplasia of CD133-expressing ductal epithelial cells in vivo. Furthermore, experiments aimed at defining the functional mechanism underlying the regulatory effect of TGF-β signaling on the expansion and maintenance of CD133-expressing PDECs revealed that antagonists of TGF-β signaling induced the expression of inhibitor of DNA binding 2 (Id2). TGF-β signaling inhibition enhanced the binding of Id2 protein to the promoter region of the cell proliferation repressor p16 along with the expansion of CD133-expressing PDECs. Furthermore, Id2 binding to NeuroD1 resulted in a decrease in the transcription of its target gene encoding paired box protein 6 (Pax6) and maintained CD133-expressing PDECs in the Pdx1 expression stage. Mothers against decapentaplegic homolog 3 (Smad3) knockdown or Id2 overexpression reduced the expression of Pax6 and p16. Therefore, we conclude that the inhibition of TGF-β signaling blocked the differentiation of CD133-expressing PDECs, but promoted their expansion. This finding is important for the expansion of pancreatic progenitor cells in vitro and may allow us to understand the biology underlying the maintenance of progenitor cells and regulation of their differentiation in the pancreas.
Materials and Methods
Animals
C57BL/6J mice were purchased from the College of Veterinary Medicine of Yangzhou University. SCID/NOD mice, B6.129-Tgfbr2tm1/Nju mice (TGFβRIIflox/flox mice), B6.FVB-Tg1Tuv mice (Pdx1-Cre mice), CAG-EGFP (GFP) mice, and R26-mTmG (Red) mice were obtained from Nanjing Biomedical Research Institute of Nanjing University. Animal care and handling were carried out according to international laws and policies (EEC Council Directive 86/609, 1987) and approved by the animal ethics committee of China Pharmaceutical University (Nanjing, China). In Pdx1-Cre mice, exogenous Cre recombinase was cloned under the control of the Pdx1 promoter, and exon number 4 of the TGFβRII gene was “floxed” in TGFβRIIflox/flox mice. Mice with a pancreas-specific TGFβRII mutation (exon #4 deletion) were generated by mating Pdx1-Cre mice and TGFβRIIflox/flox mice. DNA of the offspring was extracted from the tail tip, and the genotype was verified with polymerase chain reaction (PCR) using the following primer pairs: Pdx1-Cre mice: 5′-CTGGACTACATCTTGAGTTGC-3′ (sense) and 5′-GGTGTACGGTCAGTAAATTTG-3′ (antisense), and TGFβRIIflox/flox mice: 5′-TATGGACTGGCTGCTTTTGTATTC-3′ (sense) and 5′-TGGGGATAGAGGTAGAAAGACATA-3′ (antisense). To detect the exon 4 deletion of TGFβRII, mouse pancreas DNA was extracted and verified using the primer pair 5′-TATGGACTGGCTGCTTTTGTATTC-3′ (sense) and 5′-TATTGGGTGTGGTTGTGGACTTTA-3′ (antisense).
In vitro 3D culture of adult PDEC colonies
To obtain single cells from an adult mouse pancreas, a 6-week-old C57BL/6J mouse was sacrificed and dissected. The whole pancreas was harvested and cut into 1 mm3 pieces in a cold dish using scissors. The tissue was washed with phosphate-buffered saline (PBS) and incubated with 2 mg/mL collagenase II (dissolved in PBS plus 0.1% bovine serum albumin (BSA) and 200 U/mL DNase I, all purchased from Sigma Aldrich, St. Louis, MO) in a 15 mL tube for digestion. During the 30-min digestion, the tissues were gently mixed every 10 min using a 20 G syringe. Digestion was terminated with the addition of 10% fetal bovine serum (FBS) to the buffer. After centrifugation for 5 min at 500 g, the cells were resuspended, screened using a 40 μm mesh, and counted under an inverted microscope.
For the colony formation assay, single cells were sorted by staining with CD133-fluorescein isothiocyanate (FITC) antibody, and the CD133+ cells were mixed with 5% Matrigel (Corning) and 1% methylcellulose (Sigma Aldrich) in Dulbecco's modified Eagle's medium (DMEM)/F12 (Gibco) supplemented with 20 ng/mL epidermal growth factor (EGF; Sigma Aldrich), 1% B27 (Invitrogen), 10 mM nicotinamide (Sigma Aldrich), 5% FBS (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). We added 2 ng/mL TGF-β (Sigma Aldrich) or the TGF-β type I receptor (TGFβRI) inhibitors SB431542 [31,32] (5 μM; Sigma Aldrich) and RepSox [33] (5 μM; Selleck) as needed. Cells were seeded into a 24-well ultra-low attachment plate (Corning) at a density of 10,000 cells/well and cultured at 37°C with 5% CO2 for 2 weeks to form colonies.
For self-renewal assessment, a serial passage assay was performed. Colonies were hand-picked, and a single cell suspension was obtained using Accutase (Sigma Aldrich). Cells were replated under the same conditions mentioned above. This process was repeated 10 times.
Animal experiments
To evaluate the effect of a TGF-β inhibitor on the replication of mouse CD133-expressing PDECs in vivo, animal experiments were performed as previously described [34]. Male C57BL/6J mice (8 weeks old, about 22 g) were randomly divided into three groups (n = 7 mice per group) and intraperitoneally injected with a single dose of the TGF-β inhibitor SB431542 (5 mg/kg body weight [BW]) or RepSox (1.4 mg/kg), or the vehicle control (50% [v/v] dimethyl sulfoxide [DMSO]), ensuring that the volume injected was lower than 100 μL. Pancreas tissues from these mice were harvested after 1 week and processed for histology, and analyzed to assess proliferation.
Real-time PCR
RNA was extracted using an RNeasy Plus Mini Kit (QIAGEN). cDNA was generated using a HiScript II 1st Strand cDNA Synthesis Kit (Vazyme). Ct values were acquired to represent the relative level of the transcripts using AceQ qPCR SYBR Green Master Mix (Vazyme) on a 480 II system (Roche). The primers used for real-time PCR are listed in Supplementary Table S1.
Protein extraction and western blotting
RIPA Lysis Buffer (Beyotime) plus 1% PMSF (Sigma) was used to dissolve the colonies. The buffer was gently mixed several times until the colonies were dissolved. After performing the standard protein extraction protocol, the protein concentration was determined using an Enhanced BCA Protein Assay Kit (Beyotime). Western blotting was performed following standard procedures. The antibodies used are listed in Supplementary Table S2.
