
Abstract
Traumatic brain injury (TBI) leads to delayed secondary injury events consisting of cellular and molecular cascades that exacerbate the initial injury. Human umbilical cord perivascular cells (HUCPVCs) secrete neurotrophic and prosurvival factors. In this study, we examined the effects of HUCPVC in sympathetic axon and cortical axon survival models and sought to determine whether HUCPVC provide axonal survival cues. We then examined the effects of the HUCPVC in an in vivo fluid percussion injury model of TBI. Our data indicate that HUCPVCs express neurotrophic and neural survival factors. They also express and secrete relevant growth and survival proteins when cultured alone, or in the presence of injured axons. Coculture experiments indicate that HUCPVCs interact preferentially with axons when cocultured with sympathetic neurons and reduce axonal degeneration. Nerve growth factor withdrawal in axonal compartments resulted in 66 ± 3% axon degeneration, whereas HUCPVC coculture rescued axon degeneration to 35 ± 3%. Inhibition of Akt (LY294002) resulted in a significant increase in degeneration compared with HUCPVC cocultures (48 ± 7% degeneration). Under normoxic conditions, control cultures showed 39 ± 5% degeneration. Oxygen glucose deprivation (OGD) resulted in 58 ± 3% degeneration and OGD HUCPVC cocultures reduced degeneration to 34 ± 5% (p < 0.05). In an in vivo model of TBI, immunohistochemical analysis of NF200 showed improved axon morphology in HUCPVC-treated animals compared with injured animals. These data presented in this study indicate an important role for perivascular cells in protecting axons from injury and a potential cell-based therapy to treat secondary injury after TBI.
Introduction
Primary Traumatic Brain Injury (TBI) occurs when a sufficient physical force is applied to the head to cause torsion and shearing of brain tissues. An ongoing secondary injury ensues which involves cellular and molecular events that result in impaired axonal integrity and function, and coma in severe cases. The prolonged time frame of the secondary injury allows for a window of opportunity to intervene therapeutically. There are currently no therapies to directly treat the underlying pathophysiology of TBI. Rather, the symptoms are treated individually and as they arise, resulting in a large financial burden on the health care system [1].
In TBI, autologous bone marrow mesenchymal stromal cells (BMSCs) are currently in early clinical trials [2] and have successfully been used to treat other diseases and are being investigated for their use in neurodegenerative diseases [3,4]. Umbilical cord-derived mesenchymal stromal cell (UC-MSC) has been investigated as a potential candidate in regenerative medicine. A subpopulation of Wharton's Jelly cells from the perivascular region of these blood vessels has been isolated and termed human umbilical cord perivascular cells (HUCPVCs) [5]. We and others have extensively characterized HUCPVCs in culture. HUCPVCs express MSC markers (CD44, CD105, CD146, CD73, CD34) and negative for hematopoietic markers, CD31 and CD45 (macrophage), when propagated in culture to maintain expression of vimentin, alpha smooth muscle actin, and pericyte markers (3G5, NG2, PDGFRβ). Investigation of the gene profile of HUCPVCs reveals their potential to secrete neurotrophic [neurotrophin-3 (NT3), nerve growth factor (NGF), and brain-derived neurotrophic factor (BDNF)] and angiogenic factors [bone morphogeneic protein (BMP1), BMP4, fibroblast growth factor (FGF1), and FGF2] [6 –9]. UC-MSC secretes factors that are conducive to survival and differentiation of immature neurons, indicating their potential to stimulate neurogenesis in vivo [10]. They also contain factors that protect against excitotoxicity and apoptosis to promote survival [9,11,12]. These data taken together point to UC-MSC and HUCPVCs being a promising candidate to study axon breakdown following injury and a potential cell therapy strategy.
The vascular system and the nervous system are mutually dependent during development [13,14]. Blood vessels secrete NT3 to guide developing neurons to their targets, target sites secrete BDNF and NGF that are necessary for central nervous system neurons [15]. Collectively, these neurotrophins are involved in survival, growth, and proliferation of neurons through both PI3K/Akt and MAPK/ERK pathways depending on the neurons and the stage of development [16]. NT3, when bound to its receptor, TrkA, activates the Akt pathway to stabilize actin in the cytoskeleton [17 –19]. In the absence of NGF, NT3 can effectively support neuron outgrowth through the TrkA receptor [15]. Although there is an overlap as well as possible crosstalk between these two pathways to promote survival, Akt activation in particular has been shown to promote survival, whereas ERK activation is generally thought to be responsible for differentiation/outgrowth [20].
Neurofilaments made up of light and medium subunits provide rigidity and stabilization to axons [21 –23]. Secondary axotomy that results from traumatic axonal injury can result in compaction or abnormalities of the neurofilamental structure and can lead to impaired transport causing abnormalities and death in more susceptible axons [24 –28]. As the main communication hub of the neuron, a dysfunctional or severed axon disrupts cell to cell synaptic signaling. Our laboratory has previously shown that the heavy neurofilament is overexpressed after injury in cerebellar neurons [29].
Given the role of vascular cells in guiding axon development and the potential of HUCPVCs to protect axons, we hypothesized that HUCPVCs can prevent axonal degeneration in cortical and sympathetic neurons by providing survival and growth cues. We further hypothesized that HUCPVCs could protect axons in vivo from degeneration and tested our hypothesis in an in vivo model of TBI.
Materials and Methods
All animal studies were approved by the Animal Care Committee at the University Health Network or St. Michael's Hospital and were performed according to Canadian Council on Animal Care Guidelines. All animals were housed in groups in a temperature- and light-controlled (12-h on/12-h off) environment and permitted access to food and water ad libitum throughout the study.
Independent Research Ethics Board approval was obtained for the collection of human umbilical cords (REB #28889, University of Toronto, Canada). Full-term newborn cords were collected from c-section births through a third party (Life Line Stem Cell, New Haven, IN). Written informed consent was obtained for each sample collection.
Cell cultures
HUCPVC cultures
HUCPVCs were freshly isolated as previously described [5,6]. BMSC (Lonza, Walkersville, MD) and fibroblasts from newborn foreskin (ATCC, Manassas, VA) were purchased commercially. Frozen stocks were thawed and plated in minimum essential medium eagle-alpha modification (αMEM) (Life Technologies, Burlington, Ontario, Canada) containing 10% serum and 1% penicillin–streptomycin and allowed to reach 70%–90% confluency before passaging. Cells were harvested by detaching them with TrypLE (Life Technologies Mississauga, Ontario, Canada), collected by centrifugation at 300g for 5 min, and plated at 2,000 cells/cm2.
