
Abstract
Embryonic stem (ES) cells have been utilized as an excellent model for the study of neural development. However, the dynamic changes of ES cell-derived neural stem cells (ES-NSCs), under the effects of prolonged cell culture and hypoxic conditions, are still obscured. In the present study, using the combination of serum-free culture of embryoid body-like aggregates (SFEB) culture and cell sorting by Sox-1, the ES-NSCs were easily isolated and showed in vitro temporal neural specification, which resulted in distinct cell fates after neural differentiation. Early passaged ES-NSCs gave rise to neurons, whereas late-passaged ES-NSCs gave rise to glial cells, similar to the in vivo dynamic changes during the neural development. Remarkably, hypoxia treatment induced the neural differentiation of ES-NSCs but did not affect the cell fate. Under hypoxic conditions, early passaged ES-NSCs showed the upregulation of neuronal markers, whereas late-passaged ES-NSCs showed the upregulation of a glial marker. In addition, the knockdown of the hypoxia-inducible factor 1α expression impaired the neuronal differentiation of early passaged ES-NSCs under hypoxic conditions. These data demonstrated the distinct effects of prolonged culture and hypoxic stimuli on the neural differentiation of ES-NSCs; prolonged culture was involved in the cell fate after neural differentiation, while hypoxia treatment efficiently promoted neural differentiation.
Introduction
The vertebrate central nervous system (CNS) development is a complex spatiotemporal process. It starts from the ectodermal monolayer and develops into a pseudostratified epithelial sheet known as the neuroepithelium. The initial development of the neuroepithelium is correlated with the onset of the expression of transcription factor Sox1, thus establishing Sox1 as the earliest and most specific marker for primordial neural progenitor [1 –3]. Sox1 belongs to the Sox family, which includes the transcription factors of cell fate decisions during neural development [4]. A previous report showed fewer ventral telencephalic neurons and the impairment of ventral neuron migration in Sox1-null mice [5]. Of note, the loss of Sox1 expression indicates the exit from the primordial neural progenitor stage to the radial glia stage [6]. This leads to the generation of the neural tube, which is the precursor of the CNS. The process then enters the neurogenesis phase until mid gestation, followed by the gliogenesis phase until birth [7 –9].
Neurogenesis is an important phase in embryonic brain formation, as new neuronal cells are generated during this phase. Since neuronal cells play a role as the primary functional units of the nervous system, any dysfunction or loss of these cells can lead to diseases or disorders of the CNS. Therefore, understanding the molecular mechanisms underlying neurogenesis is expected to aid in the development of therapies for CNS diseases. Because of the complexity and dynamic characteristics of the nervous system, in vitro studies using cell-based neurogenesis models are the preferred approach, as such models faithfully recapitulate the in vivo processes and reduce the associated complexity [1 –3].
The in vitro model for embryonic neurogenesis studies is based on the use of pluripotent stem cells, such as embryonic stem (ES) cells. ES cells have the ability to differentiate into three germ layers: the ectoderm, mesoderm, and endoderm [10]. Success in isolating and maintaining ES cells ex vivo has made ES cells the go-to cell source for developmental studies and regenerative medicine research. Of importance, ES cells are reportedly able to recapitulate neural development ex vivo [11]. In addition, previous reports have introduced efficient methods for the in vitro generation of neural cells derived from ES cells. Watanabe et al. optimized the ES cell culture conditions for neural differentiation using serum-free suspension culture to directly differentiate the telencephalic precursors from ES cells in a technique termed “SFEB culture” [12]. In this way, the in vitro generation of ES cell-derived neural cells has become a more efficient way to study neurogenesis and CNS development. Many recent advances based on serum-free culture of embryoid body-like aggregates (SFEB) culture have been reported, such as the addition of Wnt and Nodal antagonists; however, the cost effectiveness of using these chemicals is still under consideration.
Under the in vitro cultured conditions, although ES cells have the ability to undergo unlimited self-renewal, a number of adverse effects on ES cells has been reported, such as the prolonged culture and oxygen tension. For example, ES cells show a reduced ability to differentiate into the derivatives of the three germ layers, after repeated and prolonged cell passage compared with earlier passages [13]. In addition, the promotion of chromosomal abnormality acquisition by long-term culture of ES cells has been reported [13]. In ES cell-derived keratinocyte-like cells, long-term culture is reported to induce the sequential appearance of epidermal markers [14]. However, while a number of similar issues have been explored, the effects of long-term culture on the neurogenesis of ES cell-derived neural cells have not yet been clarified. To maintain a stable cell source for the study of CNS development and to ensure effective regeneration therapy, it is crucial to determine whether or not long-term in vitro cell culture leads to adverse effects on the characteristics, neural differentiation abilities, and functions of ES cell-derived neural cells.
Of further note, neurogenesis and CNS development processes occur under hypoxic conditions [15]. We previously reported that hypoxia, through the oxygen sensor protein hypoxia-inducible factor 1α (HIF-1α), is an essential microenvironment for inducing neural commitment during the period of neuroectoderm formation [16]. During neurogenesis, hypoxia exposure has been reported to enhance the generation of neuronal cells [17]. In contrast, during gliogenesis, HIF-1α has been reported to cause demethylation of the promoter for glial genes, thereby enhancing the generation of glial cells [18]. However, the effects of hypoxia on the entire neural differentiation process, from the neurogenic to the gliogenic period, have not been fully clarified.
In the present study, we proposed a simple method to isolate ES cell-derived neural stem cells (ES-NSCs) by the combination of SFEB culture and the cell sorting using Sox1 as the marker, and examined the effects of prolonged culture and hypoxic stimuli on the neural differentiation ability of ES-NSCs. We found that the cell sorting based on Sox1 marker after the SFEB induction gave the effectiveness to isolate the NSCs. Of note, the prolonged culture affected the cell fate while low oxygen tension promoted the cell differentiation level. Additionally, we found the decreased expression of HIF-1α in early passaged ES-NSCs led to the impairment of neuronal differentiation, suggesting that the role of HIF-1α is crucial for neural development under hypoxic conditions.
Materials and Methods
ES cell culture
Mouse ES cells (EB5, a kind gift from Dr. Niwa, RIKEN Institute) and Sox1-green fluorescent protein (GFP) mouse ES cells (46C, a kind gift from Dr. Smith, University of Cambridge [1]) were maintained as previously described [9]. In brief, ES cells were cultured on a gelatin-coated dish (Sumitomo Bakelite, Tokyo, Japan) at 37°C in ES medium in the presence of 0.1% fetal bovine serum (FBS; Gibco, Waltham, MA) and 2,000 U/mL LIF (Merck Millipore, Tokyo, Japan). The ES medium consisted of Glasgow Minimum Essential Medium (Gibco), 2 mM glutamine (Gibco), 10% KnockOut Serum Replacement (Gibco), 0.1 mM nonessential amino acids (Gibco), 1 mM pyruvate (Sigma Aldrich, St. Louis, MO), and 0.1 mM 2-mercaptoethanol (Wako, Osaka, Japan). The medium was changed the next day. ES cells were passaged every 3 days.
Neural differentiation of ES cells was induced with SFEB culture [12,19]. In brief, ES cell colonies were dissociated, and 3 × 106 cells in 8 mL ES medium were seeded into 10-mm bacterial-grade dishes. For the analysis of neural-committed cells, we induced neural differentiation with SFEB culture method [20], as this method produced uniform ES cell aggregates compared with the SFEB method. For SFEB culture, 3 × 105 cells per 150 μL ES medium were seeded into low-cell-adhesion 96-well plates (Sumitomo Bakelite). The day ES cells were seeded to differentiate was defined as differentiation day 0. ES cell aggregates were generated spontaneously in a suspension culture within a day. ES medium was half changed to fresh ES medium on day 3. On day 5, differentiated ES cells were subjected to a fluorescence-activated cell sorter (FACS) analysis.
The FACS analysis
After 5 days of culture, SFEB-cultured cell aggregates were washed with phosphate-buffered saline (PBS) and dissociated into single cells with 0.5 mL of 0.25% Trypsin-EDTA (Life Technologies, Waltham, MA). After neutralization with 2 mL of ES cell medium, the cells were then centrifuged at 1,000 rpm for 5 min at 4°C and suspended in 1 mL of 1% FBS/PBS. The suspended cells were subjected to FACS with MoFlo™ XDP (Beckman Coulter, Inc., Brea, CA). The same SFEB-timed differentiating mouse EB5 cell aggregates were used as the GFP-negative control gate, because EB5 cells do not express GFP.