Co-immunoprecipitation
Co-immunoprecipitation (Co-IP) was performed to determine the binding of endogenous Id2 and NeuroD1. First, 14-day-old colonies were hand-picked, and the colonies were then dissolved using RIPA Lysis Buffer (Beyotime). The Co-IP experiment was then performed strictly following the manual for Protein A/G PLUS-Agarose (SC-2003; Santa Cruz). Id2 antibody (Supplementary Table S2) was used to precipitate the endogenous Id2-NeuroD1 complex, and immunoblotting was performed to detect Id2 and NeuroD1.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) experiments were performed strictly following the manual for the ChIP Assay Kit (P2078; Beyotime). To detect the binding of c-myc to the Id2 promoter region, total DNA of PDECs from 14-week-old colonies was Ch-IPed with c-Myc antibody (Supplementary Table S2). A 389 bp region of the Id2 promoter was amplified using the primers 5′-TCTGTTCCGCTGTGGCACGTATG-3′ (sense) and 5′-AAAGCTCTGTGTGGAGAAACAGTA-3′ (antisense), which contain three E-boxes. To detect the binding of Smad3 to the Foxa2 promoter region, PDECs were Ch-IPed with Smad3 antibody (Supplementary Table S2). The binding sites were predicted using JASPAR 2016, and five primer pairs (∼100 bp product) were used for amplification: S1: 5′-GGCGGTACGCGCCTTCGGATTGA-3′ (sense) and 5′-CCTGGGATCAAGCCACCCTTGGAGA-3′ (antisense), S2: 5′-CAGCCGCTCACGTCATTGC-3′ (sense) and 5′-CGTGGGGGCGGGCGGGGGGAGA-3′ (antisense), S3: 5′-CCGCTAGCTTCCCGCCGCCT-3′ (sense) and 5′-GAGACTCGCCGTCGGGCGTGCACT-3′ (antisense), S4: 5′-GGGTTCCGGCCTCCAGGGTTCTTCG-3′ (sense) and 5′-CTCTCTCCCGCGGCCTGC-3′ (antisense), and S5: 5′-AGAGGAAGTGGAGTCCAGAGAGGG-3′ (sense) and 5′-CCCTCTTAAAAAAAAAAGTCAGCAA-3′ (antisense). PCR products were analyzed by agarose electrophoresis.
Immunofluorescence
Frozen pancreas tissue sections were boiled in sodium citrate buffer (Solarbio) in a microwave oven for 5 min for antigen retrieval. After washing twice with PBS for 5 min, the frozen sections were blocked with 5% BSA and stained with a primary antibody at 4°C overnight. Following incubation, the sections were washed twice with PBS and treated with a secondary antibody for 1 h at 37°C. We used 4′,6-diamidino-2-phenylindole (DAPI; 0.1 μg/mL; Sigma Aldrich) for nuclear staining. The slides were washed twice with PBS and observed under an inverted fluorescence microscope (Olympus).
To stain the colonies, single colonies were hand-picked and seeded in a 96-well plate. The staining procedure was carried out as described above. After staining, the individual colonies were transferred to a cover slip for imaging under a laser confocal microscope (Zeiss). All the antibodies used are listed in Supplementary Table S3.
Plasmid construction and lentivirus infection
To study the mechanism by which TGF-β signaling affects self-renewal maintenance and proliferation, a Smad3-shRNA lentivirus plasmid and an Id2-overexpression lentivirus plasmid were constructed. In detail, shRNA targeting Smad3 mRNA (NM_016769.4) was generated using the following primer pairs: 5′-GATCCGTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGACTTTTTTG-3′ (sense) and 5′-AATTCAAAAAAGTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGACG-3′ (antisense). The primers were annealed and cloned to the plvx-shRNA2 lentivirus vector (Takara). For Id2-overexpression lentivirus plasmid construction, mouse pancreas cDNA was used to generate an open reading frame (ORF) of Id2 mRNA (NM_010496.3). The following primer pair was used for the construction: 5′-CCGCTCGAGATGAAAGCCTTCAGTCCGGT-3′ (sense) and 5′-CGGGATCCTTAGCCACAGAGTACTTTGCTATCA-3′ (antisense). The ORF of Id2 mRNA was then cloned into the plvx-IRES-Zsgreen1 lentivirus vector (Takara). Lentivirus packaging was performed following standard procedures using the helper vectors psPAX2 and pMD2.G (Addgene). The titers of the lentivirus were measured following standard procedures.
Flow cytometry
To determine the CD133+ subpopulation percentage in the PDEC colonies, the colonies were hand-picked, and a single cell suspension was obtained using Accutase. After they were washed twice with PBS, the cells were stained for 20 min on ice using CD133-FITC antibody or an isotype control. After they were washed twice with PBS and resuspended in PBS, the CD133+ cells were detected using C6 (Becton, Dickinson and Company).
For ex vivo pancreas culture, the pancreas was dissected from the mouse following the above-mentioned procedure to generate single cells. CD133 and Ki67 double-positive cells were detected following a standard intracellular staining procedure using Cytofix/Cytoperm solution (Becton, Dickinson and Company). CD133-PE and Ki67-percp antibodies were used for staining, and isotype antibodies were used as a negative control.
For cell sorting, single cells were generated from an adult mouse pancreas using the above-mentioned procedure. The cells were first incubated with CD16/32 antibody for blocking, followed by CD133-FITC or isotype antibody staining for 20 min on ice. After they were washed twice, cells were resuspended in PBS/0.1% BSA/DNase I. Then, CD133+ and CD133− subpopulations were sorted using a BD Aria-special order research product followed by 3D culture or RNA extraction.
An initial gating to exclude cell debris and doublets was included in all analyses and sorting experiments. The purity of the sorted population was more than 95%. All antibodies used for flow cytometry are listed in Supplementary Table S4.
In vitro differentiation assay
To induce the differentiation of pancreatic progenitor-like colonies into β-like cells, an in vitro differentiation assay was performed. Colonies from the third passage were collected using a pipette and transferred to an ultra-low attachment plate. The colonies were maintained for 3 days in Roswell Park Memorial Institute (RPMI) medium supplemented with 0.2% FBS and 100 ng/mL Activin A (Stage I). The colonies were incubated for 5 days in the presence of 500 nM phorbol 12,13-dibutyrate (PdBU), 100 ng/mL fibroblast growth factor 10 (FGF10), and 2% FBS (Stage II), followed by incubation in DMEM supplemented with 1% B27 and 50 ng/mL Noggin (Stage III). The colonies were eventually treated with DMEM containing 2 μM retinoic acid and 0.25 μM Sant-1 for 6 days (Stage IV), followed by incubation for 4 days in DMEM supplemented with 1% B27 and 1 μM Compound E (Stage V). Finally, the colonies were picked for immunostaining.
In vivo assessment of multilineage potential
To assess the multilineage potential in vivo, PDEC colonies were transplanted under the kidney capsules of SCID-NOD mice. In detail, 14-day-old colonies cultured in the 3D system were hand-picked and used to obtain a single cell suspension (∼2 × 106 cells). The cells were centrifuged at 500 g for 5 min to obtain a cell pellet. The mice were anaesthetized, and a small incision was made on the left abdomen. The kidney was gently pushed outside the abdominal cavity, and a small cut was made on the capsule using the tip of a 30-gauge needle. The cells were injected with a 1-mL syringe under the capsule through the cut made with the needle. The kidney was gently pushed back, and the incision was sutured. The mice were sacrificed after 35 days. The engrafted kidney was dissected out and fixed in formalin. Paraffin embedding, hematoxylin-eosin staining, and immunohistochemistry were performed following standard methods.