Sympathetic neuron cultures
Sympathetic neuron cultures were established in Campenot chambers to grow axons in a different environment from the cell body. Sympathetic neurons were isolated from P1 to P3 Sprague–Dawley rat pups as previously described [30]. Briefly, superior cervical ganglia were removed and placed in ice cold UItraCulture (UC; Lonza) supplemented with
Cortical neuron cultures
To evaluate the effects of HUCPVCs in a system that was more relevant to central nervous system neurons, we used cortical neurons isolated from E16 to E17 Sprague–Dawley rat embryos. Briefly, E16 pregnant rats were deeply anesthetized to remove the embryos, and mothers were sacrificed immediately following embryo retrieval. Heads were dissected and placed in ice-cold HBSS. Under a dissecting microscope, the skulls were peeled back and meninges removed. The cortices were dissected and placed in ice-cold HBSS. The cortices were minced and digested with a papain solution for 30 min at 37°C. The cells were centrifuged at 1,000 rpm for 2 min and resuspended in neurobasal plating media. Cells were established for 6 days. To injure the cortical cultures, the cells were washed three times with and placed in a hypoxic chamber for 90 min. After injury, treatment consisted of either adding 7,000 term HUCPVCs, or the conditioned media to the cultures. After 2 days, cultures were fixed and immunostained for βIII-tubulin to quantify axon degeneration, as described below.
ERK and Akt inhibition
PD98059, an inhibitor of ERK (Enzo Life Sciences, Burlington, Canada), and LY294002, an inhibitor of Akt, (Enzo Life Sciences) were used to examine potential contributing mechanisms of HUCPVC activity on axons. The inhibitors were each suspended in 1 mL of dimethyl sulfoxide (DMSO; Sigma-Aldrich) to make stock solutions of 1 mM. Following the 90-min NGF withdrawal injury, 50 μM of either inhibitor was added along with HUCPVCs to the axon compartment of Campenot chambers. Twenty-four hours later, another 50 μM was added to the axon compartment. Cultures were fixed for analysis 2 days after injury.
Immunocytochemistry and cellular labeling
At 10 DIV, sympathetic neuron cultures were fixed and immunolabeled as previously described [32,33]. Briefly, cells were fixed in 4% paraformaldehyde (PFA), permeabilized in 0.2% Igepal (Sigma-Aldrich), and blocked for 1 h in 0.5% bovine serum albumin, and 6% normal goat serum in phosphate-buffered saline (PBS). Primary antibodies used were mouse anti-βIII-tubulin (New England Biolabs, Boston, MA), rabbit anti-βIII-tubulin (Abcam, Cambridge, MA), and were incubated for 48 h at 4°C at a dilution of 1:500 in a blocking buffer. Secondary incubation was performed with Alexa 555 goat anti-rabbit or anti-mouse (1:500; Cell Signaling, Burlington, Ontario, Canada) for 1 h at room temperature. Hoechst nuclear counterstaining was used at a dilution of 1:2,000 for 3 min at room temperature. Negative controls' immunostaining was performed simultaneously with the omission of primary antibodies. In initial experiments HUCPVCs were prelabeled with CellTrackerTM Green CMFDA dye (Life Technologies) to visualize interactions with neurons in live cultures. Lyophilized powder was dissolved in DMSO to yield a 10 mM stock concentration. A total of 100,000 cells were resuspended in a 1:500 dilution of CellTracker in serum-free basal media and incubated at 37°C for 45 min. The cells were centrifuged and resuspended in basal media to wash away excess dye and resuspended in 200 μL of Ultraculture media before coculturing. HUCPVCs were prelabeled using PKH26-red (Sigma-Aldrich) according to the manufacturer's instructions.
RNA isolation, reverse transcription-polymerase chain reaction, and qPCR for gene array
RNA samples were prepared using the RNAeasy Kit (Qiagen) according to the manufacturer's instructions, followed by DNase treatment to eliminate traces of genomic DNA. RNA was reverse transcribed into cDNA using the RT2 First-Strand Kit (Qiagen) according to the manufacturer's instructions. For quantitative polymerase chain reaction (qPCR), commercial Human RT2 ProfilerTM PCR Arrays (PAHS-041Z; SABiosciences) were used. The experiment was performed according to the manufacturer's protocol on the Rotor-gene 6000 (Corbett) using 5 ng total cDNA per reaction. Data normalization was performed with four out of the five endogenous control genes present on the PCR array (ACTB, B2M, GAPDH, HPRT1, and RPLPO). Each replicate cycle threshold (Ct) was normalized to the average Ct of four endogenous controls on a per plate basis. All assays were done in triplicate for three independent HUCPVC lines at passage 4. The cutoff for Ct values was set at 28, and genes with Ct ≥28 were considered not detected.
Conditioned media and enzyme-linked immunosorbent assay
HUCPVC, BMSC, or fibroblast was plated in serum-containing conditions and allowed to reach seventy percent confluency. Media were aspirated and cells washed twice with PBS (without Ca2+ or Mg2+) (Sigma-Aldrich) and replaced with serum-free basal media. For UC-conditioned media, cells were expanded to 70% confluency in serum containing αMEM and then replaced with UC. Cells remained in this media for 72 h before collection, the media were centrifuged at 3000 rpm for 10 min and filtered through a polyvinylidene fluoride 0.2 μm syringe-driven filter (EMD Millipore, Etobicoke, Ontario, Canada), aliquoted and stored at −80°C until needed. For conditioned media from chambers containing neurons cocultured with either HUCPVC; media were collected before fixing or lysing cells, centrifuged at 3000 rpm for 10 min, aliquoted, and stored in −80°C until needed. Aliquots of conditioned media were removed and thawed to room temperature immediately before loading on enzyme-linked immunosorbent assay (ELISA) plates (RayBiotech, Norcross, GA).
Fluid percussion injury
Male Sprague–Dawley rats (200–300 g) were anesthetized with 2% isoflurane delivered in laboratory-grade air. A thermal heating blanket at 37°C was placed under the rats to maintain normothermia. A burr hole (∼2–3 mm diameter) was drilled in the right lateral hemisphere midway between the bregma and lambda sutures. A polyethylene-modified Luer Lock was fixed to the opening in the rodent's skull with cyanoacrylate and dental acrylic. The fitting was filled with 0.9% isotonic saline and attached to tubing that was connected to the fluid percussion injury (FPI) device. The rats were then subjected to a 2.0 atmosphere extradural impact. The craniotomy was sealed with bone wax and the skin sutured before recovery. Sham animals underwent the craniotomy but not the FPI.
HUCPVC infusion
FPI animals were anesthetized 1.5 h postinjury with isoflurane. Previously isolated, expanded, characterized, and cryopreserved stocks of HUCPVCs were thawed and passaged once before infusion. Seventy-percent confluent plates were washed once with PBS and adherent cells were treated with TrypLE to dissociate them. The cell suspension was centrifuged to harvest the cells. The cell pellet was resuspended in HBSS such that the concentration was 1.5 million cells per 0.5 mL. Animals were infused with 0.5 mL of cell suspension through tail vein injection and allowed to recover before returning them to the animal housing facility.