Neural stem cell culture
Sox1+ cells were sorted and plated on 96-well low-adhesion culture dishes (9 × 103 cells/well; Sumitomo Bakelite) and then recultured in 100 μL of Neural Stem Cell medium (NSC medium) consisting of KnockOut DMEM/F-12 (Life Technologies), 2 mM GlutaMAX Supplement (Life Technologies), 1 × StemPro Neural Supplement (Life Technologies), 0.1% (v/v) penicillin/streptomycin (100 U/mL penicillin, 0.1 mg/mL streptomycin; Life Technologies), 20 ng/mL basic fibroblast growth factor (bFGF) (Wako), and 20 ng/mL epidermal growth factor (EGF) (PeproTech, Rocky Hill, NJ). After 4 days, the formed neurospheres were dissociated with 2 mL Accutase (Nacalai Tesque, Kyoto, Japan) and pipetting every 15 min at 37°C. The neurospheres were treated with Accutase for 30–45 min. The cells were then centrifuged for 1,000 rpm for 5 min at room temperature. The dissociated cells (2–4 × 105 cells/4-well culture dish) were then seeded onto culture dishes coated with 5 μg/mL poly L-ornithine (Sigma Aldrich) and 7 μg/mL laminin (Corning, Bedford, MA). Unless stated otherwise, the cells were maintained in NSC medium at 37°C in a humidified atmosphere (20% O2, 5% CO2). The cells were passaged or used for differentiation experiment in an 80%–90% confluent state.
For early and mid-passaged differentiation experiment, neural differentiation was induced by withdrawal of growth factors (EGF and FGF) from 80% to 90% confluent NSC cultures. Differentiation day 0 was defined as the day on which the growth factor was removed from the medium. NSC medium was half changed to fresh medium every 3 days, and differentiation was performed for 5 days.
For neural trilineage characterization and late-passaged differentiation experiments, 80%–90% confluent late-passaged ES-NSCs (P5-P10) were differentiated for 5 days with B27 differentiation medium consisting of Neurobasal medium (Life Technologies), 2 mM GlutaMAX Supplement, and 1 × B27 neural supplement with vitamin A (Life Technologies). Unless stated otherwise, the culture was maintained under normoxic conditions (37°C, 20% O2, 5% CO2). B27 differentiation medium was half changed to fresh medium every 3 days, and differentiation was performed for 5 days. For hypoxia treatment, ES-NSCs were exposed to 3% oxygen tension with a hypoxic incubator balanced with nitrogen gas (ASTEC, Tokyo, Japan).
The reduced expression of HIF-1α in early passaged (P1) ES-NSCs
The knockdown study of HIF-1α in this study was performed by introducing short-interfering RNA (siRNA). Specifically, 80%–90% early passaged ES-NSC (P1) was transfected with either 5 nM of FlexiTube HIF-1α siRNA (Qiagen) or All-Star Negative control siRNA (Qiagen) with 0.5 μL ScreenFect siRNA reagent (Wako) on differentiation day 0. The transfected cells were differentiated by means of withdrawal of growth factors and cultured under 3% oxygen tension (37°C, 5% CO2) for 2 days before being harvested for gene expression analyses.
Gene expression analyses
Total RNA was purified using Sepasol-RNA 1 Super G (Nacalai Tesque), and 1 μg of first-strand complementary DNA was synthesized using a Reverse Transcription (RT)-PCR Kit with genomic DNA remover (Toyobo, Osaka, Japan) according to the manufacturer's instructions. Semiquantitative RT-PCR was performed using KOD-plus (Toyobo) with 22–35 cycles of denaturation (94°C, 10 s), primer annealing (68°C, 30 s), and extension (72°C, 30 s) with a GeneAmp PCR system 9700 (Applied Biosystems, Foster City, CA). PCR products were electrophoresed in 3% agarose gel and then visualized with 0.5 mg/mL of ethidium bromide (Life Technologies). The strength of each band was quantified using the ImageJ software program (version 1.51H,
The Primers Used for Quantitative Polymerase Chain Reaction
Immunostaining
ES-NSCs (1 × 105 cells/well) were seeded on eight-well plates. Culturing was performed under maintenance conditions for 1–2 days, and then the medium was switched to differentiation medium. After 2 or 5 days of differentiation culture, cells were fixed with 4% paraformaldehyde (PFA) for 25 min and then washed with PBS or 0.05% Tween20/PBS three times for 20 min. Permeation was achieved through incubation of 0.3% Triton X-100/PBS twice for 15 min. Blocking was performed with blocking solution (2% Skim milk/PBS) for 1 h followed by incubation of primary antibody (diluted in blocking solution) overnight at 4°C. Cells were then washed with PBS or 0.05% Tween20/PBS three times for 20 min. Alexa-488 or Alexa-546-conjugated goat anti-mouse or anti-rabbit immunoglobulin G (IgG) or mouse immunoglobulin M (IgM) antibody (Invitrogen, Waltham, MA) were used as secondary antibodies to visualize signals. 4′,6-diamidino-2-phenylindole (DAPI) (1:1,000) was used for counterstaining nuclei. Finally, a plate was mounted with SlowFade Gold Antifade Reagent (Molecular Probe, Carlsbad, CA) and photographed with the Olympus BX51 fluorescence microscope (Olympus, Tokyo, Japan). For O4 immunostaining, the permeation step was replaced with PBS to maintain the surface cellular structure.
The antibodies used in this study are listed in Table 2. The relative fluorescence intensity or mean fluorescence intensity was quantified using the ImageJ software program (NIH).
The Antibodies Used for Immunostaining
IgM, immunoglobulin M.
Terminal deoxynucleotidyl transferase dUTP nick end labeling assay
Apoptosis was detected by an in situ Apoptosis Detection Kit (TaKaRa, Shiga, Japan). A terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed according to the manufacturer's protocol and then followed up with immunocytochemistry. As a positive control, cells were treated with DNaseI 10 μg/mL for 10 min at room temperature. In brief, ES-NSCs were seeded into eight-well plates and differentiated upon confluency. After 5 days of differentiation, cells were fixed with 4% PFA for 25 min and then washed with PBS three times for 10 min, followed by blocking with 2% skim milk in 0.05% Tween20/PBS for 60 min. Cells were then washed with 0.05% Tween20/PBS three times for 5 min and permeated with 100 μL Permeation buffer (TaKaRa) on ice for 5 min. Cells were washed with PBS twice for 5 min and then labeled with 50 μL labeling reaction mixture (consisting of 5 μL TdT enzyme +45 μL labeling safe buffer; TaKaRa) at 37°C for 60 min. The chamber was then washed with 0.05% Tween20/PBS twice for 5 min and reblocked with blocking buffer for 30 min at room temperature, followed by incubation with the primary antibody for 90 min at room temperature. After incubation, cells were washed four times with 0.05% Tween20/PBS for 5 min. Alexa-488 or Alexa-546-conjugated goat anti-mouse or anti-rabbit IgG or mouse IgM antibody were used to visualize signals. Nuclei were counterstained with DAPI. Finally, the plate was mounted with SlowFade Gold Antifade Reagent and photographed with an Olympus BX51 fluorescence microscope.
Western blotting
Cultured ES-NSCs were differentiated for 2 days. On a designated day, cells were harvested and lysed in low-salt buffer (10 mM HEPES, pH 7.9; 10 mM KCl; 0.1 mM EDTA; 1 mM dithiothreitol; and protease inhibitor mixture; Nacalai Tesque) on ice for 10 min, followed by the addition of 0.5% Nonidet P-40 and centrifugation at 15,000 rpm for 2 min at 4°C to isolate the nuclear pellet. The nuclear pellets were then suspended in high-salt buffer (20 mM HEPES, pH 7.9; 500 mM NaCl; 1 mM EDTA; and 1 mM dithiothreitol; protease inhibitor mixture), kept on ice for 15 min, and then centrifuged at 15,000 rpm for 10 min at 4°C. The supernatant was used as the nuclear extract sample for HIF-1α detection.