Dual luciferase assay
To study the DNA binding effect of Id2, the promoter region (the sequence was obtained from GeneCopoeia, product ID: MPRM23644) of mouse Cdkn2a was cloned into the pGL3-basic vector (Addgene). The following primer pairs were used: 5′-CTAGCTAGCCCCACACATTTGAATTCGTGGAGTT-3′ (sense) and 5′-CCCAAGCTTGCTCTCCTCGAGTTCGCTTTCCTCG-3′ (antisense). The binding site was predicted using Jaspar 2018 (
Statistical analysis
Student's t test was used to determine whether differences were significant between two groups, and ANOVA was used to analyze the differences among multiple groups. Dunn's multiple comparisons for one-way ANOVA and Fisher's least significant difference for two-way ANOVA were used. The level of significance was set at *P < 0.05, **P < 0.01, and ***P < 0.001. Data are expressed as ± SD or ± SEM. GraphPad prism 7 was used for all calculations.
Results
CD133-expressing PDECs are responsive to TGF-β receptor signaling
In our previous study, we presented a 3D system for the cultivation of mouse pancreatic progenitor-like colonies in vitro [14], which was a methylcellulose-based semisolid medium. Methylcellulose is a kind of long-chain polysaccharide that can form compact grid in the liquid suspension medium, while restricting the movement of single cells, yet soft enough to allow colony formation. To further investigate the colonies were single cell derived, we sorted CD133+ cells of CAG-EGFP (GFP) mice and R26-mTmG (red) mice and cultured them in the 3D system. As shown in Supplementary Fig. S1A, all the colonies were single colored. And the colonies cultured in the 3D system showed increased expression of progenitor and duct markers and almost no endocrine and acinar markers (Supplementary Fig. S1B–D). To investigate the gene expression pattern of the colonies, we performed high-throughput sequencing (HTS) [25] and found that the pancreatic progenitor-like colonies were highly enriched in TGF-β signaling-related proteins (Fig. 1A). Real-time PCR confirmed the HTS results (Fig. 1B). As most progenitor-like colonies comprised CD133-expressing cells [11,12,14], we determined whether CD133-expressing PDECs were responsive to TGF-β signaling. As expected, the freshly sorted CD133+ PDECs from the mouse pancreas were enriched in TGF-β signaling-related factors (Fig. 1C). Moreover, CD133 and TGF-β receptors were co-expressed in the 3D culture system (Fig. 1D, E) as well as under in vivo conditions (Fig. 1F, G). The percentages of CD133+/TGFβRI+ cells in the 3D culture system and under in vivo conditions were 41.85% and 7.13%, respectively, by flow cytometry analysis (Supplementary Fig. S1E, F). These results suggest that the CD133-expressing PDECs are responsive to TGF-β receptor signaling.
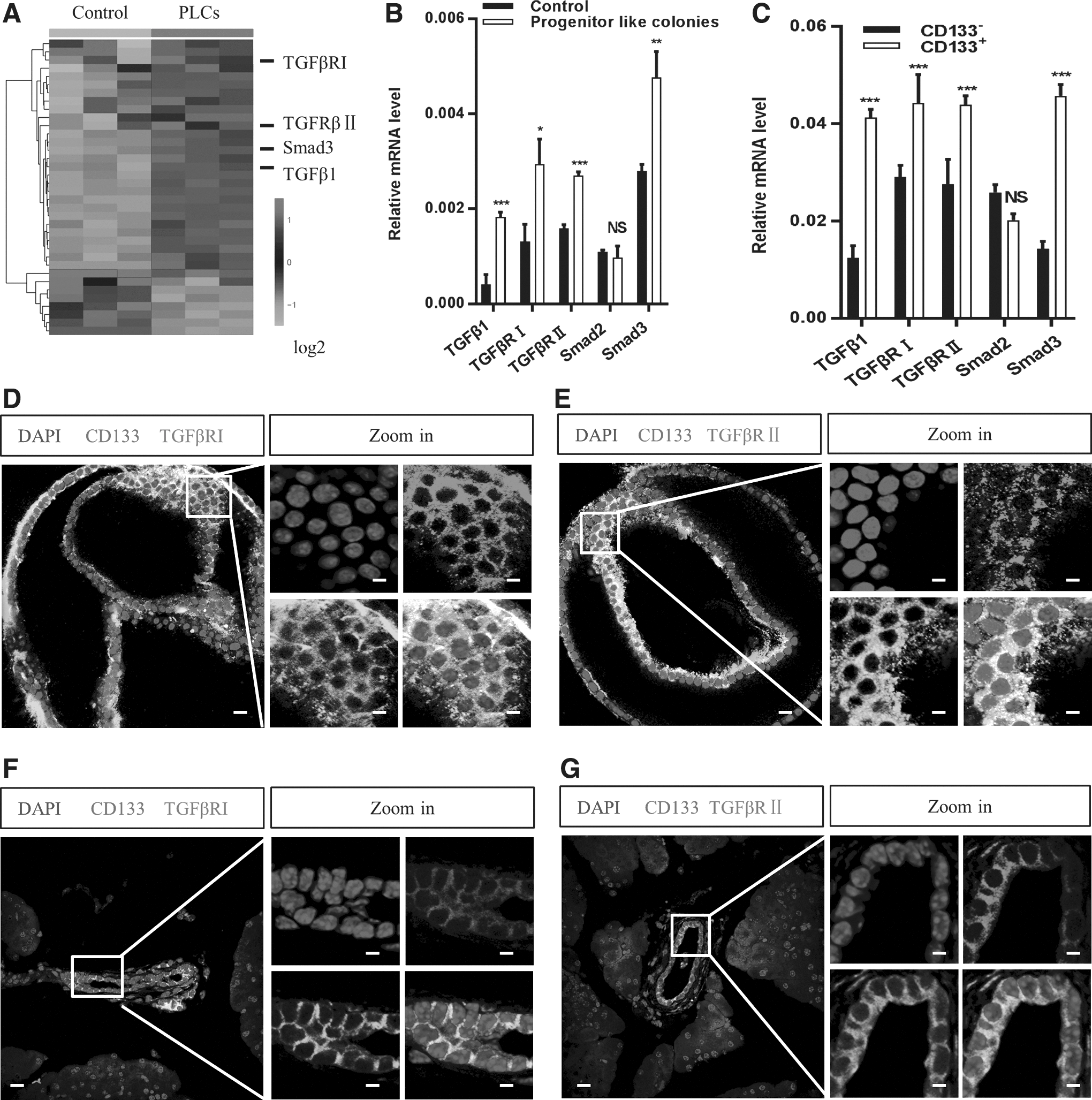
Expression of TGF-β signaling-related genes in CD133-expressing PDECs.