Immunohistochemistry
At 24 and 48 h postinjury animals were sacrificed and transcardially perfused with saline followed by 4% PFA. Brains were removed, cryopreserved in 30% sucrose PBS solution, and embedded in optimal cutting temperature medium (OCT) before 20 μm coronal sectioning on a cryostat. Immunofluorescence labeling of the heavy neurofilament (NF200) was used to qualitatively assess axonal length and morphology. Briefly, sections were blocked with 10% normal goat serum (NGS) for 1 h at room temperature, a rabbit anti-NF200 antibody diluted to 1:200 was applied and incubated overnight at 4°C. Alexa 488 goat anti-rabbit secondary antibody was applied to the sections and incubated for an hour at room temperature. Images were imaged on a Nikon 90i upright fluorescent microscope.
RNA isolation, reverse transcription-polymerase chain reaction, and qPCR for in vivo tissue
Animals were decapitated under deep anesthesia, the brain excised, and placed in iced artificial cerebral spinal fluid that was continuously bubbled with laboratory-grade air (5% CO2, 20% O2). The pia matter was removed and a 1 cm2 tissue area of that injury site as well as penumbra were isolated; the cortex was isolated and were snap frozen in liquid nitrogen. RNA isolation was performed according to the manufacturer's instructions (Macherey Nagel, Ontario, Canada). An NT3 probe (
Image analysis and statistical methods
To quantify in vitro axonal degeneration, the distal ends of the axons in each track were imaged using an EVOS®FL (Life Technologies) fluorescent microscope. Using ImageJ, ∼100 individual axons per experiment, per condition with at least three microtubule breaks, or two blebs, at the leading edge were counted by a blinded researcher [32,34]. Intact axons were also counted, and the degeneration was expressed as a percentage of the total axons counted. Post-hoc analysis of pair-wise comparisons was performed using the Holm–Sidak method.
Results
HUCPVCs interact with axons in sympathetic neuron cocultures
We cultured HUCPVCs with mass cultures of sympathetic cervical ganglia (SCG) neurons to determine whether HUCPVCs interact with neuronal cell bodies and/or their axons. HUCPVCs were found to interact with the axons of SCG neurons in mass cultures at 3 DIV, but not with the cell bodies (Fig. 1A). BMSCs were also found to interact with the axons but not with the cell bodies (data not shown). To determine whether HUCPVCs interact with axons in a compartmented environment we grew sympathetic neurons in Campenot chambers, which allowed isolation of axons from cell bodies (Fig. 1B).
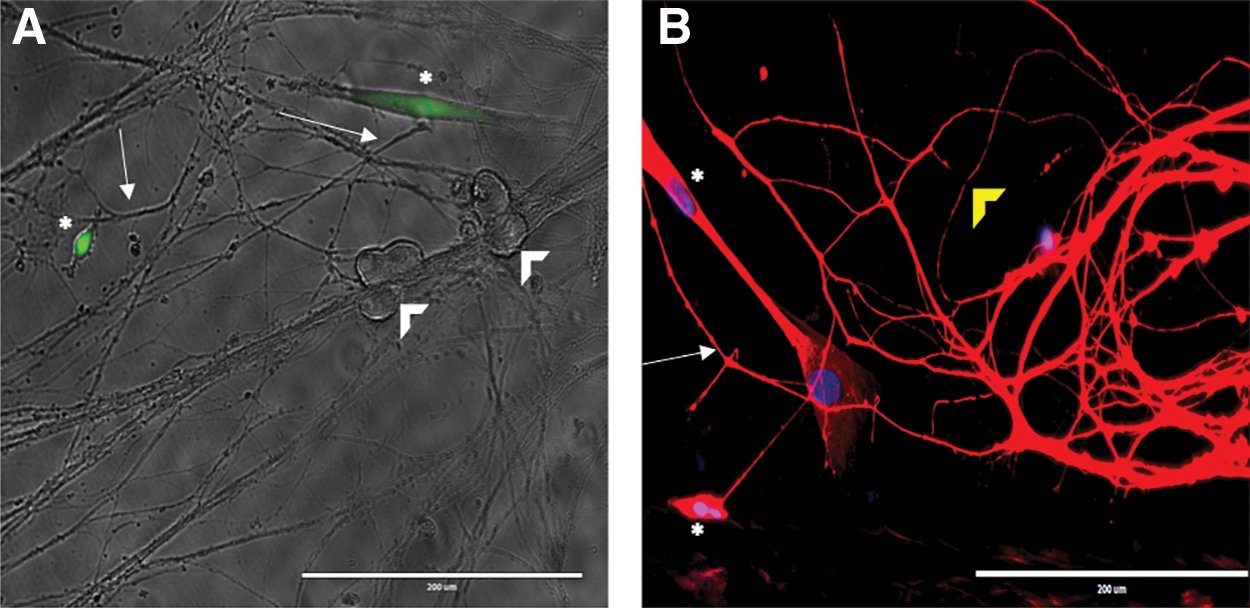
HUCPVCs interact with axons in sympathetic neuron cocultures.
HUCPVCs express genes encoding neurotrophins and neural growth/development factors
To determine whether HUCPVCs express genes encoding neurotrophic factors or other neuronal prosurvival cues, we examined the expression profiles of growth factor genes using a commercially available qPCR-based array. We analyzed the gene expression profiles of three independent HUCPVC lines expanded to passage 5–6 in normoxic conditions. Eighty-four growth factor-encoding transcripts were profiled, along with five housekeeping genes. Sixty-nine growth factor transcripts were expressed in the three HUCPVC lines (Table 1). Seventeen of 30 growth factors identified in the HUCPVC lines were either neurotrophins, neural growth or neural development factors, including glial cell-derived neurotrophic factor (GDNF) and midkine. The three neurotrophins identified were BDNF, NGF, and NT3. NT3 and BDNF had strong positive expression in HUCPVC lines (Ct = 25.97 and Ct = 26.12, respectively) and NGF had a Ct value of 27.66 in the HUCPVC lines. Transcripts encoding several key angiogenic factors, and inflammatory cytokines were also expressed, such as vascular endothelial growth factor (VEGF), leukemia inhibitory factor (LIF), and BMP1/4.
Polymerase Chain Reaction Gene Array Analysis of Growth Factors Expressed by Human Umbilical Cord Perivascular Cells
Genes are highlighted in bold.
Seventeen neural growth factors were identified in HUCPVCs, including neurotrophins, angiogenic factors, and survival factors (n = 3 lines).
SD, standard deviation; HUCPVC, human umbilical cord perivascular cell; Ct, cycle threshold.
