
Abstract
After intra-articular injection, synovium-derived mesenchymal stem cells (SMSCs) can adhere to damaged cartilage (a process called homing) and then repair the cartilage defect. Nonetheless, the main obstacle of the current method is the insufficient homing ratio of SMSCs, which fails to meet the requirements for cartilage repair and thereby greatly limits the therapeutic effect. In this study, the optimal homing time of SMSCs was determined by evaluating the SMSC homing efficiency at 1, 3, 7, 14, and 28 days after injury using a rat cartilage defect model. The ability of platelet-derived microparticles (PMPs) to promote SMSC homing was evaluated by cartilage/subchondral bone cell adhesion, transmembrane migration, and intra-articular cell distribution assays. SMSCs had an optimal homing efficiency in the very early stage (1 day) after cartilage injury. We found that PMPs, which were abundant in the synovial fluid at this early stage, were responsible for this augmented SMSC homing. An ex vivo cell adhesion assay revealed that the coincubation of SMSCs with PMPs at a 1:50 ratio markedly enhanced cell adhesion to cartilage and the subchondral bone surface. The transmembrane cell migration assay yielded similar results. Further in vivo homing assays revealed that PMPs possess excellent homing capacity, which they transferred, to some extent, to SMSCs by coating the cell surface. We measured the expression of homing-related genes in SMSCs exposed to PMPs and identified several upregulated genes. Moreover, platelet-specific adhesion molecules, particularly GPIIb/IIIa, CXCR4, ITGβ1, and ITGα2, were determined to play a critical role in the homing of SMSC/PMP complexes. This improvement in SMSC homing increased the volume of regenerated tissue in the cartilage defect. In conclusion, PMPs significantly promoted the homing of SMSCs to cartilage, which facilitated cartilage regeneration. These data suggest a safe and promising strategy for improving the outcome of stem cell therapy.
Introduction
Osteoarthritis (OA)
Synovium-derived mesenchymal stem cells (SMSCs), which exert immunomodulatory function and undergo robust chondrogenic differentiation, have been applied by direct intra-articular injection (DIAI) for the treatment of early- to middle-stage OA [3]. This strategy is simple, but the efficiency of cell resettling onto cartilage lesions (termed “homing”) is quite low, which greatly limits the effectiveness of cartilage repairment. Strategies to stimulate MSC homing to cartilage lesions have become a research focus [4,5], but no satisfactory strategy has been found.
The timing of MSC injection is a matter of concern. Mokbel et al. found that the early intra-articular injection of MSCs after cartilage trauma led to a better therapeutic effect than the delayed injection and speculated that this difference might be associated with MSC homing [6]. In fact, in the early stage after joint injury, a complex sequence of events involving multiple mechanisms, including coagulation, acute inflammation, remodeling, and regeneration, occurs in the joint cavity microenvironment [7]. These events occur in specific time windows and in sequence with some overlap. This situation presents an excellent opportunity to identify the stimulatory factors and to elucidate the specific mechanism of MSC homing by using the time window as a clue.
In this study, we established a rat model and MSC tracer imaging and found that MSC homing was significantly enhanced at the very early stage (1 day) after cartilage injury and then rapidly decreased. In comparison with the inflammation time window of 1–4 days and the later regeneration time window, this timing is more consistent with the platelet activation period [8]. This result led us to focus on the role of platelets in MSC homing. Chemokines are the typical focus of cell migration and homing research [9], but platelets also play an important role in leukocyte recruitment [10,11].
Recent studies revealed that a platelet can act as a bridge to mediate endothelial cell (EC) and MSC adhesion in blood vessels [12,13]. The articular cavity, however, differs from the endovascular cavity in the synovial fluid (SF) component, the cartilage surface, and the cell migration and adhesion mechanisms. In particular, intact platelets are almost nonexistent in SF [14]. This fact prompted us to concentrate on another factor, platelet-derived microparticles (PMPs), which are 0.1–1 μm particles shed from the plasma membrane of platelets undergoing activation, stress, or apoptosis [15,16].
There is increasing recognition of the role of PMPs in broad biological functions, ranging from wound healing and tumor growth to inflammation and regeneration [17]. PMPs have been found to play roles in leukocyte recruitment and the initiation of intravascular inflammation [11]. In OA, most PMPs exist in inflammatory SF and can activate synoviocytes, thus participating in the amplification of joint inflammation. Whether PMPs in specific joint microenvironments affect the cartilage homing of MSCs has not been reported. In this study, we examined PMPs as a possible stem cell delivery system for cartilage regeneration and preliminarily identified the underlying mechanism.
Materials and Methods
Isolation and culture of SMSCs
All animal experiments were conducted according to the animal protocol approved by the Third Xiangya Hospital of Central South University. Approximately 5–10 mg synovial tissue was isolated from 10 knee joints of Sprague-Dawley (SD) rats (3 weeks old, male). The synovium was digested overnight at 5% CO2 in 95% humidity at 37°C with 0.2% type I collagenase (Life Technologies) in Dulbecco's modified Eagle's medium (DMEM) (10567014; Gibco) supplemented with 10% fetal bovine serum (FBS, 10270106; Gibco). After digestion, the cells were collected by centrifugation, washed twice, resuspended in DMEM supplemented with 10% FBS and 1% penicillin–streptomycin (15140122; Gibco), plated in a 100-mm dish, and allowed to attach for 48 h at 37°C. Nonadherent cells were removed carefully by changing the medium.
SMSC identification
Isolated primary SMSCs were identified by morphology, cell surface markers, and trilineage differentiation ability. OriCell rat MSC differentiation medium (Cyagen) was utilized to induce the differentiation of 1 × 106 second-passage SMSCs toward the osteogenic, adipogenic, and chondrogenic lineages according to the reagent instructions.
For osteogenic differentiation, MSCs were cultured in osteogenic differentiation medium for 4 weeks and subsequently stained by Alizarin Red. For adipogenic differentiation, MSCs were cultured in adipogenic differentiation medium A (induction medium) for 3 days and in medium B (maintenance medium) for 1 day. After four cycles, MSCs were cultured in medium B for 7 days before Oil Red O stain. For chondrogenic differentiation, MSCs were centrifuged to form a cell pellet and cultured in chondrogenic differentiation medium for 2 weeks. Pellets were formalin fixed for Alcian blue stain.
1 × 105 second-passage SMSC suspension was blocked with 1% bull serum albumin (BSA) for 20 min, incubated with anti-rat CD34 (MA5-17901), CD73 (MA5-15537), CD45 (17-0461-82), and CD90 antibodies (ab225), or isotype control antibodies for 15 min at room temperature, washed twice, and suspended in 300 μL phosphate-buffered saline (PBS) buffer and then analyzed by flow cytometry (BD FACSCalibur) and FlowJo™ v10.