Inhibition of TGF-β signaling plays a pivotal role in the control of the proliferation of CD133-expressing PDECs
Based on our finding that CD133-expressing PDECs were responsive to TGF-β signaling, we evaluated the influence of TGF-β signaling on CD133-expressing PDECs in vitro and in vivo. We performed colony formation assays and compared the clonogenic ability of CD133-expressing PDECs in the presence of either vehicle, TGF-β1 or TGFβRI inhibitor (SB431542 and RepSox). The presence of SB431542 and RepSox increased the colony formation ability of these cells, but we did not observe similar results in the TGF-β1 and vehicle groups (Fig. 2A, B). We compared the clonogenic ability of CD133-expressing PDECs derived from TGFβRIIwt/wt and Pdx1-cre+ TGFβRIIflox/flox (TGFβRIInull/null) mice and observed similar results in TGFβRIInull/null mice (Fig. 2C, D). The colony size and cell number from total colonies increased in the TGFβRI inhibitor-treated group (Fig. 2E and Supplementary Fig. S2A) and in the TGFβRIInull/null mouse group (Fig. 2F and Supplementary Fig. S2B). Fluorescence-activated cell signaling (FACS) analysis revealed that the number of CD133-expressing PDECs in TGFβRIInull/null mice increased about fivefold compared with that in TGFβRIIwt/wt mice (Supplementary Fig. S2C), and that the number of CD133+Ki67+ cells increased by about 13-fold in TGFβRIInull/null mice compared with that in TGFβRIIwt/wt mice (Supplementary Fig. S2D).
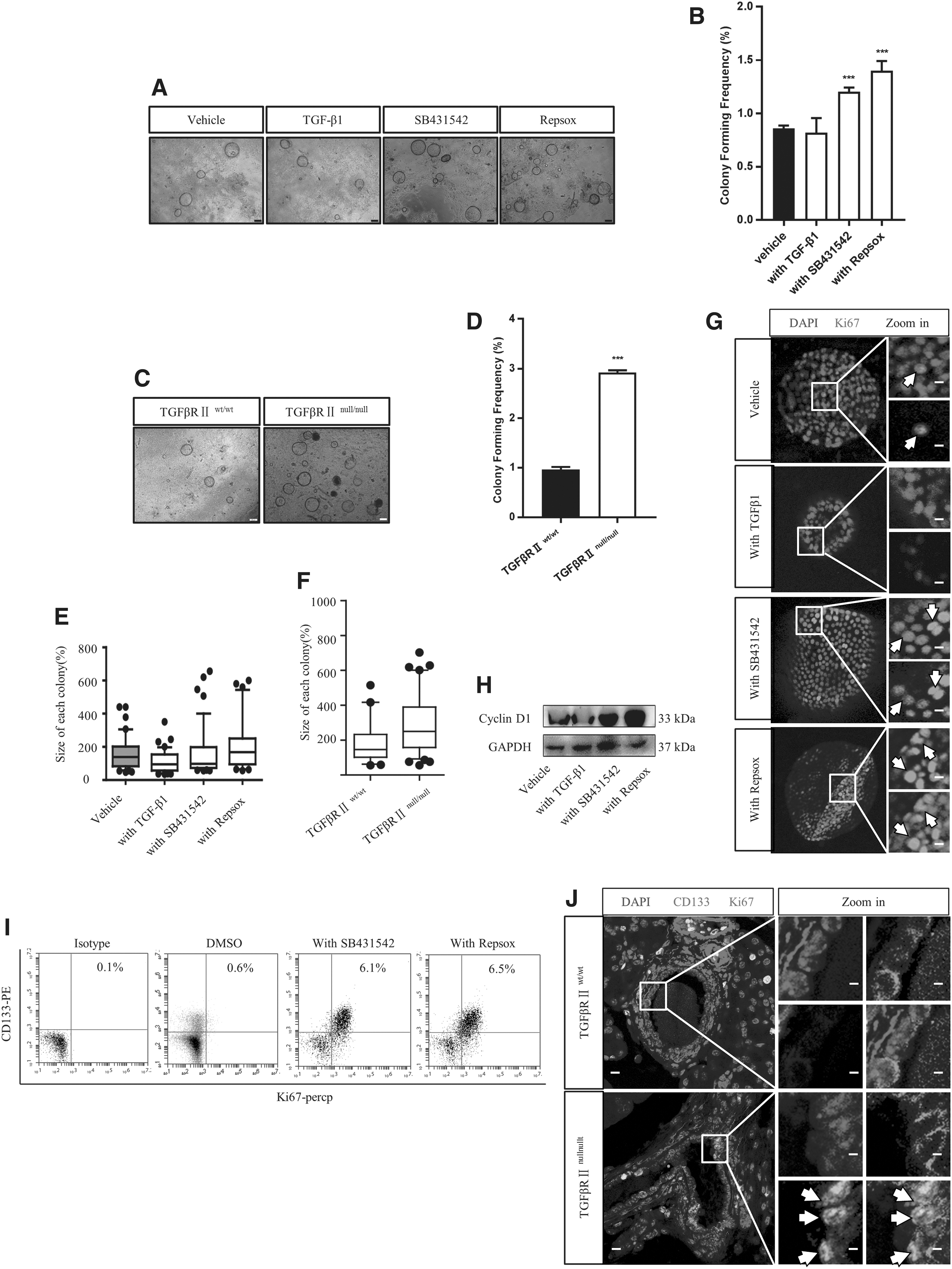
Inhibition of the TGF-β receptor promoted the proliferation of CD133-expressing PDECs. TGF-β inhibitors, SB431542 (5 μM) and RepSox (5 μM), were added to the culture medium, and the colony formation ability of CD133-expressing PDECs was analyzed under a microscope
To determine whether the high cell number was associated with increased proliferation, cells treated with or without TGFβRI inhibitors were analyzed for the expression of proliferation markers Cyclin D1 and Ki67. These two markers showed increased expression following TGFβRI inhibitor treatment at both the mRNA and protein levels (Fig. 2G, H and Supplementary Fig. S2E, F). The injection of the inhibitors under the pancreas capsule resulted in an increase in the number of CD133+Ki67+ cells in the TGFβRI inhibitor-treated groups, as seen in the FACS analysis (Fig. 1I). The increase in the number of CD133+Ki67+ cells in TGFβRIInull/null mice, as confirmed by immunostaining (Fig. 2J), demonstrates that TGF-β signaling inhibition enhances the proliferation of CD133-expressing PDECs.
TGF-β signaling inhibition enables the in vitro expansion of CD133-expressing PDECs for several generations
To determine whether TGF-β pathway inhibition regulates the self-renewal ability of CD133-expressing PDECs, we performed a serial replating experiment. Freshly sorted CD133-expressing PDECs were plated in the presence of vehicle, TGF-β1, SB431542, or RepSox. Cultures were maintained for 14 days, and the primary colonies formed were dissociated and replated to evaluate the potential change in the abundance of colony-forming cells among the total cell population. Compared with the vehicle- and TGF-β1-treated cells, the cells treated with SB431542 or RepSox showed a threefold increase in the number of colonies in secondary cultures (Fig. 3A). Moreover, TGFβRI inhibitor-treated cells were present in higher numbers in secondary cultures (Fig. 3B). The CD133-expressing PDECs derived from TGFβRIInull/null mice showed about a threefold increase in the number of colonies (Fig. 3C) and were present in higher numbers in secondary cultures compared with those obtained from TGFβRIIwt/wt mice (Fig. 3D). These results demonstrate that TGF-β pathway inhibition enhances the self-renewal capacity of CD133-expressing PDECs in primary cultures.