HUCPVC rescue axon degeneration in an in vitro sympathetic neuronal injury model
Results obtained from the real-time PCR analysis indicated that numerous neurotrophic and neural growth factor genes were expressed in the HUCPVC suggesting a role in neuroprotection. Individual axons were analyzed, and degeneration was defined as when axons exhibited at least three breaks in their tubulin immunostaining. NGF withdrawal in the axon compartments resulted in 66 ± 3% of the axons showing evidence of tubulin breakdown (Fig. 2B, F) (p < 0.005, compared with control n = 8). Coculture with HUCPVCs after the NGF washout resulted in less degeneration, 34.8 ± 3% (p < 0.0005, compared with injury n = 6) (Fig. 2C, F); control chambers had 20 ± 4% axonal degeneration (Fig. 2A, F). This rescue was not observed when BMSCs, 57 ± 2% degeneration (p < 0.0005, compared with HUCPVCs, n = 4) (Fig. 2D, F) or fibroblasts 63 ± 4% degeneration (p < 0.005, compared with HUCPVCS, n = 4) (Fig. 2E, F) were added. HUCPVC-conditioned media resulted in slight rescue (54 ± 4% axon degeneration, NS) when compared with controls. These results indicate that HUCPVCs are able to rescue NGF-induced axon degeneration (Fig. 2F).
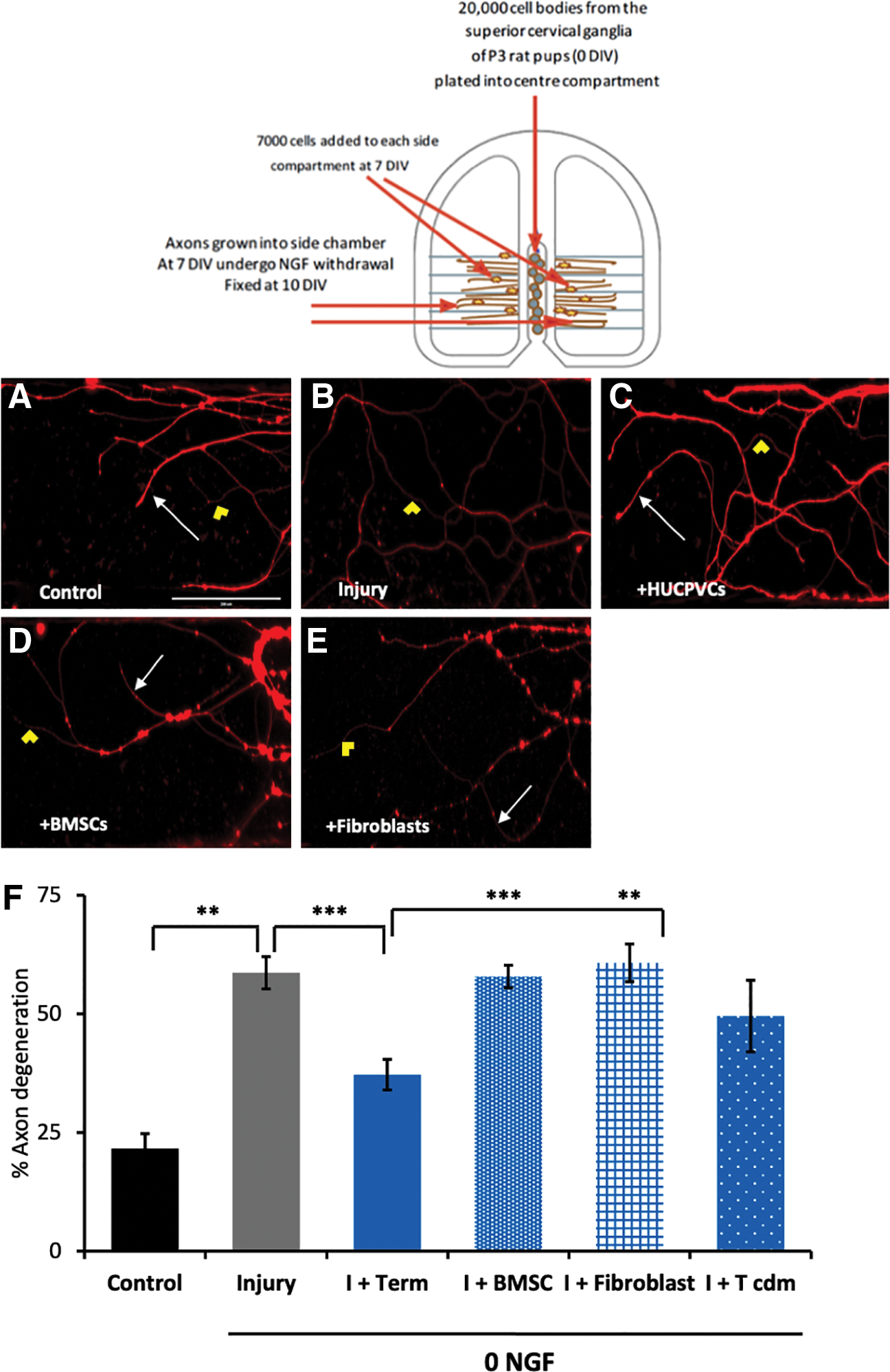
HUCPVCs protect sympathetic axons from degeneration. Sympathetic axons were grown in Campenot chambers, and axons were allowed to grow out the side compartments. HUCPVCs were added to the axon compartments and allowed to remain in coculture for 3 days. βIII-tubulin staining in the axon compartments indicates regions of tubulin breakdown and axon degeneration as well as intact axons.
Inhibition of Akt but not ERK attenuates HUCPVC-mediated protection of sympathetic axons in vitro
To determine whether HUCPVCs were rescuing axon degeneration by promoting survival cues or growth cues in sympathetic axons, we assessed the role of Akt and ERK by inhibiting the respective pathways in HUCPVC—sympathetic coculture Campenot chambers. Control conditions exhibited 22 ± 3% degeneration while NGF withdrawal significantly increased degeneration to 59 ± 3% (p < 0.05). HUCPVCs cocultured with sympathetic axons reduced degeneration to 37 ± 3% (p < 0.05) relative to NGF withdrawal. Inhibition of Akt using LY294002 resulted in a significant increase in degeneration compared with HUCPVC cocultures (48 ± 7% degeneration; p < 0.005) and similar to the degeneration levels due to NGF withdrawal (no significant difference) (Fig. 3). Inhibition of ERK with PD98059 resulted in 25 ± 1% degeneration and was comparable to HUCPVC cocultures (no significant difference). Given that the inhibitors were dissolved in DMSO, we added DMSO along with HUCPVCs to injured chambers, and degeneration was reduced to 21 ± 4%. These results suggest that HUCPVCs exert their protective effects by activating an Akt-mediated pathway suggesting a prosurvival cue.