SMSC labeling
SMSCs were dual labeled with bioluminescent and fluorescent signals. 1 × 107 SMSCs were infected with 1 × 108 transducing unit (TU) lentivirus expressing luciferase (GeneChem, Shanghai, China) for 8 h and cultured in DMEM for 48 h, then labeled with 0.1% CellTracker CM-DiI (Thermo Fisher) at 37°C for 5 min, and incubated at 4°C for 15 min. The dual labeling was examined with a fluorescence microscope (Olympus, Japan) and a bioluminescence imaging system (BLIS, PerkinElmer).
SMSC homing assays
To explore the correlation between homing and time, a full-thickness cartilage defect (2 mm in diameter) was created in the middle of the right femoral trochlear groove of 1-month-old SD rats. At 1, 3, 7, 14, and 28 days after the surgery, 1 × 106 luciferase/CM-DiI-labeled MSCs were resuspended in 200 μL of serum-free medium (SFM, 1033201; Gibco) and injected into the right knees of rats (n = 5 in each group). The rats were free to move. At 24 h after the injection, the joint was opened (the quadriceps muscle was flipped to the distal position) and rinsed with PBS to remove nonadherent cells (Fig. 2a). Cell distribution was observed by BLIS (luciferase) and fluorescence microscopy (CM-DiI).
Full-thickness defects with a depth of 2 mm (penetrating the subchondral bone), partial-thickness defects with a depth of 0.3 mm (intact subchondral bone), and sham defects were created to explore the effect of cartilage damage depth on SMSC homing. At 1 day after the surgery, SMSCs were injected and evaluated as described above (Fig. 2d).
To investigate the effect of defect surface or SF at different times on homing, the distal femurs (with cartilage defects) and SF were removed at 1 and 7 days after cartilage injury. The distal femurs were placed in 24-well plates, and 1 × 106 MSCs were added dropwise into the defect and cultured for 2 h. The number of adhered cells was calculated using BLIS (Fig. 3a). Meanwhile, 1 × 106 MSCs were pretreated for 1 h at 37°C with 1-day SF, 7-day SF, or DMEM and then injected into the joints at 7 days after cartilage damage (Fig. 3b). Cell homing was evaluated as described above.
SMSC adhesion assay
The SF was collected at 1, 3, and 7 days after cartilage injury. The 1-day SF was further separated into the cell, microparticle (MP), and supernatant components by differential centrifugation (5 min at 500g for cells and 45 min at 20,000g for MPs). A total of 5 × 104 CM-DiI-labeled SMSCs were pretreated for 1 h at 37°C with serum-free DMEM, 1-/3-/7-day SF, or the cell/MP/supernatant component, placed onto osteochondral slices with full-thickness cartilage defect, and allowed to attach at 37°C for 30 min. The surfaces were washed gently with PBS to remove the nonadherent cells. The adherent cells were counted in six random fields per filter (20 × magnification) under a fluorescence microscope.
Fluorescence-activated cell sorting analysis
Cells and SF components (1, 3, and 7 days) were removed by centrifugation at 4,000g for 10 min, and the supernatant was centrifuged at 21,000g for 60 min. Pellets containing MPs were resuspended in PBS, and the MPs were counted by flow cytometry in the presence of a fixed number of 1 μm calibration beads (1 × 106/mL, BD Biosciences). To identify PMPs, the MPs were incubated with a phycoerythrin (PE)-conjugated anti-CD41 (platelet surface marker) monoclonal antibody (sc-12773). CD41-positive particles smaller than 1 mm were counted. To demonstrate the presence of PMPs on the surface of SMSCs, the cells were incubated with PMPs, stained with the PE-conjugated anti-CD41 antibody and analyzed by fluorescence-activated cell sorting (FACS).
PMP isolation
Platelets were isolated from healthy donors and activated for 5 h at room temperature with 1 mM CaCl2 and 5 μg/mL collagen (Horm; Nycomed Arzenmittel, Munich, Germany) and centrifuged at 1,200g for 10 min. The supernatant was centrifuged at 21,000g for 60 min at 4°C in ultra-clear conical centrifuge tubes (Beckman) using a Sw55Ti rotor in a Beckman centrifuge. The concentration of PMPs was determined by FACS after staining with the PE-conjugated anti-CD41 antibody and a fluorescein isothiocyanate-conjugated anti-CD61 antibody (11-0611-82). The PMPs were stained for 20 min at room temperature with 10 μM dioctadecyloxacarbocyanine perchlorate (DiO, D4292). The concentration of PMPs was 2 × 107/mL.
SMSC-PMP adhesion and migration assays
A total of 5 × 104 CM-DiI-labeled SMSCs were mixed and incubated in suspension at 37°C for 30 min with PMPs at PMPs/SMSCs ratios of 10:1, 50:1, and 200:1. Nonadhered PMPs were removed by washing with PBS. Then, SMSCs were placed onto cartilage/subchondral bone surfaces and allowed to attach for 30 min at 37°C. The cartilage/subchondral bone surfaces were pretreated with PMPs before the adhesion assay.
Migration of SMSCs was assessed using Transwell chambers with 6.5-mm-diameter polycarbonate membrane filters with 8-μm pores (Becton Dickinson, Franklin Lakes, NJ). A total of 5 × 104 SMSCs and PMPs (PMPs/SMSCs ratios of 10:1, 50:1, and 200:1) in 200 μL of serum-free DMEM were added to the upper chambers, and the lower chambers were filled with DMEM containing 10% FBS as the chemoattractant. The cells were incubated in the Transwell system for 24 h at 37°C and 5% CO2. Cells that migrated to the lower surface of the filter were stained with crystal violet.
Electron and confocal microscopy
CM-DiI-labeled SMSCs were incubated at 37°C for 1 h with DiO-labeled PMPs in suspension at PMPs/SMSCs ratios of 10:1 and 200:1. Scanning electron microscopy (SEM) was performed as described previously to observe PMPs binding to SMSCs. Serial Z-stacked (0.5 m per section) confocal imaging was performed with a Zeiss LSM-510 Meta Confocal Microscope (Carl Zeiss, Jena, Germany), and image analyses were performed in LSM510 software and Image-Pro Plus (Media Cybernetics, Bethesda, MD).