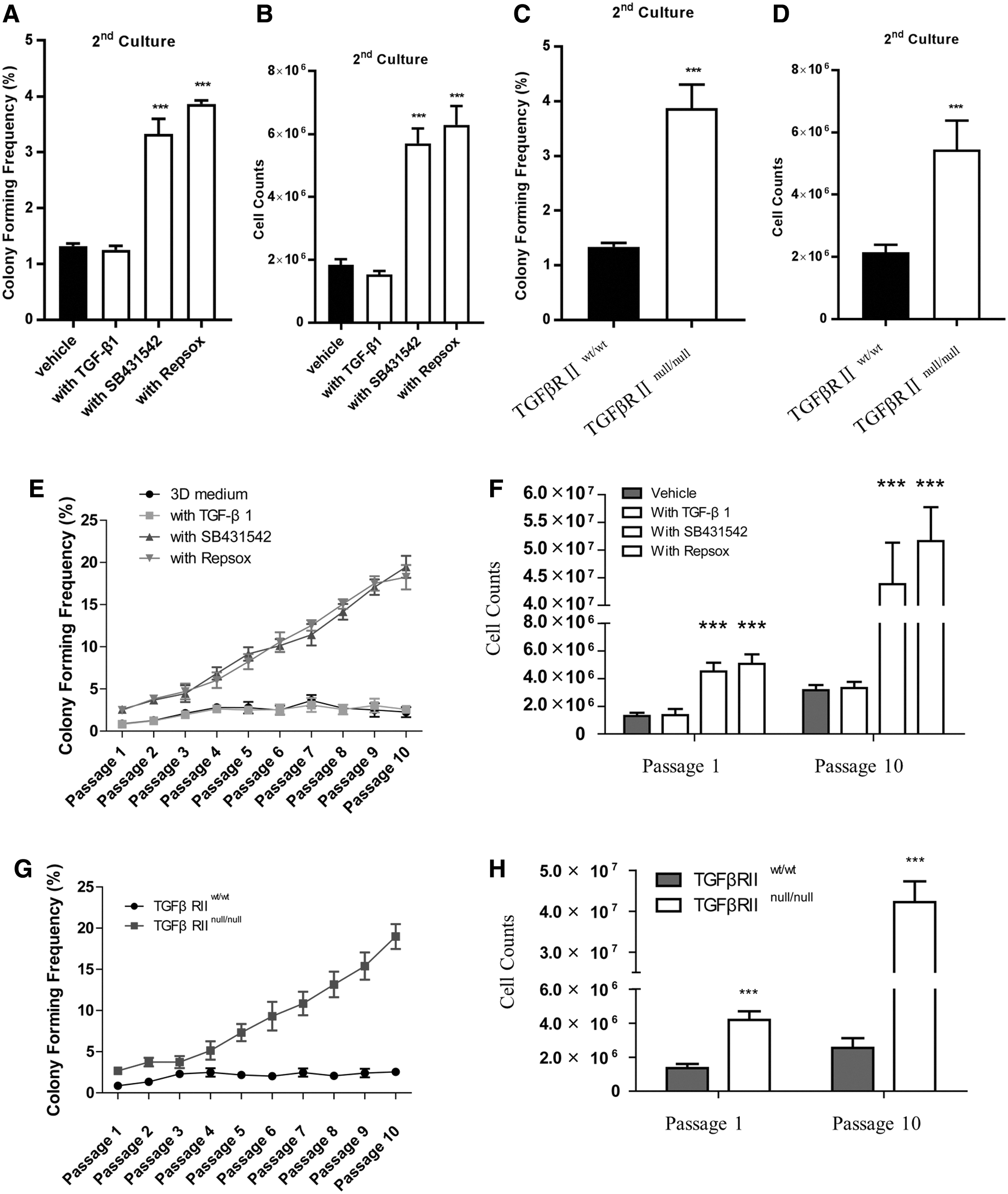
TGF-β pathway inhibition facilitated the in vitro expansion of CD133-expressing PDECs for several generations.
We determined whether these cells can retain their self-renewal capacity for a long time after TGF-β pathway inhibition. The colonies obtained from the primary cultures treated with an exogenous TGFβRI inhibitor were dissociated and serially replated for an additional 10 generations in the presence of vehicle, TGFβ1, SB431542, or RepSox. The continuous exposure of colonies to exogenous SB431542 or RepSox induced an exponential increase in the number of colony-forming progenitor cells and led to a 10-fold expansion in the number of colony-forming cells over 10 weeks. TGFβ1 did not immediately inhibit the self-renewal capacity of primary colonies, but gradually suppressed this property over a series of passages (Fig. 3E, F). The self-renewal capacity was higher for the cells obtained from TGFβRIInull/null mice than that for cells cultured from TGFβRIIwt/wt mice (Fig. 3G, H). These results demonstrate that the primary colonies obtained after TGF-β pathway inhibition exhibited a long-term potential to generate progenitor cells and that the effects of TGFβRI inhibitors on primary colonies were not immediately reversed in subsequent cultures.
Inhibition of TGF-β signaling in CD133-expressing PDECs results in the maintenance of the multilineage potential of colonies when derived from single cells
To test whether the colonies derived from single cells were multipotent in vitro, the cells from primary cultures treated with or without TGFβRI inhibitors were analyzed for the expression of progenitor cell markers using RT-qPCR. SB431542 or RepSox treatment enhanced the expression of the early pancreatic progenitor marker Pdx1, but not the late markers Pax6 and NeuroD1 (Fig. 4A). Western blot analysis confirmed the results of RT-qPCR (Fig. 4B). However, SB431542 or RepSox treatment did not induce insulin gene expression (data not shown). To investigate the multilineage differentiation potential of the colonies, we cultured them in a differentiation medium. As shown in Fig. 4C and D, SB431542- or RepSox-treated cells expressed terminally differentiated endocrine (C-peptide, glucagon) or acinar (Amylase) lineage markers following cultivation in the differentiation medium. Moreover, the mRNA expression levels of endocrine (Ins1, Ins2, and GCG) and exocrine (Amylase and CelA1) cell markers were almost undetected in the control cultures (Supplementary Fig. S3A), and the CD133+/insulin+ cells were only detected in the differentiation medium (Supplementary Fig. S3B). These results indicate that TGF-β pathway inhibition supports the expansion of endocrine and acinar progenitor cells from CD133-expressing PDECs and that the colonies could subsequently differentiate into endocrine and acinar cells in the absence of exogenous TGFβRI inhibitors.