HUCPVCs protect axons through an AKT-mediated pathway. Sympathetic axons were grown in Campenot chambers, and axons were allowed to grow out the side compartments. HUCPVCs (±PD98059 [ERK inhibitor] or LY294002 [AKT inhibitor]) were added to the axon compartments and allowed to remain in coculture for 3 days. βIII-tubulin staining in the axon compartments indicates regions of tubulin breakdown and axon degeneration as well as intact axons. Inhibiting the AKT pathway in cocultures with HUCPVCs resulted in an increase in degeneration. n = 4, *p < 0.01, **p < 0.001. Color images are available online.
HUCPVCs rescue axon degeneration in an in vivo cortical injury model
Sympathetic neurons are able to grow long axons up to 3 feet resulting in very different environments than the cell body [35]. This makes them a great model to isolate the cell body and axons to study axonal trafficking and Wallerian degeneration. However, to assess whether HUCPVCs have the same potential to rescue cortical neurons we used an ischemic (oxygen glucose deprivation [OGD]) injury model (Fig. 4A). Under normoxic conditions, 39 ± 5% degeneration was seen in cortical axons (Fig. 4B, F). OGD resulted in increased degeneration (58 ± 3%, p < 0.05) (Fig. 4C, F). The addition of HUCPVCs in cocultures reduced degeneration to 34 ± 5% (p < 0.05) in OGD conditions (Fig. 4D, F), but conditioned media did not reduce degeneration to 53 ± 3% (no significant difference) (Fig. 4E, F).
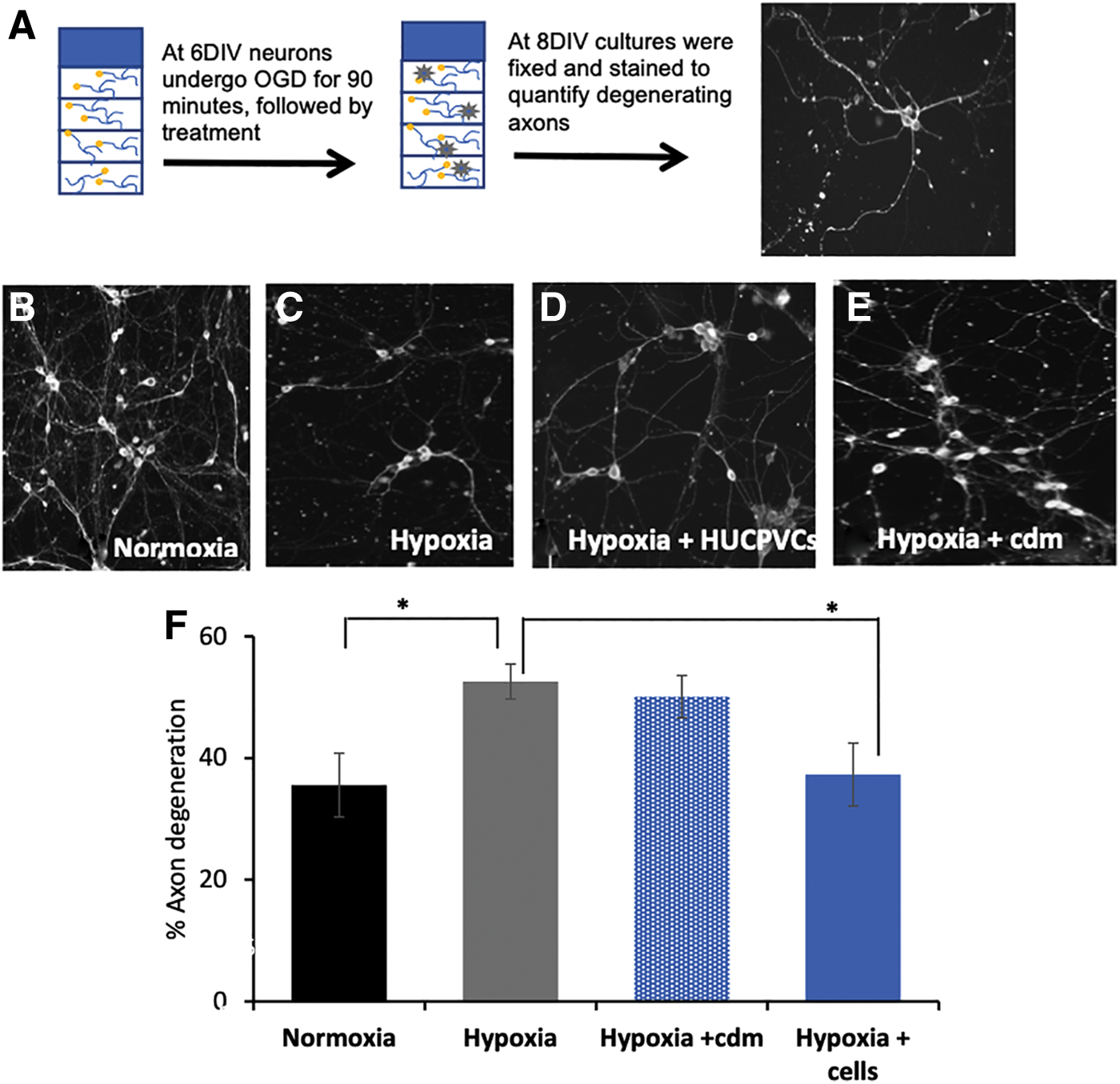
HUCPVCs protect cortical axons from degeneration.
HUCPVCs secrete NT3 and other neural growth factors
We profiled the secretome of HUCPVCs using ELISA to determine whether HUCPVCs secrete neurotrophins or neural growth factors that protect axons from degeneration. BMP4, basic fibroblast growth factor (bFGF), and LIF have been shown to be secreted by HUCPVC [9,12]. We investigated the expression of NGF, BMP4, LIF, bFGF, and NT3 in HUCPVCs cocultured with injured axons. NGF had the lowest gene expression of the three neurotrophins identified in the gene array. NGF was not detected in HUCPVC-, BMSC-, and fibroblast-conditioned media when cultured for 24 or 48 h (data not shown).
BMPs are involved in neurogenesis [36], axon guidance [37], and cell differentiation [38]. After injury, BMP4 stimulates NPC in the brain to release trophic factors [39] and BMP is needed to potentiate neurotrophin activity. BMP-4 was detected in HUCPVC-conditioned media (22 pg/mL/7 × 103 cells). BMP4 was detected in the conditioned media from control axons (106.08 ± 107.27 pg/mL). This concentration was increased in injured axons (531 ± 304.75 pg/mL) and remained elevated in axon cocultures with HUCPVCs 946.10 ± 258.06 pg/mL (Fig. 5).