The total protein concentration was measured with a Bradford assay (Bio-Rad, Richmond, CA) according to the manufacturer's instruction. Fifty milligrams of nuclear extract were separated by 10% sodium dodecyl sulfate/polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride (PVDF) membrane (EMD Merck Millipore, Darmstadt, Germany). The membranes were then blocked with 5% skim milk in Tris-Buffered Saline with 0.5% Tween20 (TBST) for 1 h at room temperature. The membranes were incubated overnight at 4°C with anti-HIF-1α mouse monoclonal (1:1,000; Novus Biologicals, CO; or 1:1,000; Abcam, Cambridge, UK). The next day, the blots were washed with TBST three times and then incubated for 1 h at room temperature with horseradish peroxidase-conjugated secondary antibody (anti-rabbit IgG antibody at a dilution of 1:10,000; Santa Cruz Biotechnology, Santa Cruz, CA). The membranes were then washed three times, and enhanced chemiluminescence (Merck Millipore) was used for detection with a luminescent image analyzer (ImageQuant LAS4000; GE Healthcare, Chicago, IL). After development, blots were reprobed with goat anti-Lamin B (1:3,000; Santa Cruz Biotechnology) to confirm that the samples contained equal amounts of protein. The band densities of the target proteins were then analyzed with the ImageJ software program (NIH). The relative expressions of target proteins were normalized with the total-LaminB expression.
Statistical analyses
All of the experiments were performed at least three times. Data are presented as mean ± standard deviations. An unpaired Student's t-test or Tukey–Kramer test after a one-way analysis of variance was used for the statistical analyses with the GraphPad Prism 7.0 software program (GraphPad Software, San Diego, CA). A statistical probability of P < 0.05 was considered significant (*<0.05, **<0.01).
Results
Sox1 as the marker to isolate ES-NSCs induced by the SFEB method
We first differentiated mouse ES cells with the SFEB method, which is an efficient culture system developed for the neural induction of ES cells [12]. After 5 days of differentiation in SFEB without additional exogenous developmental signals, we observed the messenger RNA (mRNA) upregulation of Sox1 and the neural stem cell marker, Nestin, and endoderm marker, FoxA2, as well as the downregulation of pluripotent marker, Nanog (Fig. 1A), suggesting that we had successfully induced the neural differentiation of ES cells.
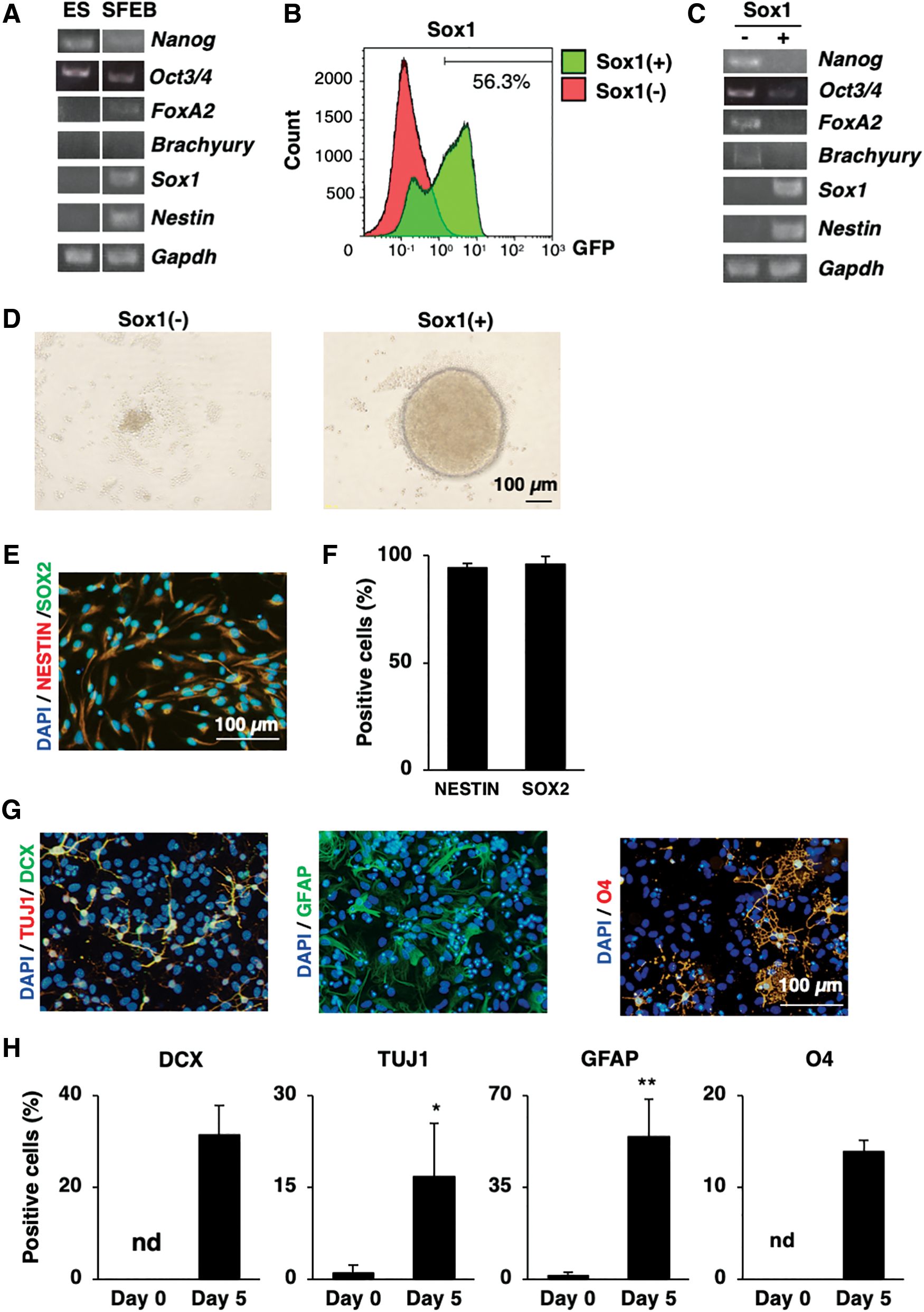
Sox1 as the marker to isolate neural ES-NSCs induced by the SFEB method.
The SFEB-derived population reportedly does not totally comprise neural cells [12]; in addition, the transcription factor Sox1 is identified as the earliest neuroectodermal cell marker [1], which also marks the primordial neural progenitor stage [6]. Therefore, we used a reporter cell line (Sox1-GFP ES cells) to isolate the committed neuroectodermal cells (Sox1-positive cells). Through this approach, we confirmed that 56.3% of cells inside the ES cell aggregates were Sox1-positive cells (Fig. 1B).
We then performed cell sorting between Sox1-GFP-positive and Sox1-GFP-negative fractions. Interestingly, while the GFP-negative faction expressed pluripotent markers, namely Nanog and Oct3/4, on the contrary, the GFP-positive cell fraction expressed the neuroectodermal cell markers, Sox1 and Nestin, without any expression of other germ layer cell markers, including Nanog, Oct3/4, FoxA2, and Brachyury (mesoderm), compared with those in the GFP-negative cell fraction (Fig. 1C). These data implied that neuroectodermal cells were specifically isolated from whole differentiating ES cells based on their Sox1 expression. Interestingly, when the isolated cell fractions were recultured in the NSC maintenance medium for 4 days, only Sox1+ cells, but not Sox1− cells, were able to generate the neurospheres (Fig. 1D), suggesting the role of Sox1 in initially maintaining NSCs in vitro.
NSCs arising from the neuroectoderm with self-renewability, are double-positive for Nestin and Sox2 markers, and display trineural differentiation abilities (neurons, astrocytes, and oligodendrocytes) [21,22]. We, therefore, next examined the characteristics of ES-NSCs isolated by the combination of the SFEB method and Sox1 marker. The NSC population was able to be maintained on adhesion culture during long-term culture (after seven passages), and 95% of cells were positive for Nestin and Sox2 antibodies (Fig. 1E, F). Next, the neural differentiation ability of NSCs was examined using differentiation medium with B27 supplementation for 5 days. The differentiated cells were positive for the immature neuron marker doublecortin (DCX) (24.1% ± 2.4%), the neuronal marker Tuj1 (13.4% ± 0.8%), the astrocytes marker glial fibrillary acidic protein (GFAP) (48.6% ± 7.9%), and the oligodendrocytes marker O4 (9.6% ± 2.5%) (Fig. 1G, H). Taken together, these data revealed that the ES-derived Sox1+ cells possessed NSC characteristics.