Inhibition of the TGF-β pathway maintained the multilineage potential of CD133-expressing PDECs. CD133-expressing PDECs were cultured with TGF-β1 (2 ng/mL), SB431542 (5 μM), or RepSox (5 μM). Real-time PCR and western blot analyses were performed to determine the mRNA
To test the in vivo potential of TGFβRI inhibitor-treated cells, we transplanted 2 × 106 SB431542-treated colonies under the renal capsules of SCID/NOD mice to assess lineage development and transplanted 2 × 106 SB431542-treated CD133− cells as a control group. The mice were sacrificed 35 days after transplantation. Hematoxylin and eosin staining of sections of the grafts revealed circumscribed tissue with considerable pleomorphism and without any evidence of invasion into the adjacent kidney parenchyma (Fig. 4E). These observations indicate that only SB431542-treated colonies were engrafted and survived for at least 35 days after their transplantation under the kidney capsules of SCID/NOD mice (control data not show). We also examined the grafts for the expression of pancreatic endocrine and exocrine tissue markers. In one mouse, a large graft examined 35 days after transplantation of SB431542-treated cells showed scattered islands of tissue, which were stained positive for the β-cell marker insulin and the acinar cell marker amylase (Fig. 4F, G). Thus, the donor cells had given rise to both endocrine and acinar cells in vivo. These results show that CD133-expressing PDECs expanded in the presence of a TGF-β pathway inhibitor have the ability to differentiate into endocrine and acinar lineages in vivo.
Inhibition of TGF-β signaling induces the expression of Id2 by decreasing p-Smad3 expression in CD133-expressing PDECs
Smad3 acts downstream of TGF-β signaling and is overexpressed in pancreatic progenitor-like colonies (Fig. 1A). Following stimulation with TGF-β1, Smad3 is phosphorylated and translocates into the nuclear region to regulate the transcription of target genes. As found in other studies [35], TGFβRI inhibition reduced the absolute level of p-Smad3 (Fig. 5A). Similar observations were recorded in the pancreas of TGFβRIInull/null mice (Supplementary Fig. S4A). To test the expansion and maintenance of CD133-expressing PDECs following TGF-β signaling inhibition by decreasing p-Smad3 levels, we used a lentivirus Smad3 short-hairpin RNA (shRNA) to knock down Smad3 expression. Smad3 knockdown dramatically increased the colony formation ability and total cell counts (Fig. 5B and Supplementary Fig. S4B, C). The RT-qPCR and western blot results showed that Smad3 knockdown upregulated the expression of Cyclin D1 and pancreatic progenitor-related genes, but downregulated the expression of endocrine progenitor-related genes (Fig. 5C and Supplementary Fig. S4D). These results were consistent with those observed following TGFβRI inhibitor treatment.
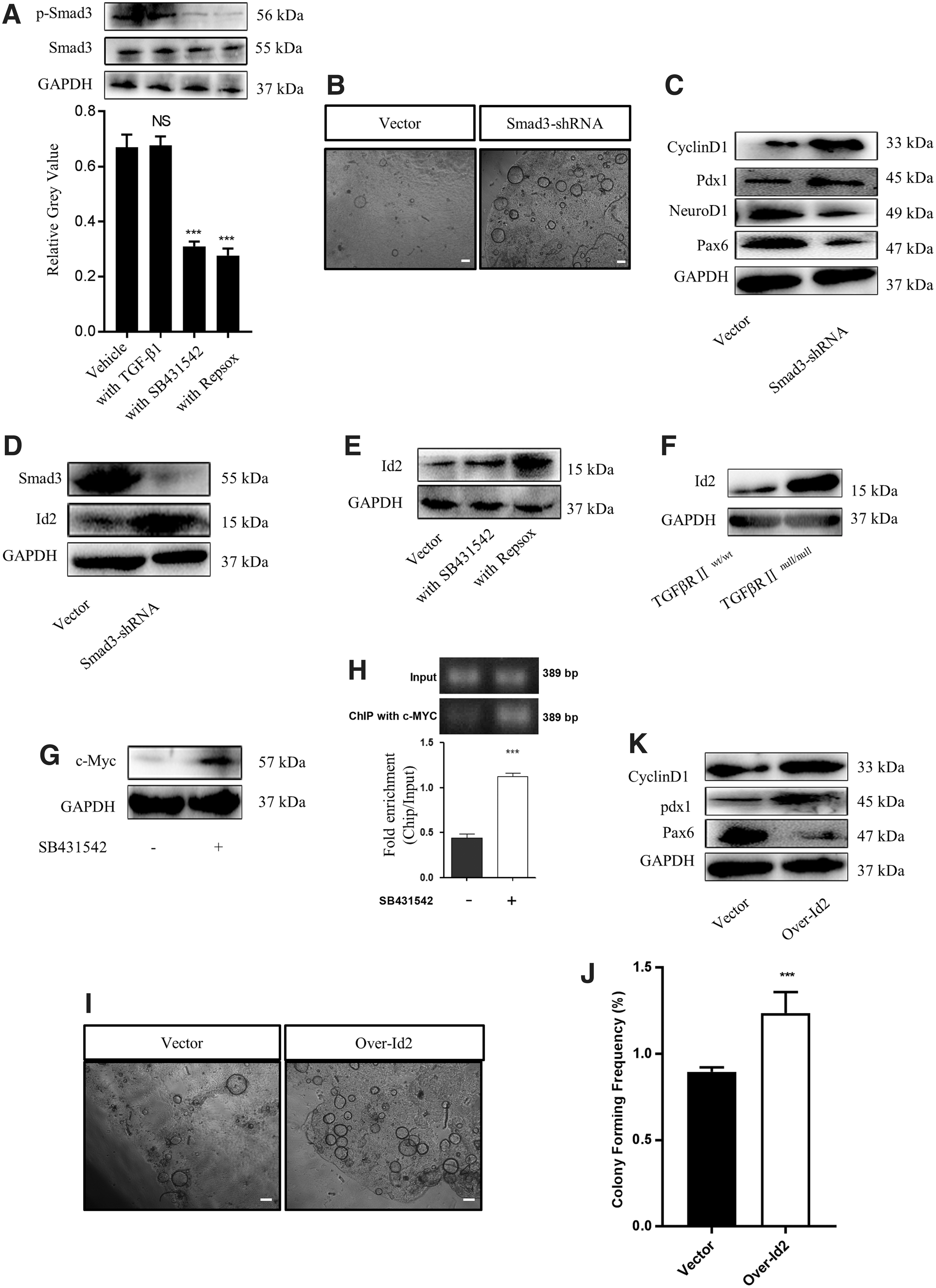
Inhibition of TGF-β signaling induced the expression of Id2. CD133-expressing PDECs were cultured with TGF-β1 (2 ng/mL), SB431542 (5 μM), or RepSox (5 μM), and western blot analysis was performed to evaluate the expression level of Smad3 and p-Smad3
We investigated the downstream effectors of p-Smad3 and found that TGF-β signaling inhibition, either with inhibitors or Smad3 knockdown, caused an increase in Id2 mRNA and protein levels (Fig. 5D, E and Supplementary Fig. S4E). This phenomenon was observed even in TGFβRIInull/null mice (Fig. 5F and Supplementary Fig. S4F). As previously reported [36,37], TGFβR1 inhibition was accompanied with an increase in c-Myc expression (Fig. 5G and Supplementary Fig. S4G). Using a ChIP strategy, we found that TGFβR1 inhibition improved the binding of c-Myc to the promoter of Id2 (Fig. 5H). This observation explains why TGF-β signaling inhibition caused an increase in Id2 expression. To determine whether the increase in Id2 expression was associated with the expansion and maintenance of pancreatic progenitor-like cells, we overexpressed Id2 in CD133-expressing PDECs and found that the overexpressed Id2 dramatically increased the colony formation ability and total cell counts (Fig. 5I, J and Supplementary Fig. S4H), upregulated the expression of cyclin D1 and the progenitor-related gene Pdx1, and downregulated Pax6 expression (Fig. 5K and Supplementary Fig. S4I). Together, these results reveal that TGF-β signaling inhibition improved the proliferation and maintenance of the stemness characteristic of CD133-expressing PDECs through the p-Smad3-Id2 axis.