Survival factors expressed by HUCPVCs alone and in coculture with sympathetic neurons. HUCPVCs were cultured in serum-free media for 72 h to assess the conditioned media for survival factors. Sympathetic axons were grown in Campenot chambers, and axons were allowed to grow out the side compartments. HUCPVCs were added to the axon compartments and allowed to remain in coculture for 3 days, the media from the side compartment were assessed for survival factors. Human enzyme-linked immunosorbent assay analysis of conditioned media from HUCPVCs grown in UltraCulture or from side compartments of sympathetic cervical ganglia axons. All OD values were corrected to UltraCulture alone concentration values. NT3 was expressed by HUCPVCs when cultured alone but was not detected in coculture with injured axons, suggesting it may be taken up by injured axons. Error bars are SEM of n = 4. NT3, neurotrophin-3. Color images are available online.
LIF is an important prosurvival and differentiation factor [9,12] and was found to be secreted by HUCPVCs (12 pg/mL/7 × 103 cells). We detected low levels in control axons (7.27 ± 12.30 pg/mL), but none in the injured axon compartments (−52.17 ± 32.63 pg/mL). Although not statistically significant, LIF expression was increased in the conditioned media containing injured axons and cocultured with HUCPVCs (28.62 ± 51.85 pg/mL) (Fig. 5).
bFGF plays a role in neurogenesis [40] and was secreted by HUCPVCs (82 pg/mL/7 × 103 cells). bFGF was secreted by control axons (587.38 ± 220.84 pg/mL) and injured axons (554 ± 307.38 pg/mL; not statistically significant). bFGF was also increased when injured axons were cocultured with HUCPVCs (765.04 ± 467.34 pg/mL) (Fig. 5).
In the absence of NGF, NT3 can effectively support neuron outgrowth through the TrkA receptor [15]. NT3 was detected in HUCPVC-conditioned media (122 pg/mL/7 × 103 cells). No NT3 was detected in control axon compartments (−35.92 ± 8.85 pg/mL), injured axons (−40.60 ± 1.54 pg/mL), or when cocultured with HUCPVCs (−40.99 ± 1.64 pg/mL) (Fig. 5).
Axon morphology is preserved with HUCPVC administration in vivo
We were interested in determining whether the protective effects of HUCPVCs seen in vitro would be reflected in an in vivo model of TBI. PKH26-labeled HUCPVCs were injected systemically and were found primarily in the peripheral organs, such as the lungs and spleen, but not in brain tissue sections at 24 h postinjury and cell injection (Fig. 6).

HUCPVCs are found in the lungs and spleen 24 h after infusion. Animals were subjected to a moderate FPI, injected with 1.5 × 106 PHK26-labeled cell HUCPVCs 1.5 h postinjury and sacrificed at 24 h.
Axonal length and morphology were assessed qualitatively on sections that were in the injury penumbra (Fig. 7B). At 24 h postsurgery, sham cortices had long and continuous NF200 expression in axons. Injured cortices lacked continuous axons in all areas assessed (Fig. 7A). HUCPVC-treated animals demonstrated compact neurofilament deposits and a larger number of continuous axons than observed in injured sections. At 48 h, injured animals demonstrated compacted and more continuous filamentous structure than at 24 h. Cortices from TBI animals treated with cells demonstrated longer and a larger number of continuous NF200-positive axon structures than injured cortices at 48 h. Overall, HUCPVC administration compared with injury resulted in an improved neurofilament structure at 24 and 48 h (Fig. 7).
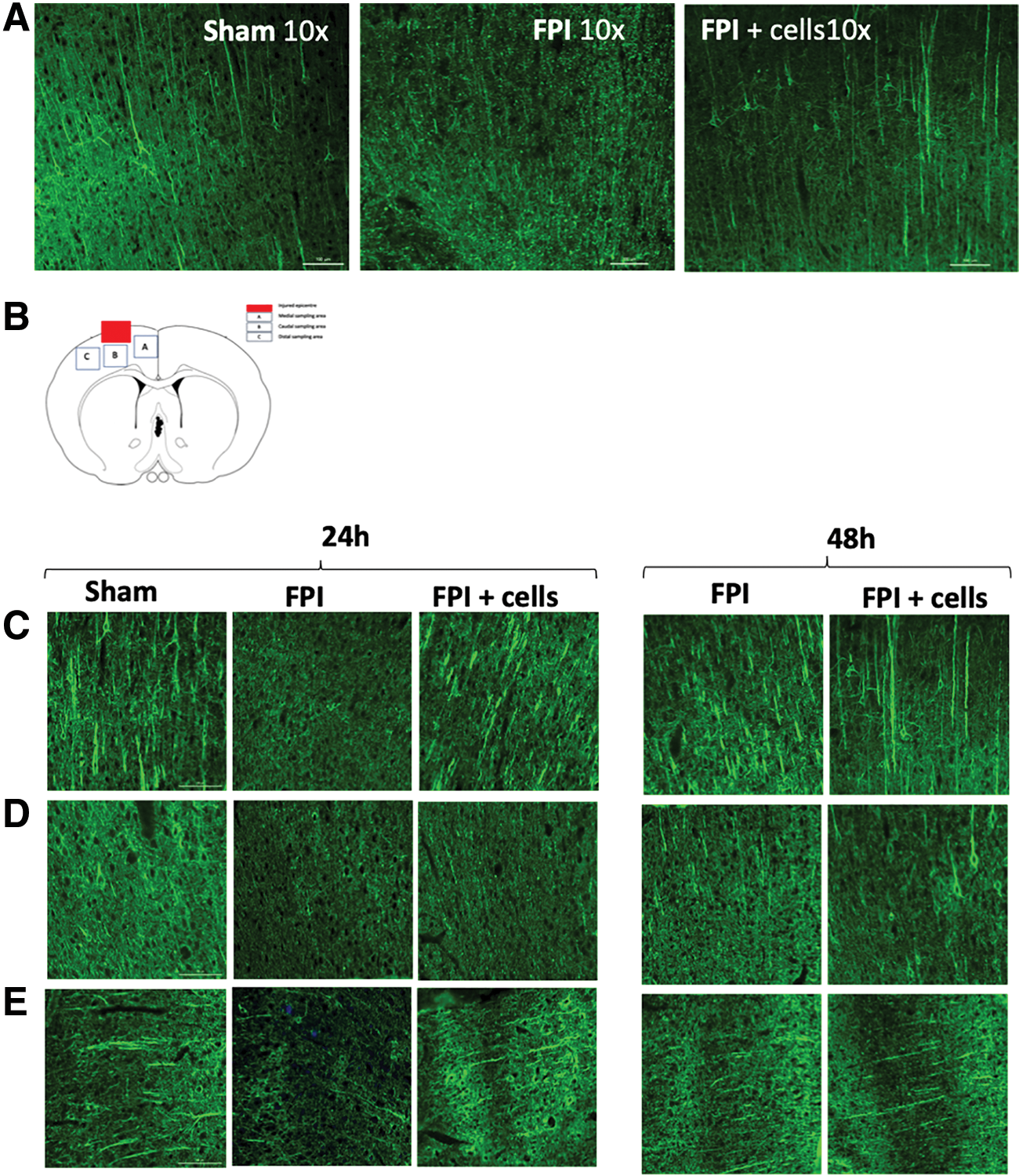
HUCPVCs preserve axon morphology after modeled traumatic brain injury.