RNA isolation and sequencing
Total RNA was isolated from untreated or PMP-treated SMSCs (3 h at 37°C, three replicas) using TRIzol reagent (Life Technologies, Gaithersburg, MD). Total RNA was tested using NanoPhotometer® spectrophotometer (IMPLEN, CA), Qubit®3.0 Fluorometer (Life Technologies, CA), and Agilent 2100 RNA Nano 6000 Assay Kit (Agilent Technologies, CA). Then, the total RNA was converted to cDNA following the manufacturer's instructions (Superscript Choice cDNA), and the products were purified and enriched with PCR to create the final cDNA library.
After the libraries were constructed, paired end run was performed on HiSeq 2500 platform (Illumina Inc., CA). The clean reads number was 46668064 and read length was 150 bp (n = 3). Evaluation of gene expression was performed using fragments per kilobase of transcript per million mapped reads. The transcripts with P < 0.05 and log2-fold change ≥1.0 were considered for differential gene expression using DEGseq program.
Blocking platelet-derived adhesion factors
We blocked the key platelet-derived adhesion factors (PDAFs) on PMPs to explore the effect of PDAFs directly from PMPs on SMSC homing. The SMSC-PMP complex was incubated with antibodies against GPIIb/IIIa (PA5-79527), GPIb (PA5-86799), P-selectin (MA5-16922), CXCR4 (PA5-19856), INTβ1 (MA5-31981), and INTα2 (MA5-32306) in suspension at 37°C for 1 h before injection. The in vivo homing assay was performed as described previously.
Histological evaluation
Seven days after creating a full-thickness cartilage injury, 1 × 106 untreated or PMP-treated (50:1) SMSCs (resuspended in 200 μL SFM) were injected into the knee joint. Four weeks after injection, the rats were euthanized. Cartilage regeneration was evaluated histologically at the macroscopic level with Safranin O/Fast green staining (Thermo Fisher), and the regenerated tissue area was calculated and assessed using the modified International Cartilage Repair Society (ICRS) histological scoring systems.
Statistical analysis
Data are expressed as the mean ± standard deviation in the text and figures, unless otherwise noted. Statistical significance was determined by Student's t test, analysis of variance (ANOVA), or multiple comparison tests in SPSS 17.0 software (SPSS, Inc., Chicago, IL). Differences with P < 0.05 were considered statistically significant.
Results
SMSC isolation and identification
After 14 days of primary culture, primary SMSCs transformed into colony-forming units of fibroblasts and had a typical spindle shape (Fig. 1a). Surface antigen analysis revealed that most of the cells expressed CD90 (99.66%) and CD73 (98.92%) but that few expressed CD34 (1.34%) or CD45 (0.36%) (Fig. 1b–f). The cells were successfully induced to undergo osteogenic, adipogenic, and chondrogenic differentiation (Fig. 1g–i).
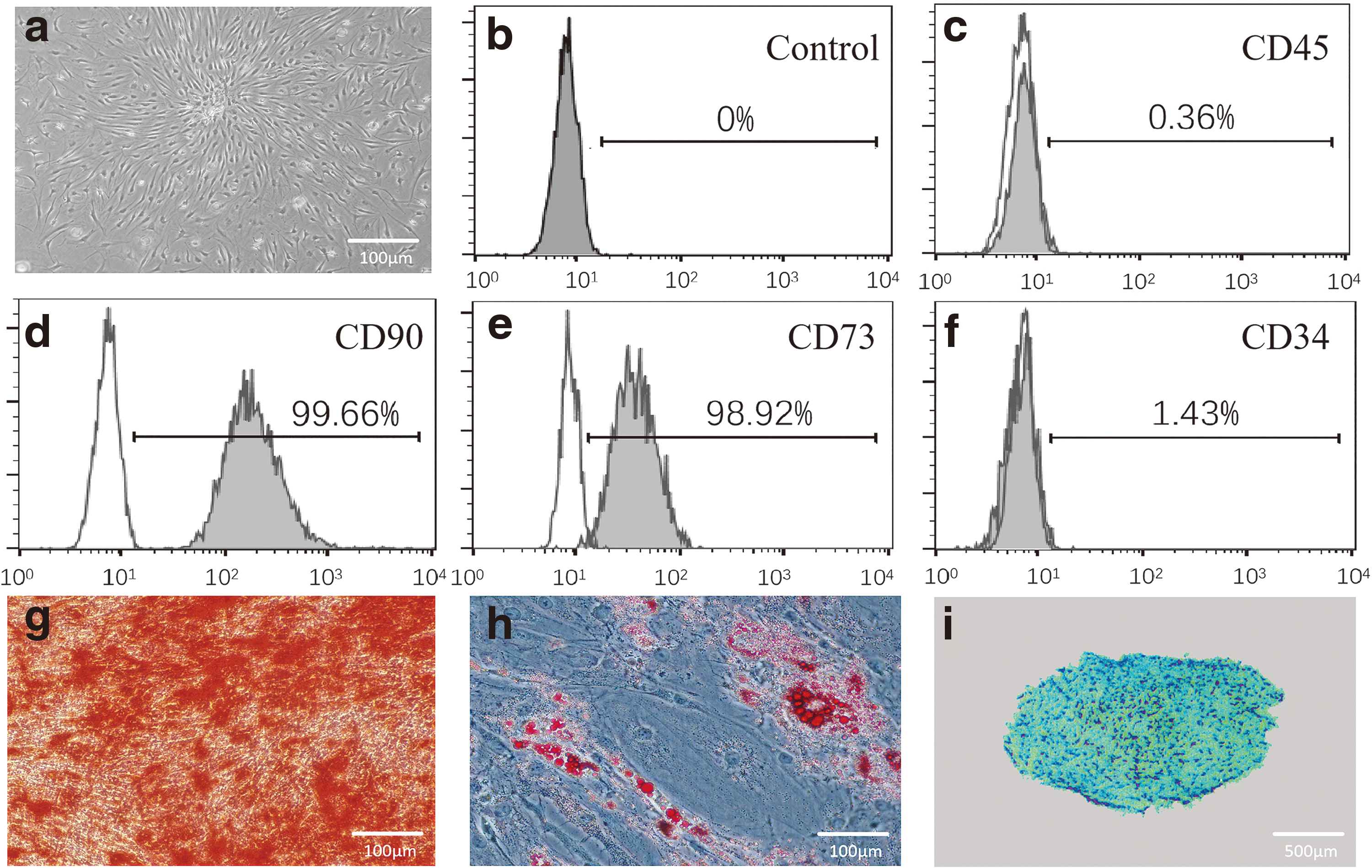
MSC identification.