In vitro long-term adherent culture reduced the expression of neuronal markers and affected the fate of ES-NSCs in spontaneous differentiation
While NSCs undergo temporal specification during development [23,24], it is reported that prolonged culture may affect the biological characteristics of stem cells [13,25], including those of fetal brain-derived NSCs [26]. In the present study, we hypothesized that in in vitro culture conditions, NSCs show similar dynamical changes as what is observed in vivo. Therefore, we define the early and late-passaged ES-NSCs equivalent to the time of in vivo neural development of mouse (Supplementary Fig. S1) and examined how long-term culture affects the ES-NSC characteristics by comparing early and late-passaged ES-NSCs.
We first examined the effects of long-term culture on the expression of NSC markers in undifferentiated ES-NSCs. Immunostaining of both of early and late-passaged ES-NSCs revealed that both populations were positive for Nestin and Sox2, with no significant difference in the expression (Fig. 2A–C). Notably, late-passaged ES-NSCs showed a 4.9-fold decreased Sox1 mRNA expression and 8.4-fold decreased Sox1 protein expression compared with the early passaged ES-NSCs (Fig. 2C–E). In addition, it was recently reported that insulin signaling through insulin-like growth factor 2 mRNA-binding protein (igf2bp2; IMP2) mediates the neuron/glial change in embryonic brain-derived neural progenitor cells during prolonged culture [27]. Therefore, we checked the expression of IMP2 and IGF-2 in early and late-passaged ES-NSCs. As expected, we observed a 1.26-fold downregulation of IMP2 and 15.28-fold downregulation of IGF-2 expression in late-passaged ES-NSCs compared with early passaged ES-NSCs (Fig. 2F).
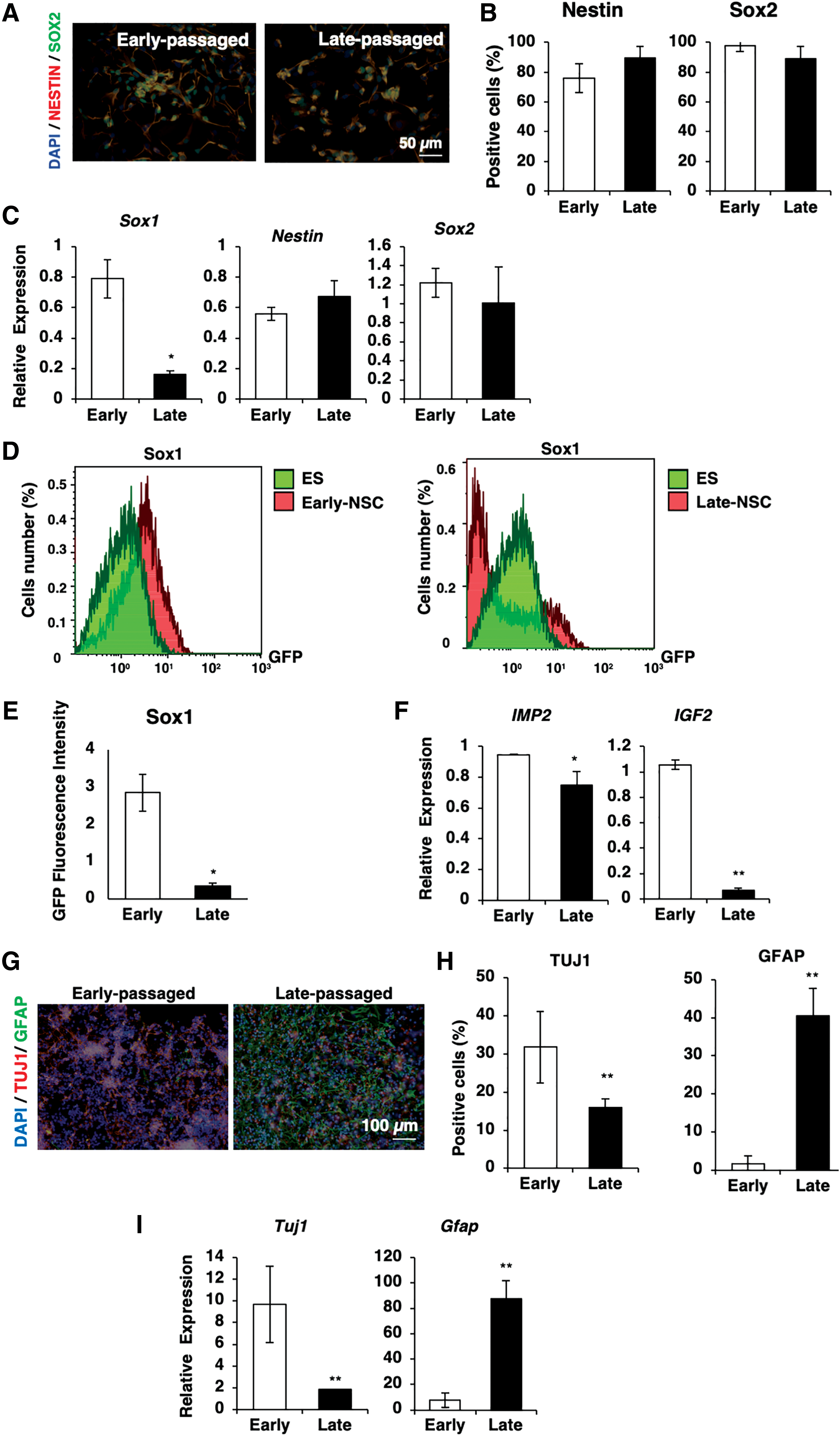
In vitro long-term adherent culture reduced the expression of neuronal marker and affected the fate of ES-NSCs in spontaneous differentiation.
Next, we examined the effects of prolonged in vitro cell culture on the spontaneous differentiation ability of ES-NSCs. Early passaged ESCs and late-passaged ES-NSCs were spontaneously differentiated without any addition of growth factors, such as bFGF or EGF, in the culture medium. Quantitative polymerase chain reaction (qPCR) and immunostaining revealed different differentiation profiles between early and late-passaged ES-NSCs after 5-day differentiation. Of note, the early passaged ES-NSCs tended to differentiate into neuronal lineages, whereas late-passaged ES-NSCs predominantly differentiated into glial lineages (Fig. 2G–I).
Taken together, these data showed that in vitro long-term culture affected the fate of ES-NSCs under spontaneously differentiated conditions. Early passaged ES-NSCs, which showed a high expression of Sox1 and Tuj1, gave rise to neuronal lineages, whereas late-passaged ES-NSCs with a decreased expression of Sox1 and Tuj1 predominantly gave rise to glial lineages. The changes in the differentiation profiles were similar to those observed in vivo, whereas during mid gestation, neural progenitors change from a neurogenic to gliogenic nature [17].
Hypoxia promotes the neuronal differentiation of neural committed cells without effects on the cell fate of ES-NSCs after differentiation
Hypoxia has been reported to prevail in the embryonic and fetal brain [15,28,29]. Therefore, we next investigated the effects of hypoxia on the neurogenesis of neural committed cells using SFEBq-induced ES spheroids.
First, we examined the effects of hypoxia treatment on the apical stemness of ES spheroids by analyzing the expression of N-cadherin, a marker of the apical side of neuroepithelium, and phospho-histone3 (pH3), a marker of actively dividing cells [12]. We found the reduced expression of N-cadherin and pH3 in hypoxia-treated spheroids, implying that the SFEBq-induced ES spheroids were unable to maintain their apical stemness under hypoxic conditions (Fig. 3A, B). We next examined the effects of hypoxia on the neurogenesis of SFEB-induced ES spheroids by analyzing the expression of DCX and Map2, which have been reported to mark immature neuron and neuronal cells, respectively [30]. As expected, we found that the expression of DCX was reduced while that of Map2 was enhanced in SFEB-induced ES spheroids under hypoxic conditions (Fig. 3C, D).