HUCPVC administration after modeled TBI was associated with an increase in NT3 expression
From the qPCR array, HUCPVC expressed NT3 at higher levels than NGF (Table 1) and ELISA analysis indicated that HUCPVCs secrete NT3 when cultured alone but not in coculture with axons, suggesting that it may be taken up by axons (Fig. 5). We therefore assessed NT3 expression at acute time points after modeled TBI and cell injection. NT3 expression was downregulated 0.5-fold at 12 h post FPI and upregulated two-fold with cell treatment at 12 h when compared with sham controls. At 24 h, expression was still downregulated 0.2-fold and upregulated 3-fold with cell treatment. These data show that HUCPVCs were associated with an upregulation of NT3 at acute time points and was associated with protection of axonal morphology (Fig. 7, 8).
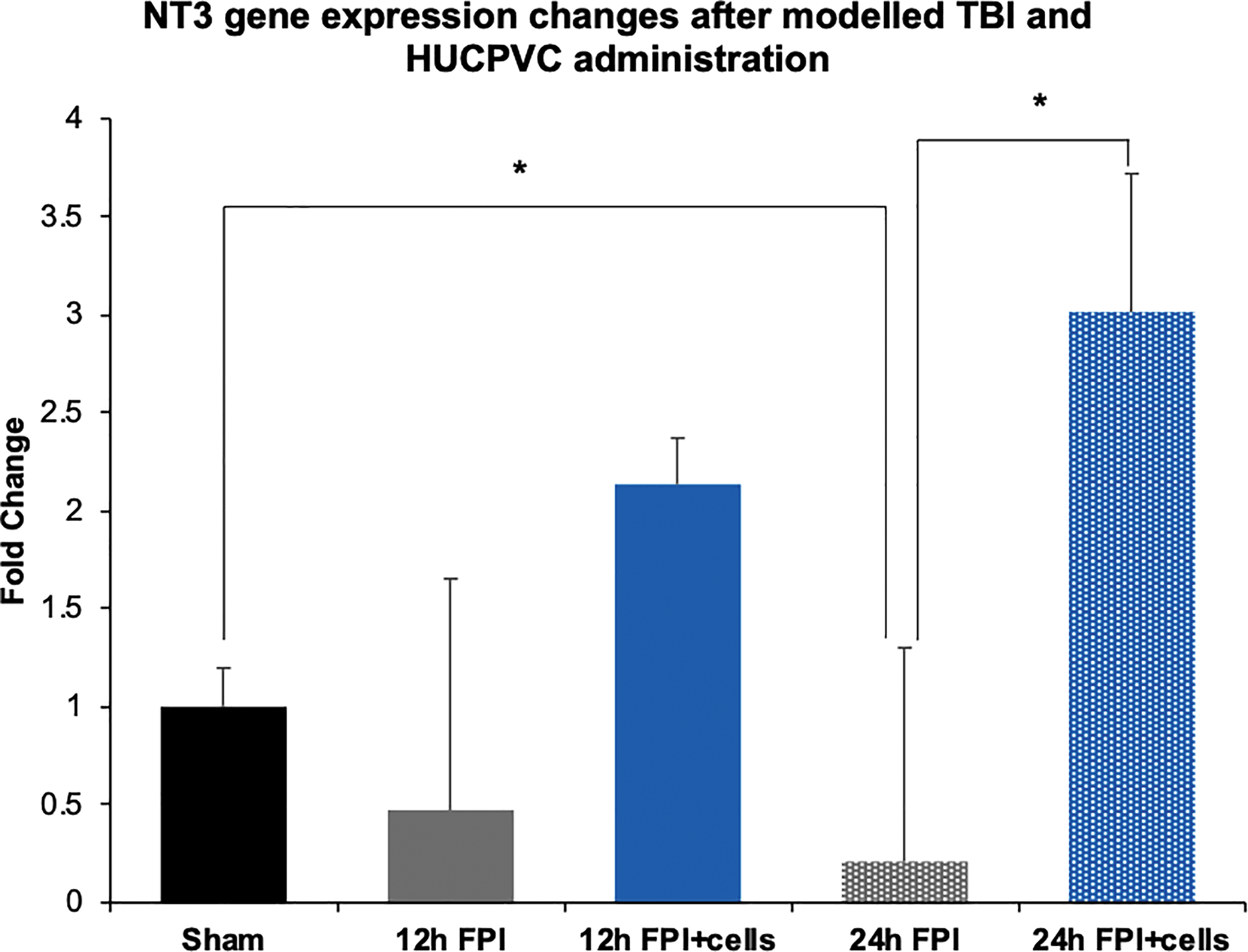
NT3 gene expression changes after FPI and HUCPVC administration. Animals were subjected to a moderate fluid percussion injury, injected with HUCPVCs 1.5 h postinjury, and sacrificed at 12 or 24 h postinjury for NT3 gene expression analysis in the injured cortex. Injury resulted in a decrease in NT3 expression and HUCPVCs were associated with an increase in NT3 expression. (n = 3, *p < 0.05). Color images are available online.
Discussion
MSC therapies have shown evidence of improved neurological outcome after trauma or stroke in clinical trials, and the signaling cascades have been partially investigated in rodent models. Cell therapy is a multifaceted approach with the potential to address numerous aspects of disease, including signaling to vascular, neurologic, and immunologic targets. These studies support the notion that factors secreted by MSCs are largely responsible for recovery and protection through angiogenesis, neuroprotection, and immune modulation [3,41 –43]. Numerous growth factors are secreted by MSCs and may provide the protection and/or regeneration seen in rodent models [44 –48]. In this present study at least three different lines were used for in vitro studies and produced consistent results. The cells used for in vivo studies were shown to be positive for CD44, CD105, CD146, CD73, CD34, and negative for CD31 and CD45 [8].
Data presented in this study support several novel conclusions. First, we demonstrate that HUCPVCs express multiple genes encoding neural growth, development, and survival factors, including three neurotrophic factors, indicating a potential for HUCPVCs in neural growth, survival, or differentiation. Second, HUCPVCs interact preferentially with axons in sympathetic neuron cocultures. HUCPVCs did not interact with the cell bodies of sympathetic neurons, but rather extended processes to neighboring axons. Third, HUCPVCs rescued injured sympathetic and cortical axons from degeneration while no rescue of axon degeneration was found when injured sympathetic axons were cocultured with BMSCs or fibroblasts. HUCPVCs secrete NT3 when cultured alone, indicating a potential mechanism by which they rescue axon degeneration independent of the NGF pathway, as no NGF was found to be secreted by HUCPVCs. Finally, NT3 expression was found to be reduced after TBI whereas HUCPVC treatment was associated with an increase in NT3 expression within 12 and 24 h of injury. We did not examine the presence of BDNF in the conditioned media from the different cell types, since BDNF is not a prosurvival cue for sympathetic neurons. Sympathetic neurons lack the prosurvival TrkB receptors and but express p75NTR, inducing death upon BDNF-induced activation [49]. The mechanism by which the HUCPVCs rescue axon degeneration may be initiated by any combination of the growth factors identified in this study, or by a separate mechanism altogether. HUCPVC-conditioned media did not result in significant rescue of sympathetic or cortical neurons. To this effect, we tested the pleiotrophic properties of HUCPVCs to target axon structural abnormalities that are present after a rodent model of TBI.