Inhibition of TGF-β signaling improves the binding of Id2 to the promoter of p16 and Neuro D1 in CD133-expressing PDECs
As TGF-β signaling inhibition improved the proliferation and maintained the stemness of CD133-expressing PDECs through the p-Smad3-Id2 axis, we investigated the factors acting downstream of Id2.
p16 is a cell cycle suppressor. While p16 expression is downregulated in rapidly growing cells, it is overexpressed in quiescent cells. We found a binding site of Id2 in the p16 promoter region with Jaspar2018 (Supplementary Fig. S5A). We hypothesized that Id2 could bind to this site and negatively regulate p16 transcription. As expected, the dual-luciferase assay results showed that the overexpression of Id2 in 293T cells resulted in a decrease in the transcriptional activity of the p16 promoter, and a mutation in the binding site negatively affected Id2 gene transcription (Fig. 6A). Further experiments showed that TGFβRI inhibitors (Fig. 6B, C), Smad3 knockdown (Fig. 6D, E), or Id2 overexpression (Fig. 6F, G) decreased the mRNA and protein levels of p16 in CD133-expressing PDECs. Thus, the inhibition of TGF-β signaling improved the expansion of CD133-expressing PDECs through the Id2-p16 axis.
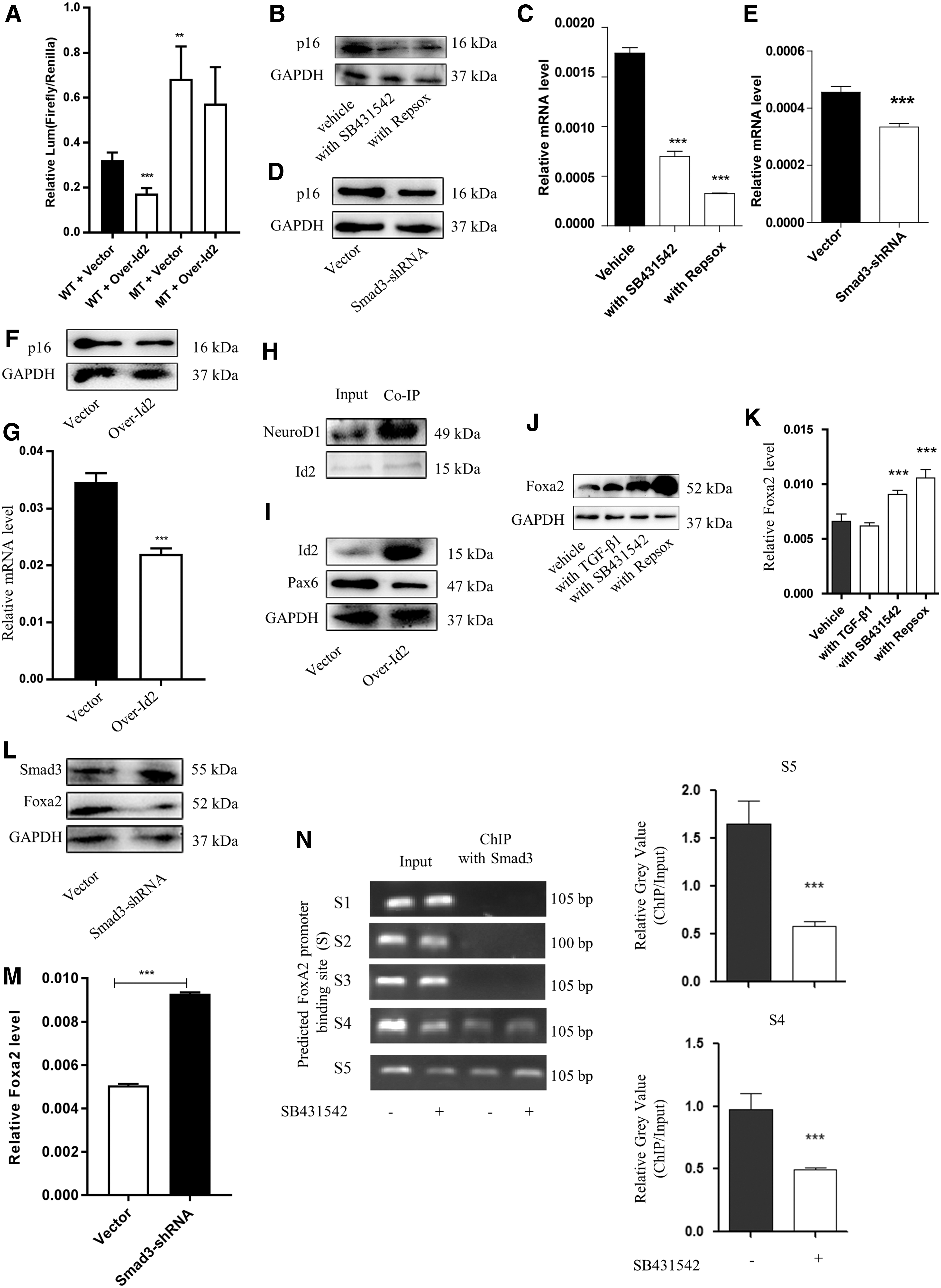
Inhibition of TGF-β signaling improved the binding of Id2 to the promoter of Cdkn2a (p16).
We evaluated the factors acting downstream of Id2, which is involved in the maintenance of the stemness of CD133-expressing PDECs. Id2 was reported to bind to NeuroD1 and inhibit the transcription of Pax6 in neuroendocrine cells [38,39]. To determine whether Id2 could bind to NeuroD1, downregulate Pax6 expression, and maintain CD133-expressing PDECs in an undifferentiation stage, we used co-immunoprecipitation (Co-IP) and overexpression strategies in CD133-expressing PDECs. As expected, we detected the binding of endogenous Id2 to NeuroD1. An increase in the Id2 level was found to downregulate the expression of Pax6 (Fig. 6H, I).