Timing of SMSC homing to cartilage defects
SMSCs showed the most obvious homing toward cartilage defects in the early stage (1 day) after joint injury, and homing decreased gradually thereafter. With the increase in the postinjury interval, SMSCs tended to gather irregularly in the suprapatellar capsule, lateral recess, and cruciate ligament. CM-DiI-labeled SMSCs formed a limited monolayer on the cartilage lesion surface in the 14-day group and were sparse in the 28-day group (Fig. 2b). There was no significant difference in the total SMSCs bioluminescence signal within the knee joint between the groups (Fig. 2c). Regarding different types of cartilage injury, we found that full-thickness cartilage lesions significantly stimulated SMSC homing compared with superficial layer cartilage lesions (Fig. 2d).
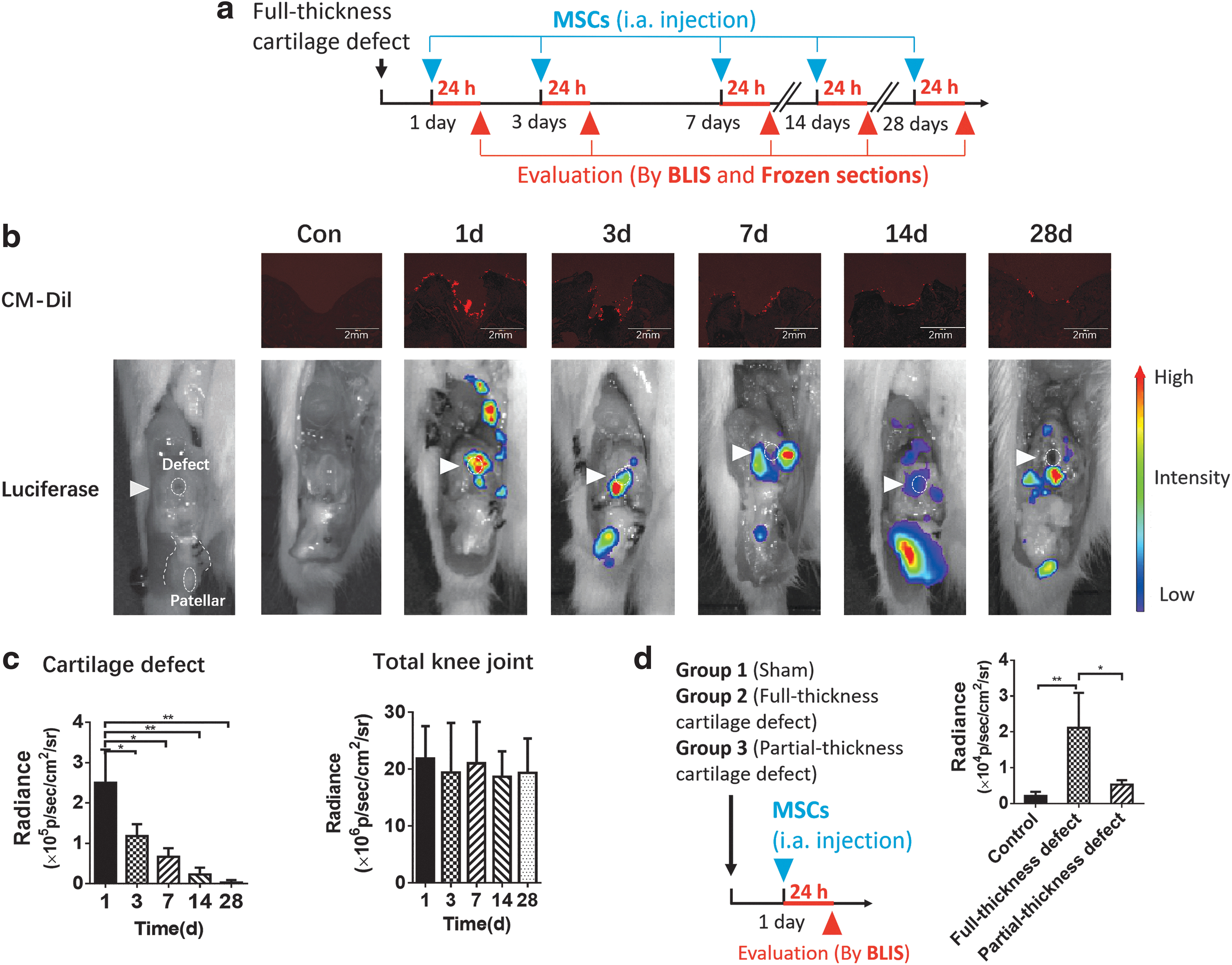
Timing of SMSC homing to cartilage defects. At 1, 3, 7, 14, and 28 days after the surgical procedure, dual-labeled SMSCs were injected into the right knees of SD rats (n = 7 joints in each group).
Furthermore, the SF early after injury (1-day SF) enhanced the homing of SMSCs to cartilage defects, while the defect surface had no obvious effect at different times after injury (Fig. 3a, b). This finding was further confirmed by an in vitro adhesion assay. The 1-day SF significantly improved the ability of SMSCs to adhere to cartilage slices (Fig. 3c), and this effect was mainly attributed to MPs in the 1-day SF (Fig. 3d). There were considerably more MPs in 1-day SF than in 3-day or 7-day SF (Fig. 3e), and most of the MPs in 1-day SF were derived from platelets (Fig. 3f, g).