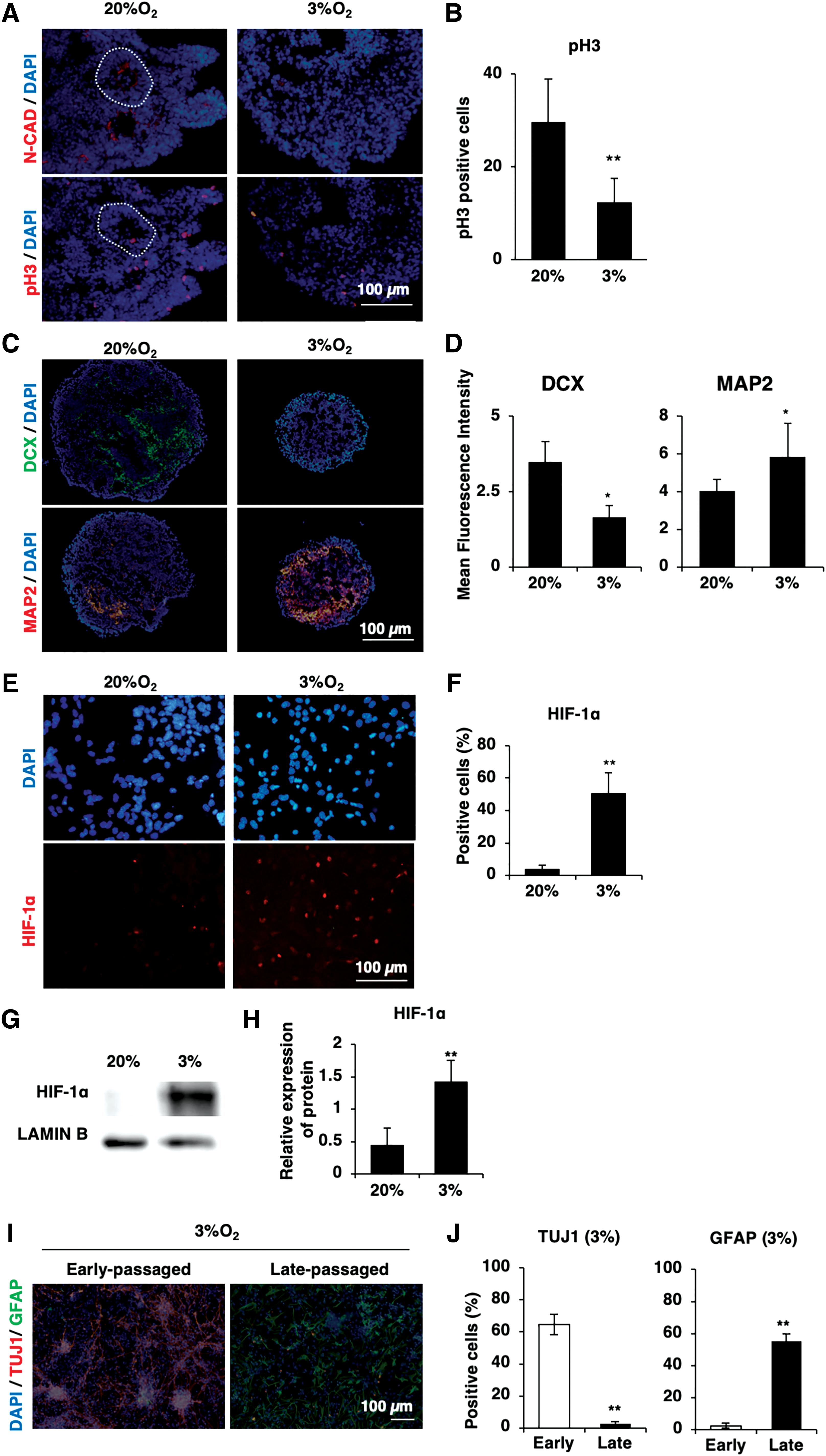
Hypoxia promotes the neuronal differentiation of neural committed cells without effects on the cell fate of ES-NSCs after differentiation.
To clarify the effects of hypoxia on ES-NSCs, we next examined the responses of ES-NSCs to hypoxic stimuli. First, we examined the expression of HIF-1α in early and late-passaged ES-NSCs. As expected, both early and late-passaged ES-NSCs demonstrated the upregulation of HIF-1α upon treatment with hypoxic stimuli (early passaged: Fig. 3E, F; late-passaged: Fig. 3G, H). Next, we examined the effects of hypoxia treatment on the spontaneous neural differentiation of early and late-passaged ES-NSC. We found that hypoxia treatment showed no effects on the fate of either early or late-passaged ES-NSCs; under hypoxic conditions, early passaged ES-NSCs differentiated to neuronal lineages, while late-passaged ES-NSCs differentiated to glial lineages (Fig. 3I, J).
Collectively, these results suggested that hypoxia accelerates the neuronal differentiation of neural committed cells in SFEBq-induced ES spheroids. In addition, hypoxia treatment showed no effects on the cell fate of ES-NSCs after spontaneous neural differentiation.
Hypoxia promotes glial differentiation of late-passaged ES-NSCs
To analyze whether or not the external stimuli might change the glial fate of late-passaged ES-NSCs, we examined the differentiation ability of late-passage ES-NSCs in the presence of B27 supplementation, which is reported to promote neuronal formation [31]. Interestingly, even in a neuronal supportive environment, a decrease in the Tuj1-positive cell number was still observed among late-passaged ES-NSCs, whereas the number of GFAP-positive cells was increased, suggesting that late-passaged ES-NSCs showed the characteristics of glial progenitors (Fig. 4A, B). To determine whether or not the predominantly glial commitment noted in late-passaged ES-NSCs was due to the possibility that neuronal cells undergo apoptosis, a TUNEL assay was performed with neuronal or glial markers in differentiated cells. No coexpression of TUNEL-positive cells with neuronal or glial markers was noted, thus eliminating the possibility of apoptosis of neuronal cells (Supplementary Fig. S2).
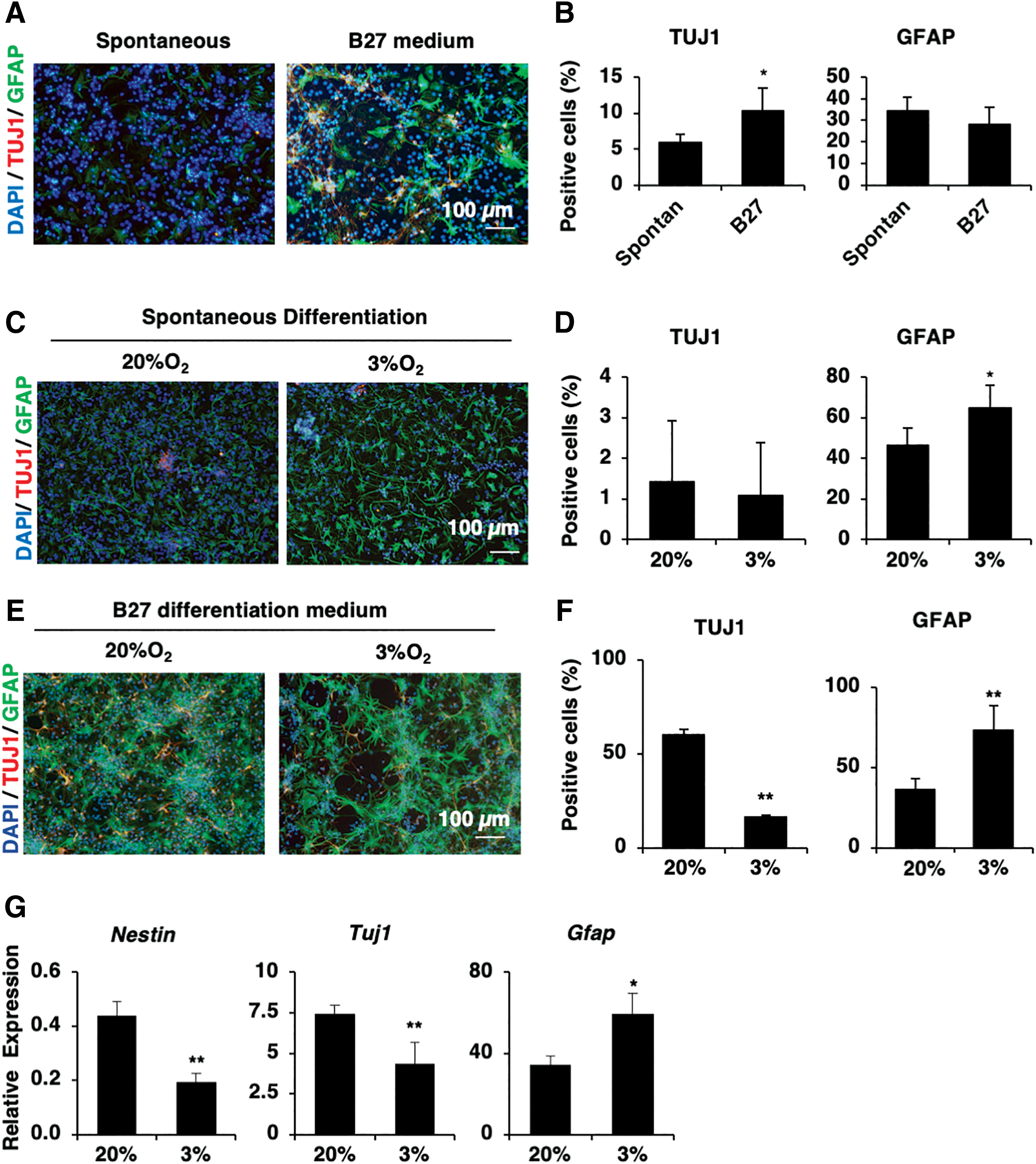
Hypoxia promotes glial differentiation of late-passaged ES-NSCs.