NT3 can activate the TrkA receptor and support growth in the absence of NGF [15]. In basal conditions, HUCPVCs secreted NT3; however, no NT3 was detected in cocultures with injured axons. We interpret these findings as evidence that NT3 expressed by HUCPVCs was taken up by the axons. Recently, it has been shown that NT3 is not transported to the cell body once bound to TrkA in sympathetic neurons, but rather NT3/TrkA endosomes mediate axon growth locally [50]. When NT3 is bound to its neurotrophin receptor, TrkC, signaling involving Akt activation (phosphorylation) has been shown to stabilize the actin skeleton by inhibiting phosphorylation of GS3K, which would otherwise cause disassembly [19]. This notion would be supported by our results demonstrating that inhibition of Akt phosphorylation results in axon degeneration. We performed several experiments with increasing concentrations of anti-NT3 blocking antibody to axon compartments (Promega, Madison, WI; 20–100 ng/mL). Although it was possible to visually isolate the ends of axons and count degeneration in 20–30 axons in each of the anti-NT3-treated and HUCPVC coculture experiment wells (compared with over 100 in other treatment conditions), the axons with this treatment condition had morphological changes with the ends of the axons consistently clumped together and folded back onto themselves. Further study to determine whether NT3 is involved in HUCPVC-mediated rescue of sympathetic axon degeneration is warranted because of the changes in axon morphology and the resultant decrease in axons counted observed in these experiments. MSCs have been shown to increase neurotrophin expression in spinal cord injury, in particular BDNF and NT3, when injected intraperitoneally [51].
Other survival factors that could contribute to the axonal protection effects of HUCPVC include BMPs, such as BMP4 that are crucial in establishing neurite outgrowth and spinal cord patterning, demonstrating involvement in both sympathetic and cortical neurons. Together with LIMK1, BMPs regulate actin dynamics by deactivating actin-destabilizing proteins [39,52]. FGF (bFGF) and LIF are important survival factors for neurons. In human embryonic stem cells, activation of the Akt pathway rather than the ERK pathway were shown to be crucial for survival through neurotrophins [53].
Encouraged by the promising results from the sympathetic neuron culture, we sought to determine the effects of HUCPVCs on cortical neurons in a more central nervous system (CNS)-relevant model. NT3, NGF, and GDNF signaling are common to both sets of neurons but BDNF is not a prosurvival cue in sympathetic neurons, as they are in cortical neurons [49] The gene array performed on HUCPVCs demonstrated their ability to express BDNF transcripts. Although secretion of BDNF by the HUCPVCs may play a role in cortical axon rescue, it is more likely that a combination of prosurvival and neurotrophic factors influence outcome in cortical neurons. The fact that conditioned media derived from HUCPVCs did not rescue degeneration in OGD cultures suggests that an injury cue is required to produce factors that promote axon survival. This notion is supported by our in vivo findings whereby HUCPVCs improved cortical axon morphology despite evidence indicating that many MSCs get trapped in peripheral organs, such as the lung, liver, and spleen [8,54,55]. At 24 h, although not quantified, we found the systemically injected PKH26-labeled cells in the lungs and spleen but not in the brain. Given the number of cells we observed in the lungs, we do not expect them to migrate to the brain and we do not expect them to differentiate into other cell types.
In this study, we only sought to determine the potential effects of HUCPVCs on axonal structure after TBI; the upregulation of NT3 after injury and treatment in contrast to the downregulation of this gene after injury supports our hypothesis that HUCPVCs can maintain axonal health through NT3. However, it remains to be seen how they exert their protective effects after injury if they themselves do not migrate to the brain. The mechanism of action is likely dependent on the route of administration, and in studies where cell transplantation is key, the MSCs may exert their effect through direct signaling with the injured cell types [51]. In this study, intravenous administration may result in distal paracrine effects or indirect immunomodulatory effects that promote endogenous interactions between pericytes and neuronal axons could be a potential mechanism by which the HUCPVCs exert their protective effects [8]. Our in vitro studies illustrated a clear difference between the two MSCs and requires further investigation. In addition to the therapeutic potential of HUCPVCs, they can be used to understand signaling that occurs with perivascular cells and axons to maintain health. Our data show that NT3 is important to stabilize axons after TBI and support axon health.
Neurofilaments are the most abundant cytoskeletal filaments in myelinated axons and are an indication of axonal caliber [22,56]. Compaction of neurofilament are thought to be responsible for impaired transport. The response of neurofilament to injury is highly dependent on the injury and can include proteolysis, loss, and compaction [57]. It is likely that all these events occur simultaneously at 24 and 48 h after injury contributing to the highly variable reports in the literature. Immunohistochemistry revealed that in injured animals, there were areas of neurofilament accumulation (NF200), but not accompanied with maintenance of axonal length, administration of HUCPVCs was associated with preservation of axonal length and less compaction of NF200, and is consistent with other studies [58]. These data suggest that HUCPVCs can support axonal health and integrity that maintains proper signaling after injury and preserve functional outcomes.
Pharmacotherapies have demonstrated effectiveness in preclinical studies including: inhibitors of N-methyl-
Conclusions
Neurotrophins play an important role in maintaining axon health, which is crucial for proper communication. HUCPVCs secrete important survival factors and through an Akt-mediated pathway promote survival of axons. White matter preservation has been shown to be a strong predictor of outcome after TBI. As such, targeted effects of HUCPVC treatment may provide protection of white matter, which in turn may potentially result in synergistic improvement in outcome after TBI.
The secondary injury events after TBI are multifactorial and include degeneration, inflammation, repair, and innate regeneration. As current therapeutic strategies generally focus on targeting single pathways, a more multifaceted approach may prove to be a better solution to treating TBI. HUCPVCs have the potential to target multiple pathways simultaneously by secreting survival and neuroprotective factors.
Footnotes
Author Disclosure Statement
All authors have read the journal's policy on disclosure of potential conflicts of interest and have none to declare. Dr. C.L. is the owner and director of the CReATe Cord Blood Bank and PeristemTM program and shareholder in Tissue Regeneration Therapeutics (Toronto, Ontario).
Funding Information
This work was supported by the Physicians Services Incorporated foundation (15-13) and by the CReATe Fertility Center.