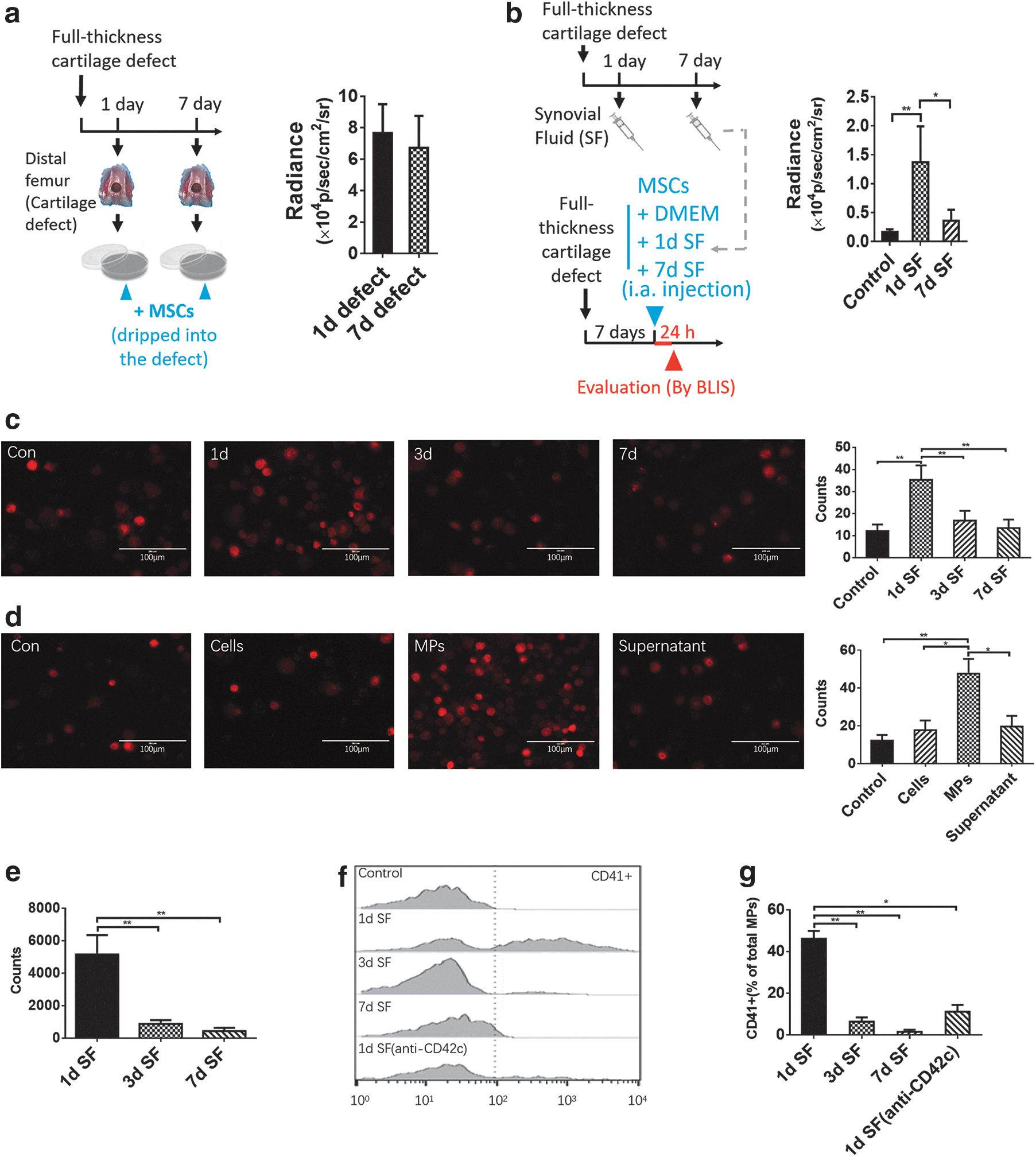
The effects of the cartilage lesion surface and SF on MSC homing.
Effects of PMPs on SMSC homing
PMPs were identified by FACS (Fig. 4a, b). After the PMPs and SMSCs were preincubated and injected into the animal model, they colocalized at the cartilage defect (Fig. 4c). PMP pretreatment markedly promoted SMSC adhesion to cartilage/subchondral bone in vitro (at the optimal ratio of 50:1) and migration in the transwell assay (Fig. 4d). Pretreating the substrate with PMPs had no obvious effect (Fig. 4d). We measured the homing of SMSCs to joints 1 and 7 days after injury and found that inhibiting endogenous intra-articular PMP generation impaired SMSC homing (Fig. 4e) and, in contrast, that pretreating SMSCs with PMPs strongly promoted this homing behavior (Fig. 4f).
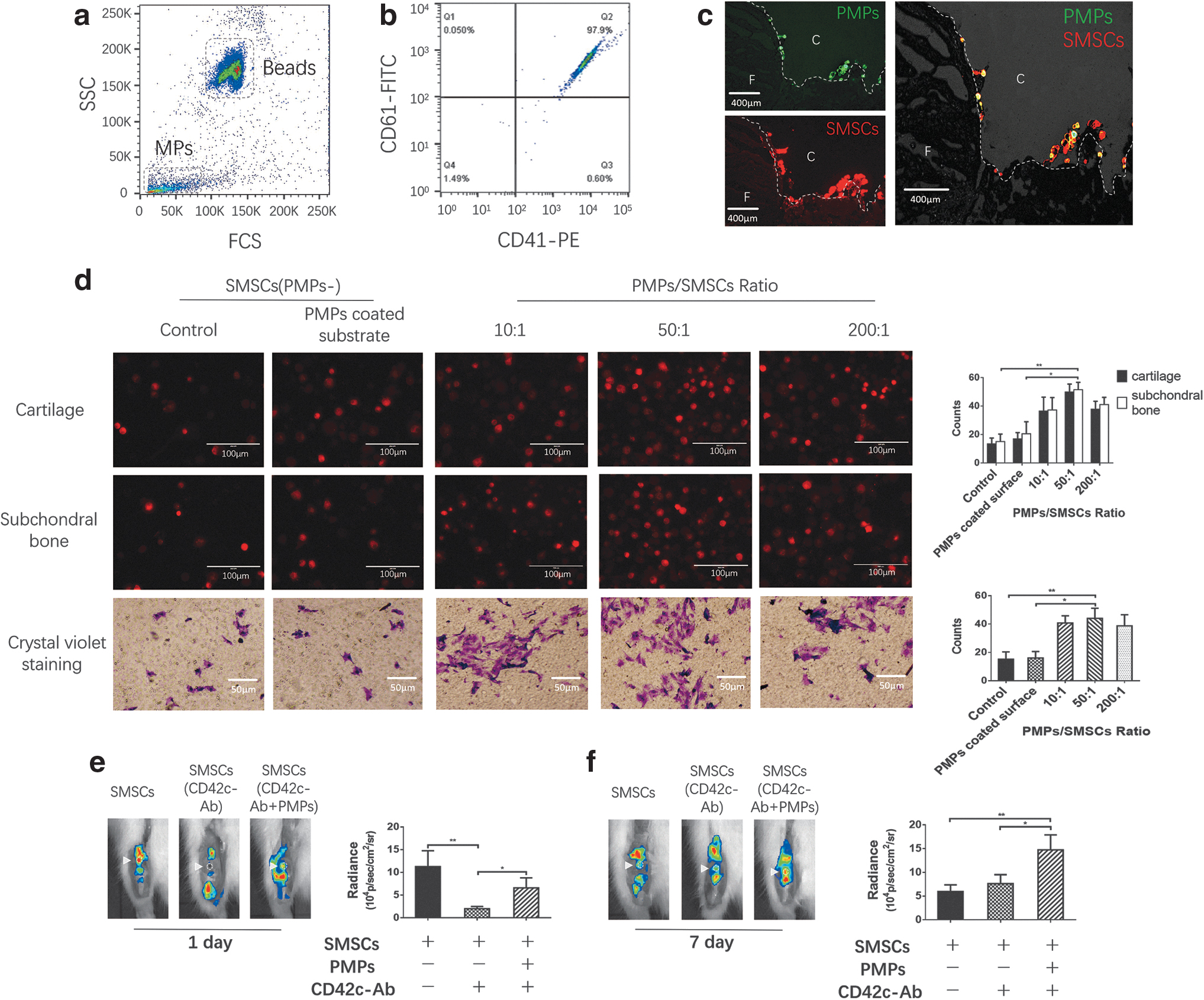
The effect of PMPs on SMSC homing.