Hypoxic induction of HIF-1α reportedly promotes astrocyte differentiation in later stages of fetal brain development [18,32]. Therefore, we speculated that although hypoxia showed no effects on the ES-NSC fate after differentiation, hypoxia treatment would ultimately promote the astrocyte differentiation of late-passaged ES-NSCs. To assess this hypothesis, we compared the expression of neuronal and glial markers in spontaneously differentiated late-passaged ES-NSCs under normoxic and hypoxic conditions. Under normoxic conditions, the late-passaged ES-NSCs also predominantly formed glial lineages after spontaneous differentiation. Compared with normoxic conditions, hypoxic stimuli increased the glial marker expression (GFAP) by 1.4-fold in late-passaged ES-NSCs (Fig. 4C, D), suggesting a greater differentiation ability of late-passaged ES-NSCs under hypoxic conditions.
We then examined the effects of hypoxia treatment on the differentiation ability of late-passaged ES-NSCs with neuronal differentiation-favored supplementation using B27. Similar to the tendency observed under normoxic conditions in the presence of B27 supplement, late-passaged ES-NSCs predominantly differentiated into glial cells, as determined by the increased number of GFAP-positive cells (Fig. 4E, F). In addition, after 5 days of differentiation, we found the downregulation of the NSC marker Nestin in late-passaged ES-NSCs under hypoxic conditions compared with normoxic conditions (8-fold decrease; Fig. 4G), suggesting that hypoxia treatment induced neural differentiation of late-passaged ES-NSCs. Of note, under hypoxic conditions, late-passaged ES-NSCs showed downregulation of the neuron marker Tuj1, but the upregulation of the early glial gene GFAP compared with normoxic conditions (60-fold increase in GFAP expression; Fig. 4G), suggesting that hypoxic stimuli accelerated the glial cell commitment and differentiation in late-passaged ES-NSCs.
Taken together, these data indicated that hypoxic stimuli promote the glial cell differentiation of late-passaged ES-NSCs.
Hypoxia accelerates the neurogenesis of early passaged ES-NSCs
Next, we examined how hypoxia treatment affects the neuronal differentiation of early passaged ES-NSCs. Under hypoxic conditions, we found significant upregulation of the immature neuronal markers, DCX (8-fold increase) and Tuj1 (2-fold increase), and the postmitotic neuronal marker Map2 (2.4-fold increase) in early passaged ES-NSCs compared with those cultured under normoxic conditions (Fig. 5A, B). Since SFEB-derived cells are reported to be mainly composed of anterior forebrain cells that generate cortical neurons [20], we then examined the expression of the cortical layer neuron marker Ctip2 under hypoxic conditions. Ctip2+ cells were found to be 2.1-fold upregulated under hypoxic conditions, suggesting that some neuronal populations generated under hypoxic conditions resemble deep-layer cortical neurons (Fig. 5C, D).
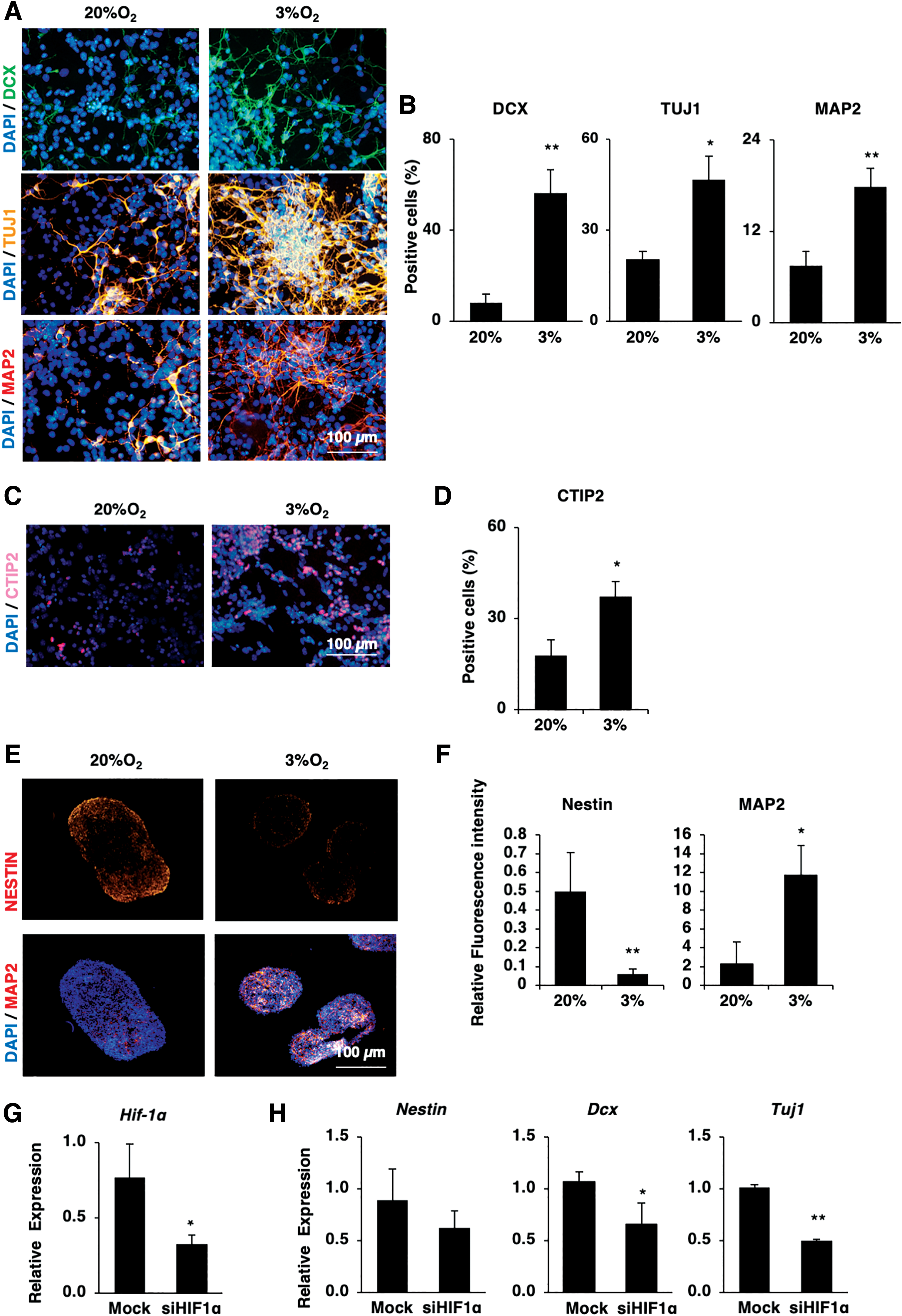
Hypoxia accelerates the neurogenesis of early passaged ES-NSCs.
In addition, we examined the effect of hypoxic conditions on neural tissue formation by performing three-dimensional (3D) culture of early passaged ES-NSCs. Similar to the results of monolayer culture, we observed the upregulation of the neuronal marker Map2 in spheres formed, cultured under hypoxic conditions (fluorescence intensity: 3.5-fold increased under hypoxic conditions compared with normoxic conditions, n = 6, P < 0.05; Fig. 5E, F). We also noted the obvious downregulation of the NSC marker Nestin (fluorescence intensity: 8.3-fold decrease, n = 6, P < 0.05; Fig. 5E, F), suggesting that hypoxic stimuli encourage the differentiation of early passaged ES-NSCs into neuronal lineages.