In this study, we found that the expression level of Pdx1 was dramatically increased after the inhibition of TGF-β signaling. We investigated the mechanism underlying the maintenance of the stemness properties of CD133-expressing PDECs following TGFβRI inhibition and found that TGFβRI inhibition reduced the level of Forkhead box protein A2 (Foxa2) (Fig. 6J, K). This observation is consistent with a previously reported finding, wherein Pdx1 expression was increased by Foxa2 [40,41]. We found that the knockdown of Smad3 expression in CD133-expressing PDECs resulted in an increase in the mRNA and protein levels of Foxa2 (Fig. 6L, M). Five binding regions were predicted between Smad3 and the Foxa2 promoter with Jaspar2018. The ChIP analysis results showed that a TGFβRI inhibitor improved the binding of Smad3 to the promoter of Foxa2 (Fig. 6N), providing an explanation for the increased Foxa2 expression following TGFβRI inhibition. Foxa2 protein is among the pioneer factors of pancreatic development and dynamically regulates Pdx1 expression. Taken together, these results explain the TGF-β signaling inhibition-mediated improvement in the self-renewal ability and proliferation of CD133-expressing PDECs.
Discussion
Inhibition of TGF-β signaling facilitated the expansion of CD133-expressing PDECs. The transplantation assay results indicated that colonies have multilineage potential to generate endocrine and exocrine cells in vivo. We found that TGF-β signaling inhibition induced the expression of Id2 along with the expansion of CD133-expressing PDECs. The binding of Id2 to the promoter region of the cell proliferation repressor p16 and endocrine-restricted transcription factor NeuroD1 decreased their expression. Our results support the hypothesis that the inhibition of TGF-β signaling may be critical for the expansion of pancreatic duct epithelial progenitors and for the maintenance of their predifferentiated state (Fig. 7).

A working model describing the role of the TGF-β pathway in the expansion and maintenance of CD133-expressing PDECs.
In general, PDECs comprise pancreatic progenitor cells [42,43]. Recently, a population of PDECs called CD133-expressing PDECs was isolated based on the specific cell surface marker CD133, and the pancreatic progenitor property was tested in vitro and in vivo [9,10]. However, it is difficult to cultivate these cells for an extended period under in vitro conditions using conventional culture protocols [13,14]. The effective expansion of progenitor cells may require mimicking of the in vivo environment to maintain a large proportion of cells as progenitor cells. The TGF-β signaling pathway has been implicated in the regulation of stem cell quiescence and activation [29,44]. To determine whether TGF-β signaling induced CD133-expressing PDEC proliferation, we evaluated the expression of TGF-β factors in these cells and found that CD133-expressing PDECs were enriched in most of the TGF-β receptors, ligands, and downstream components; these factors were absent in CD133− cells. This observation strongly suggests a role of TGF-β signaling in CD133-expressing PDECs and that these cells should be responsive to TGF-β factors.
The importance of TGF-β signaling in cell growth has been previously demonstrated [29,34,45,46]. We found that TGF-β signaling inhibition was essential for the growth of CD133-expressing PDECs in vitro and in the pancreas-specific TGFβRII-knockout mice. The small-molecule inhibitors of the TGF-β pathway induced the expression of Id2 protein. Several other studies have also linked TGF-β signaling with the expression of Id2, and two groups of researchers have shown that SMAD elements could bind to Id2 gene promoters [36,37]. In our study, we found another TGF-β signaling-related downstream factor, c-myc, which could bind to the promoter of Id2 and downregulate its expression. The overexpression of Id2 in CD133-expressing PDECs promoted cell proliferation through the inhibition of p16 expression. Induction of p16 expression reduces the self-renewal ability of progenitor cells across mammalian tissues [47 –49]. We identified the link between Id2 and p16, thereby improving our understanding of the mechanisms underlying the TGF-β pathway-mediated effects on pancreatic progenitor cell proliferation.
TGF-β signaling often mediates the in vivo maintenance of progenitor cells [29,50,51]. In this study, we observed that the inhibition of TGF-β signaling induced Id2 expression and binding to NeuroD1. Previous studies have shown that NeuroD1 can form heterodimers with E47 and bind to its E-box target [38,39]. Moreover, one of the targets of NeuroD1 is Pax6, a factor that is critical for pancreas morphogenesis and endocrine progenitor maintenance [52,53]. In agreement with this result, our data revealed the reduced expression of Pax6 after the inhibition of TGF-β signaling. In addition, TGF-β signaling inhibition was shown to induce the expression of Foxa2 along with the expansion of CD133-expressing PDECs. This observation is consistent with the result that the promoter of the Foxa2 gene contains a binding site for Smad3, an effector of the TGF-β signaling pathway that downregulates Foxa2 transcription in response to TGF-β treatment [54]. Foxa2 protein is essential for pancreatic development and regulates Pdx1 expression [55,56]. We observed Pdx1 expression induction along with an increase in Foxa2 levels, consistent with the findings reported by Bastidas PA and Mfopou JK [40,41]. These results enhance our knowledge of TGF-β signaling pathways, which are necessary for the expansion of pancreatic progenitors.
Inhibition of TGF-β signaling enhances the expansion of CD133-expressing PDECs, and the colonies maintain the characteristics of pancreatic progenitors, as demonstrated by their self-renewal capacity and multilineage differentiation potential in vitro and in vivo. This result confirmed the in vivo findings that TGF-β signaling is essential for the proliferation of pancreatic progenitors during pancreatic organogenesis [57]. Small-molecule inhibitors of the TGF-β pathway induced the production of numerous Pdx1-expressing cells that were negative for endocrine and acinar markers. After the removal of the inhibitor and the transplantation of the expanded cells in the renal capsule of a mouse, these undifferentiated epithelial cells differentiated into endocrine and acinar cells. This result indicates that the inhibition of TGF-β signaling promotes the expansion of pancreatic progenitors from CD133-expressing PDECs without the induction of in vitro differentiation. However, whether TGF-β signaling inhibitors act through the plasticity of the ductal cells or through the direct expansion of the spontaneously committed Pdx1-expressing cells is unclear. We suggest that the latter scenario is more likely because a very high number of Pdx1-expressing cells were observed in CD133-expressing PDECs before small-molecule treatment. Additional studies are warranted to elucidate the exact role of TGF-β signaling in the expansion of pancreatic progenitors from CD133-expressing PDECs.
In summary, we used a combination of cell culture and in vivo transplantation experiments to demonstrate that the inhibition of the TGF-β signaling pathway could lead to the expansion of CD133-expressing PDECs. Our findings show that the manipulation of the TGF-β signaling pathway can directly influence the survival and differentiation of pancreatic progenitor cells. These findings are not necessarily relevant to physiologic processes, but could aid in efforts to exploit TGF-β signaling pathways for tissue regeneration. The translation of these pathways to human CD133-expressing PDECs represents another important step that may eventually allow the exploitation of their potential therapeutic applications.
Footnotes
Acknowledgments
We are grateful to Xiaojun Xu and Caoyong He (State Key Laboratory of Natural Medicines, China Pharmaceutical University, 210009, Nanjing, Jiangsu, China) for critically reviewing and editing the article as well as providing some scientific advice.
This work was supported by the National Natural Science Foundation of China (grant no. 81570696, no. 31270985, and no. 31472159). Support was also provided by the Qing Lan Project.
Author Disclosure Statement
The authors disclose no conflicts.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.