Detection of PMPs on the SMSC surface
We utilized SEM, confocal microscopy, and FACS to observe the interaction between PMPs and SMSCs. After 30 min of coincubation in suspension, PMPs attached firmly to the SMSC membrane, and some PMPs underwent membrane fusion with SMSCs. As a whole, PMPs achieved high surface coverage of SMSCs. When PMPs and SMSCs were mixed at a 200:1 ratio, PMPs covered at least 18.3% of the surface area of SMSCs (Fig. 5a), and at a 50:1 ratio, the PMP fluorescence signal was detected on 62% of SMSCs (Fig. 5b). The majority of the PMP signals were distributed on the peripheral surface of SMSCs, whereas few intracellular signals were detected (Fig. 5c). Similarly, FACS analysis indicated that after coincubation at a 50:1 ratio, the platelet-specific marker was detected on the surface of 53% of SMSCs (Fig. 5d, e).
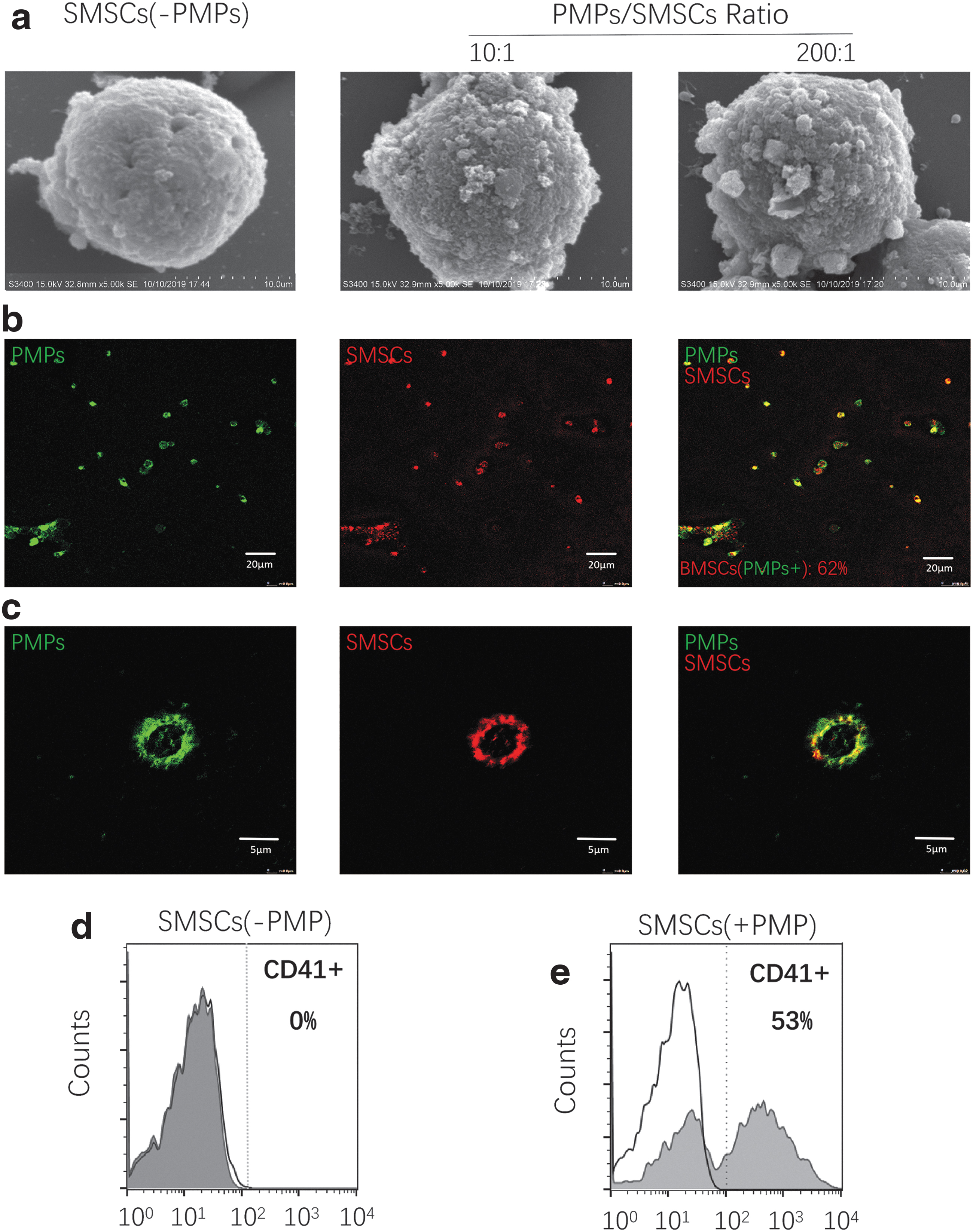
The pattern of interaction between PMPs and SMSCs.
Expression of homing-related genes
We examined a total of 15,453 genes in untreated and PMP-treated SMSCs; compared with untreated SMSCs, PMP-treated SMSCs showed the upregulation of 1,081 genes and the downregulation of 1,464 genes. Among these genes, 41 were involved in chemotaxis, migration, and adhesion, including significantly upregulated Ager, ccr10, cd24, cxcr1, cxcr2, cxcr3, ICAM-1, Sirpa, and Snai2 (Fig. 6a–c).