While HIF-1α promotes astrocyte differentiation in later stages of brain development, it is not clear how HIF-1α is involved in the early stages of brain development during neurogenesis. Therefore, to clarify the function of HIF-1α in the neuronal differentiation of early passaged ES-NSCs, its expression was diminished in those cells using siRNA. The reduced expression of HIF-1α expression was confirmed in the siHIFα-treated early passaged ES-NSCs compared with the control siRNA-treated cells (54.3% ± 12% reduction, n = 3, P < 0.05; Fig. 5G). Interestingly, we noted the impaired expression of neuronal genes in siHIFα-treated early passaged ES-NSCs compared with the control siRNA-treated cells (Dcx: 40% ± 19% reduction, and Tuj1: 50% ± 1% reduction, n = 3, P < 0.05; Fig. 5H), suggesting that HIF-1α is associated with the neuronal differentiation of early passaged ES-NSCs.
Taken together, these findings showed that prolonged culture affects the fate of ES-NSCs; early passaged ES-NSCs are neurogenic, whereas late-passaged NSCs are gliogenic. Of interest, hypoxia treatment accelerated the neural differentiation but showed no effects on the fate of ES-NSCs. In addition, it was confirmed that the hypoxic response through the upregulation of HIF-1α was closely involved during neurogenesis of ES-NSCs (Fig. 6).
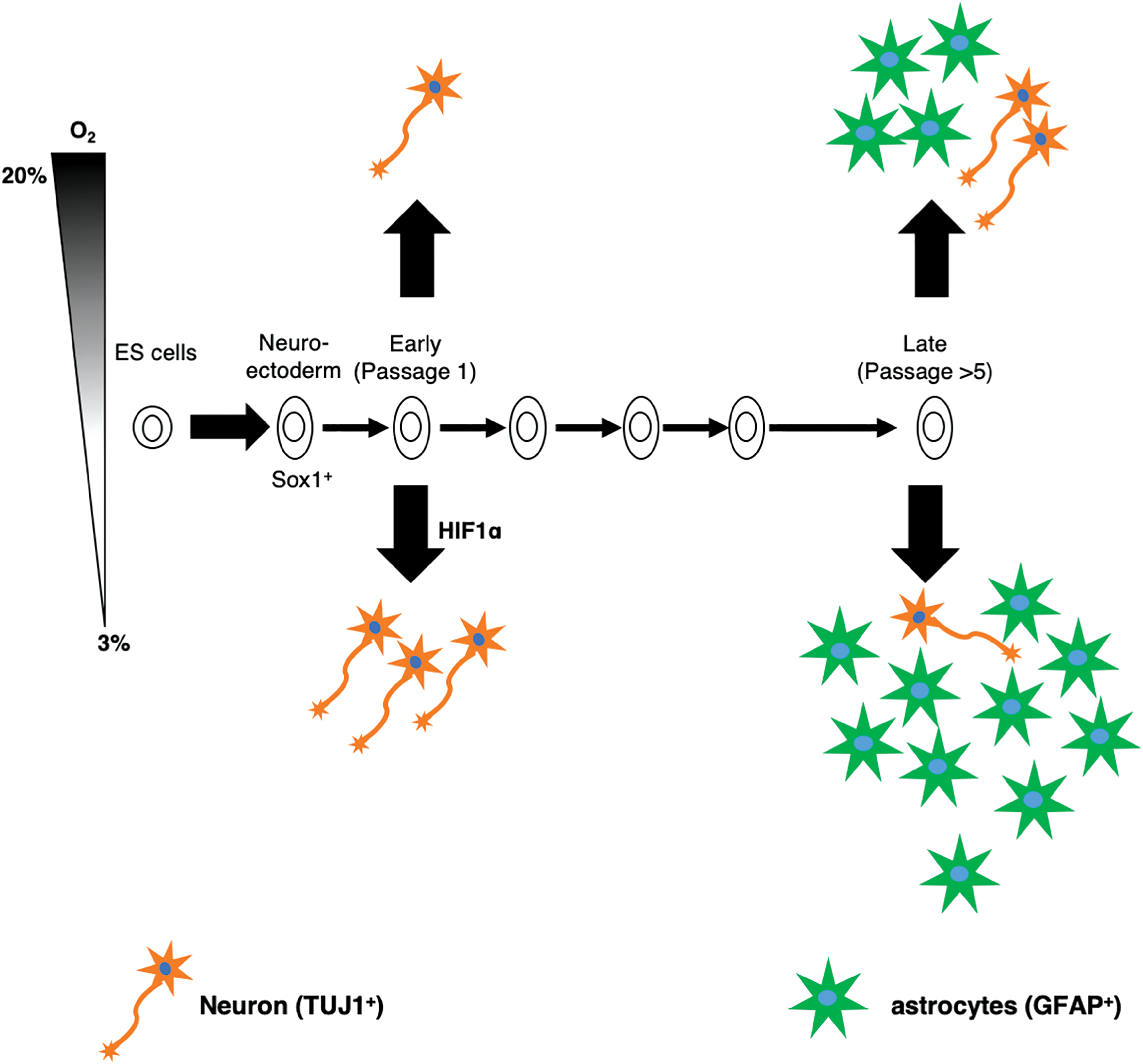
A schematic diagram summarizing this study. The feature temporal neural development of ES-NSCs in vitro. The ES-NSCs were isolated from the SFEB-induced ES aggregates, which were positive for Sox1. During in vitro culture, ES-NSCs showed the feature temporal neural specification, which was similar to that observed in vivo. Early passaged ES-NSCs tended to differentiate into neuronal lineages, whereas late-passaged ES-NSCs predominantly differentiated into glial lineages. During neural differentiation, hypoxia promotes both the neuronal differentiation in early passaged ES-NSCs and glial differentiation in late-passaged ES-NSC through the induction of HIF-1α. Color images are available online.
Discussion
In the present study, we effectively isolated the neuroectoderm cell population derived from ES cell aggregates by using a simple method combined of SFEB culture and cell sorting with Sox1 as a marker. The Sox1-positive cell population showed NSC characteristics and were analyzed for the effects of prolonged culture under hypoxic conditions. The results clearly demonstrated that the prolonged culture affected the cell fate, whereas low oxygen tension promoted the differentiation level of the SFEB-derived ES-NSCs. Early passaged ES-NSCs possessed neurogenic characteristics, whereas late-passaged ES-NSCs predominantly led to gliogenic lineages.
One effective method for inducing the neural differentiation of ES cells is the SFEB method [12]; however, SFEB-derived cells do not wholly consist of neural populations. Many recent advances have been reported to improve the SFEB culture, including the addition of developmental signals such as Wnt and Nodal antagonists at a specific time to enhance its neural differentiation. However, the complexity of the protocols and cost effectiveness are the problems that need to be considered. To simplify the protocol, we took advantage of cell-sorting technology by using Sox1 as a marker. Using this method, we previously reported the molecular mechanisms underlying the selective neural differentiation by SFEB methods related to the direct activation of Sox1 expression [16]. To rule out the possibilities of other germ layer cells in neural differentiation, we examined whether or not Sox1 is an appropriate marker for isolating the neuroectoderm, which are also primordial neural progenitors. As expected, the isolated ES-derived Sox1+ cells developed into an NSC population, with 95% of cells stained double positive for the NSC markers, Nestin and Sox2, and showing a differentiation ability toward trineural lineages. Further functional studies of isolated ES-NSCs as well as the comparison with other recent protocols are necessary to validate our protocol for a widened application.
Numerous reports have identified the dynamical changes of neural stem cells during the in vivo embryonic neural development. It is reported that in the embryonic neural development, E12.5 still retains the neurogenic potential and E17.5 is the time of neuron/glial switch [18]. After neuron/glial switch, NSCs start to differentiate into glial cells and this gliogenesis would last until early neonate period when all embryonic NSCs will become gliogenic NSC. Additionally, several reports have suggested the effects of the stage of NSC [33] or passage number [11] on the characteristics of NSCs in the in vitro culture system. However, whether the in vitro changes of NSCs similar to what naturally happens in in vivo is still obscured. In the present study, we examined the dynamical changes of NSCs during in vitro culture by the comparison of ES-NSCs at early, and late-passaged numbers, which were defined to be equivalent with the in vivo developmental stages. In specific, the mouse ES cells were isolated from E3.5 followed by the 5-day SFEB induction and 4-day in vitro expansion, which was considered as early passage (passage 1-P1) and to be equivalent with E12.5, when the neurogenic ability is retained in vivo. Continuously, the late passage was defined at P5, which was considered to be equivalent with the early neonate period (neonate day 2), when all the NSCs were supposed to become gliogenic NSCs in vivo. Interestingly, the dynamic changes of ES-NSCs in vitro were similar to what occurs in vivo, in that the early stage NSCs have neurogenic ability, whereas the late-stage NSCs are gliogenic.