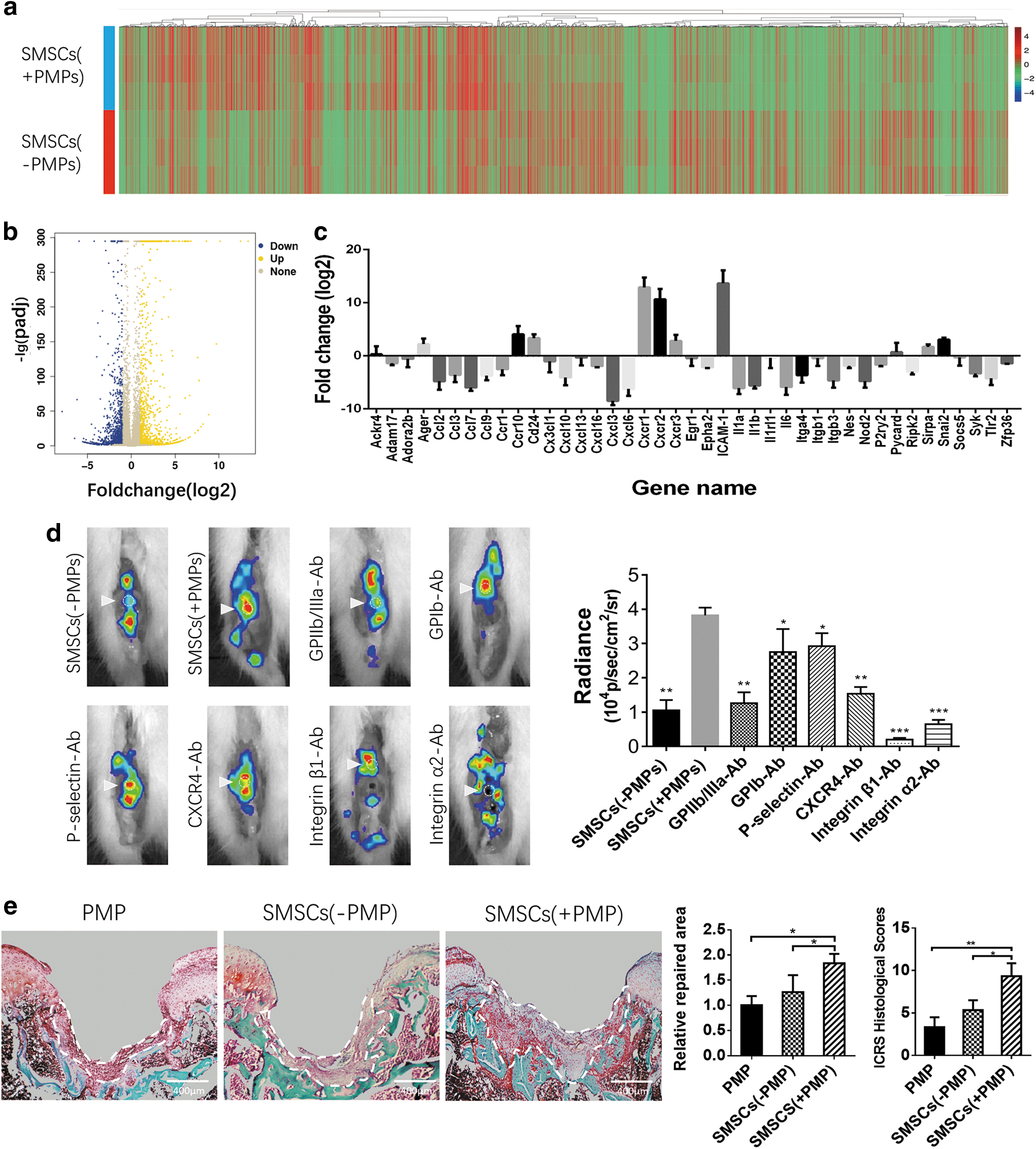
The mechanism by which PMPs affect SMSC homing
Effects of PDAFs
The homing of SMSC-PMP complexes was inhibited to varying degrees by blocking PDAFs directly from PMPs. In particular, GPIIb/IIIa, CXCR4, ITGβ1, and ITGα2 were found to play a critical role in the homing of SMSC-PMP complexes (Fig. 6d).
Histological evaluation of cartilage regeneration
Only a thin layer of regenerative tissue was produced within the cartilage defect after the injection of untreated SMSCs. In contrast, PMP-treated SMSCs generated a significantly larger volume of reparative tissue, filling the defect. Hardly, any noticeable regenerative tissue in the defect area was produced upon the injection of PMPs alone (Fig. 6e). We obtained similar results with the ICRS histological score. The regenerated tissues in all three groups showed varying degrees of Safranin O staining, and none of these tissues exhibited typical hyaline cartilage morphology (Fig. 6e).
Discussion
Increased stem cell engraftment at the injury site can enhance the therapeutic effect of cartilage regeneration. Therefore, there have been numerous research efforts to identify a more effective cell delivery system [18]. Some natural and synthetic cell scaffolds, which represent the most studied strategies, show high cell transplantation efficiency. Nonetheless, the use of these scaffolds requires a surgical intervention, and scaffolds are currently of limited clinical use.
The DIAI of MSCs is a simple and reliable method. One of the main challenges with this strategy is the ineffective cell engraftment from SF to the cartilage lesion. Some studies have focused on the systemic administration of MSCs that are targeted and trafficked to the injury site through blood circulation. This homing model is based on the mechanism of leukocyte recruitment into inflamed tissue. In this process, the intravascular chemotactic gradient and adhesion molecules expressed on the EC surface are thought to be the primary cell-attractive forces [11].
However, MSCs seem to home via a different mechanism in the articular cavity than in vessels. The composition of SF is quite different from that of blood, and its flow in the joint cavity is relatively irregular; therefore, it is nearly impossible for a chemical gradient to form. The internal surface of the joint cavity, especially the damaged cartilage surface, is complex, and the mechanism of MSC adhesion to cartilage is different from the mechanism of EC adhesion to blood vessels [14]. Therefore, it is necessary to clarify the mechanism of MSC trafficking in the joint cavity.
According to studies on chemotaxis, the transient activation of coagulation and inflammation in the microenvironment during the acute phase after join injury showed the highest correlation with MSC homing. Nonetheless, the peak time windows of these two events are not consistent (0–2 days vs. 3–9 days) [7,19]. We evaluated the time window of MSC homing and found that SMSC homing was significantly stronger at 1 day than in the subsequent inflammation phase.
Leijs et al. recently reported that the pretreatment of MSCs with some inflammatory factors enhanced the expression of adhesion and migration receptors, but no improvement in cartilage adhesion was observed [20]. This result is consistent with our finding that the cartilage adhesion of MSCs sharply decreased within 3 days, while the joint inflammation evolved, implying a correlation between stem cell homing and platelet activation. Immediately after endangium injury, platelets are some of the first blood components that recognize and adhere to the lesion site and provide homing cues for leukocytes and MSCs [10,12,13].