At the start of brain development, neurogenesis occurs to generate new neurons [34]. Subsequently, glial precursors are generated, followed by the rise of astrocytes, as these cells support neurons [34]. Finally, neural development is finalized with the formation of oligodendrocytes upon birth [34]. Of interest, by isolating neuroectoderm cells, we uncovered direct evidence that the developmental timer is intrinsically encoded within the neuroectoderm itself and might be involved in the downregulation of Sox1 expression during cell passaging. Although the underlying mechanisms regarding how Sox1 expression gradually decreases during neural development have not been clarified yet, there is evidence showing the relationship between the downregulation of Sox1 and the neuron/glial switch during neural development [6,35]. Sox1-expressing cells reportedly represent an activated neural progenitor population that gives rise to most newly born neurons [36], probably due to the promotion of neurogenic factors [35]. With age, the fraction of Sox1-expressing cells decreases in correlation with the decrease in neurogenesis [36].
Previous studies have suggested that the neuron/glial switch is regulated by IMP2, the expression of which decreases over the course of development [27]. IMP2 is known to activate IGF in glioblastoma [37]. Of note, it was recently reported that IGF promotes the expression of Sox1 during neural differentiation in the SFEBq model [38]. In the present study, along with the downregulation of Sox1, we found the decreased expressions of IMP2 and IGF2 in late-passaged ES-NSCs compared with early passaged ES-NSCs. These data suggest that the Sox1 and IMP2 expressions are signals of the neuron/glial switch in ES-NSCs.
It has been proposed that not only neurons but also the properties of glial cells contribute to unique brain cognition [39]. Glial cells include oligodendrocytes, which myelinate the CNS axons; astrocytes, the star-shaped cells that interact with neurons and synapses and control thousands of processes; and the resident immune cells, microglia [39]. In particular, astrocytes play key roles that influence the brain performance, such as maintaining brain homeostasis, inducing newly formed synapses and responding to neurotransmitters [39]. During astrogliogenesis, astrocytes change shape, with immature astrocytes showing a bipolar shape and mature astrocytes showing the typical star shape [40]. Thus far, the different functions of immature and mature astrocytes have been unclear. While previous studies have suggested that immature astrocytes contribute to the brain structure during development, mature astrocytes play the functional role of neuronal support [41]. Interestingly, we noted different shapes of glial cells derived from mid-passaged and late-passaged ES-NSCs. Mid-passaged ES-NSCs gave rise to bipolar-shaped astrocytes, whereas late-passaged ES-NSCs resulted in the formation of star-shaped astrocytes (Supplementary Fig. S3C, D). Our results suggested that the formation of immature or mature astrocytes after the differentiation of ES-NSCs is regulated by the in vitro passage, thus proposing a new approach to controlling the cell fate in astrogliogenesis.
In addition, our study highlighted the different signal pathways that regulate the mid- and late-passaged ES-NSC populations. Glial differentiation is regulated by numerous pathways depending on the secretion of cardiotrophin-1 (CT-1) from the surrounding neurons, through the LIF/JAK/STAT pathway and bone morphogenetic proteins (BMP) signaling pathway, which result in different cell shapes. Specifically, bipolar-shaped glial cells develop through the LIF signal pathway, whereas stellate-shaped glial cells develop through the BMP signal pathway [40]. Therefore, we hypothesized that the LIF signals were activated during mid-passaged ES-NSC spontaneous differentiation, thus resulting in the development of bipolar-shaped astrocytes. In contrast, the direction of late-passaged ES-NSC differentiation has changed to glial cell lineages, resulting in the reduction in the neuronal cell number and in the secretion of CT-1. Subsequently, the endogenous BMP signals become dominant, and more stellate-shaped glial cells are differentiated from late-passaged ES-NSCs (Supplementary Fig. S3).
Hypoxia is a natural, beneficial microenvironment for neural development [15,28]. We previously reported that hypoxic induction, which occurs spontaneously inside differentiating ES cell aggregates, acts as a positive regulator of neural commitment by promoting the transition of ES cell differentiation from the epiblast into the neuroectoderm state [16]. In terms of neurogenesis, previous reports have shown that hypoxia promotes neuronal differentiation [17,42], especially the midbrain-derived dopaminergic neuronal differentiation [43], through an as-yet-unknown mechanism. Consistent with these findings, we detected the upregulation of neuronal markers in early passaged ES-NSCs differentiated under hypoxic conditions, both in monolayer and 3D culture. Of note, in mid- and late-passaged ES-NSC differentiated under hypoxic conditions, we found only the upregulation of glial markers, not neuronal markers, suggesting that hypoxia may enhance the neural differentiation efficiency without affecting the direction of ES-NSC differentiation. Instead, the direction of ES-NSC differentiation might be determined more by endogenous mechanisms than the microenvironment of ES-NSCs, and these endogenous mechanisms might be persevered during cell passages in long-term culture in vitro.
In glial differentiation, HIF-1α has been reported to cause demethylation of glial genes, thereby enhancing the generation of glial cells [18]. In addition, the silencing or overexpression of HIF-1α has been reported to impair or enhance glial differentiation, respectively [32]. In this context, we found that hypoxia, through the induction of HIF-1α, upregulated the expression of astrocytic genes but not that of neuronal genes in mid- and late-passaged ES-NSCs. Furthermore, a previous study showed that knockdown of HIF-1α at the early stage of gestation is lethal to embryos due to a defect in the neural development of mouse embryos [44]. However, direct evidence of the role of HIF-1α in the neuronal differentiation of ES-derived NSCs has not yet been reported. In the present study, we showed for the first time that silencing HIF-1α in early passaged ES-NSCs led to impaired neuronal differentiation.
To clarify the underlying mechanisms, we examined several related signals such as Notch signals [27] and Wnt signals [34,35,47,48]. Unexpectedly, no significant difference between control- and HIF-1α -siRNA-cells on the expression of Hes1, a target of Notch signaling, was observed (Supplementary Fig. S4A). Since Notch signaling is known to express in oscillatory manner and what we observed might only reflect a single time point, it requires the further study of time course to conclude the role of notch signaling in neuronal differentiation under hypoxic conditions. Additionally, related to Wnt signaling, only the slight elevation of Tcf1/7 expression (1.25-fold increased) was observed under the impaired HIF-1α condition (Supplementary Fig. S4B). Our data suggested that instead of promoting neuronal differentiation, wnt signals might be involved in inhibition of neuronal differentiation in early passaged ES-NSCs. In addition, the role of wnt signals is dynamic; Kim et al. reported that depending on the phase of NSCs in the neural development, the activation of wnt signal is known to promote or inhibit neuronal differentiation [45].
In the present study, we took the advantage of mouse samples with the short gestation (18 days) to understand the overview of how prolonged culture and oxygen tension affect the whole process. We suggested that the in vitro dynamic changes of ES-NSCs happened similar to those in vivo, and prolonged culture affected the cell fate, whereas low oxygen tension only promoted the cell differentiation level. Although using mouse samples benefits the study related to the time/cost, the mouse brain is simple compared with that of human brain. Therefore, to bridge the gap between mouse and human, it is worth for further study to confirm the results using human samples such as ES or iPSC.
In summary, we established a simple method to isolate ES-NSCs by the combination of SFEB induction and cell sorting using Sox1 as a marker. By the comparison of ES-NSCs at different passaged numbers, which were equivalent to the in vivo developmental stage, we found that ES-NSCs recapitulate temporal neural development in which early stage ES-NSCs show characteristics of the neurogenic period, and late-stage ES-NSCs are predominantly gliogenic. Importantly, hypoxia treatment promotes neuronal differentiation in early passaged ES-NSCs, but then switches to promote glial differentiation in late-passaged ES-NSCs. Additionally, knockdown of HIF-1α in early passaged ES-NSCs led to the impairment of neuronal differentiation, suggesting the crucial role of HIF-1α during the neural development process under hypoxic conditions. Our study suggested that both oxygen tension and the passage number of NSCs should be carefully considered when developing neural cells derived from pluripotent stem cells for use in clinical studies in the future.
Footnotes
Author Disclosure Statement
The authors declare no conflicts of interest in association with the present study.
Funding Information
This work was supported by Grant-in-Aid for Scientific Research from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (MEXT) to O.O. and M.T.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.