Using intravital confocal imaging, Teo et al. found that 43% of the intravascular MSCs that home to an inflamed ear dermis come in contact with platelets, and MSC homing decreases when platelets are depleted [12]. Other studies have yielded similar results [13,21]. In contrast to the numerous studies on intravascular MSC trafficking, few studies have addressed the role of platelets in MSC–cartilage adhesion, perhaps because intact platelets are less likely to spontaneously end up inside the cavity. In our study, we found that cartilage defects with subchondral bone penetration and local hemorrhage significantly stimulated MSC homing in vivo compared with defects with intact subchondral bone (Fig. 3a). Therefore, we hypothesized that platelets play a positive role in MSC homing.
However, as mentioned above, various factors in the articular cavity influence MSC homing. Most studies have focused on the influence of the SF composition. For example, research shows that both the high-molecular-weight hyaluronic acid (HA) in OA-affected SF and the proteoglycan-rich matrix on the cartilage surface have inhibitory effects on MSC adhesion to cartilage [4,5]. In contrast, an appropriate concentration of magnesium has a stimulatory effect [22].
To clarify the specific mechanism, we first obtained SF 1 day after knee joint injury (1-day SF) and pretreated MSCs with this fluid before application. This approach significantly promoted MSC homing in vivo and cartilage adhesion in vitro (Fig. 3c-d). Then, 1-day SF was separated into the cell, MP, and soluble supernatant fractions, and we demonstrated that the promoting effect was mainly derived from the MPs in SF (Fig. 3e). After ascertaining the quantity and source of these MPs in 1-, 3-, and 7-day SF, we found that 1-day SF contained a large number of MPs derived from platelets (PMPs). The quantity of PMPs showed a rapid decrease in 3- and 7-day SF, in accordance with the declines in MSC homing and cartilage adhesion. Therefore, these results imply that PMPs in 1-day SF are an essential factor for promoting MSC cartilage homing.
Ed Rainger et al. summarized six potential mechanisms of platelet-mediated leukocyte recruitment [11], including the stimulation of chemokine and adhesion molecule expression by ECs, the attachment of leukocytes to the EC surface, and the binding of leukocytes in circulation to enhance recruitment. It is well established that PMPs are involved in inflammation, regeneration, angiogenesis, and immune responses and constitute a major fraction of MPs in circulation [16].
In angiogenesis, the PMPs that contain cytokines and chemokines cause rapid EC recruitment, chemotaxis, and migration [23]. PMPs induce the activity of bone-derived cells and neural stem cells and promote bone and neural regeneration [16,17].
Therefore, we investigated the effect of PMPs on the intra-articular homing of MSCs in the following study. First, using fluorescence colocalization, we found that a large number of SMSCs that homed to cartilage defects were in contact with PMPs (Fig. 4g); this result was quite similar to the finding by Teo et al. that 43% of arrested MSCs in blood vessels were in contact with neutrophil–platelet clusters [12]. These data implied that PMPs play a role in intra-articular MSC homing by some mechanism.
Then, we evaluated the effect of PMPs on the cartilage/subchondral bone adhesion, migration, and in vivo homing of MSCs. In contrast to the effect of PMPs on leukocyte recruitment, MSC adhesion showed no noticeable improvement after the cartilage/subchondral bone surface was precoated with PMPs. In sharp contrast, SMSCs preincubated with PMPs, even at a quite low PMP concentration, showed significantly enhanced adhesion (Fig. 4c). The migration assay yielded similar results (Fig. 4d). An in vivo study further confirmed the stimulatory effect of PMPs on MSC cartilage homing (Fig. 4e). In the articular cavity, two approaches for SMSC trafficking to cartilage lesions have been suggested: lateral migration from the periarticular margin and direct adhesion from SF [24].
The cells must migrate a relatively long distance from the periarticular margin before reaching the cartilage lesion. Therefore, direct cell adhesion from SF to the lesion surface is regarded as the principal method. Cell adhesion kinetics has revealed that MSCs firmly attach to cartilage within 10 h [25]. Interactions between cell surface integrins and injury-exposed matrix proteins, such as collagens and fibronectin, mediate this cell adhesion [22,26]. Some investigators have tried to increase this adhesion by priming MSCs with HA or pretreating these cells with differentiation induction media [27,28], and slight improvements have been achieved (<1.5-fold). Meanwhile, other studies tried to facilitate stem cell homing via magnetic guidance [29], a creative approach.
However, as the articular cavity has a complex surface, magnetic guidance is limited by its single orientation and the operational complexity. We found that PMPs can directly promote the migration and adhesion of MSCs to damaged cartilage surfaces. Moreover, autologous PMPs are easy to extract, simple to handle, and safe to use. No special equipment is required, so off-the-shelf products can be made for convenient use with stem cells.
We preliminarily explored the mechanisms underlying the effects of PMPs on SMSC homing. PMPs carry numerous adhesion receptors and chemokines derived from the parental platelets, and they can enhance leukocyte recruitment by stimulating circulating leukocytes or by interacting with ECs [30]. PMPs have been found to deposit chemokines, such as CCL5, on inflamed ECs while rolling on the EC surface [31].
In addition, PMPs induce chemokine and adhesion molecule expression by ECs and synoviocytes via surface-bound IL-1 [14,32]. However, PMPs directly activate circulating cells to induce systemic recruitment. Vasina et al. showed that PMPs can enhance monocyte adhesion and spreading on a fibronectin surface by increasing the expression of integrin α(M)β(2), the adhesion molecules CD14 and CD31, and the chemokine receptors CCR5 and CXCR4 [33]. Furthermore, PMPs can reportedly directly transfer functional surface receptors, for example, CXCR4, to circulating cells to promote homing [34,35].
Based on these mechanisms, we first measured the expression of homing-related genes in SMSCs after incubation with PMPs and identified several upregulated genes by RNA-seq. However, we blocked six major PDAFs [36] to evaluate the effect of PDAFs transferred directly from PMPs to the SMSC membrane since we found numerous PMPs adhered to the surface of SMSCs, covering the cells and fusing with the membrane. We discovered blocking these PDAFs impaired the homing capacity of SMSC-PMP complexes. These results indicate that PMPs might reinforce SMSC homing by multiple mechanisms, including by enhancing the homing capacity of SMSCs and by serving as an “anchor” or “bridge” to directly mediate the binding of SMSCs to the lesion surface.
However, it is not yet clear how these mechanisms synergize or which mechanism or molecule plays the most critical role in the SMSC-PMP complex homing process. Further studies are required to elucidate more detailed mechanisms.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by grants from the National Natural Science Foundation of China (81572150) and the Science and Technology Program of Hunan Province (2017SK2063).