
Abstract
Domestic cats suffer from a range of inherited genetic diseases, many of which display similarities with equivalent human conditions. Developing cellular models for these inherited diseases would enable drug discovery, benefiting feline health and welfare as well as enhancing the potential of cats as relevant animal models for translation to human medicine. Advances in our understanding of these diseases at the cellular level have come from the use of induced pluripotent stem cells (iPSCs). iPSCs can differentiate into virtually any cell type and can be derived from adult somatic cells, therefore overcoming the ethical implications of destroying embryos to obtain embryonic stem cells. No studies, however, report the generation of iPSCs from domestic cats [feline iPSCs (fiPSCs)]. Feline adipose-derived fibroblasts were infected with amphotropic retrovirus containing the coding sequences for human Oct4, Sox2, Klf4, cMyc, and Nanog. Isolated iPSC clones were expanded on inactivated mouse embryonic fibroblasts in the presence of feline leukemia inhibitory factor (fLIF). Retroviral delivery of human pluripotent genes gave rise to putative fiPSC colonies within 5–7 days. These iPS-like cells required fetal bovine serum and fLIF for maintenance. Colonies were domed with refractile edges, similar to mouse iPSCs. Immunocytochemistry demonstrated positive staining for stem cell markers: alkaline phosphatase, Oct4, Sox2, Nanog, and SSEA1. Cells were negative for SSEA4. Expression of endogenous feline Nanog was confirmed by quantitative polymerase chain reaction. The cells were able to differentiate in vitro into cells representative of the three germ layers. These results confirm the first generation of induced pluripotent stem cells from domestic cats. These cells will provide valuable models to study genetic diseases and explore novel therapeutic strategies.
Introduction
Inducing pluripotency is a process where differentiated cells are reprogrammed into a pluripotent or embryonic stem cell-like state [1]. These induced pluripotent stem cells (iPSCs) are then able to differentiate into derivatives of all three germ layers, generating great promise for their use in regenerative medicine and disease modeling, while avoiding the ethical implications of using embryonic stem cells (ESCs) [2,3]. They can also be derived from patients with disease causing genetic mutations, allowing patient-specific iPSC lines to be developed for in vitro drug testing. For example, iPSC lines harboring mutations known to cause hypertrophic cardiomyopathy (HCM) effectively recapitulate the disease at the single-cell level, allowing discovery of novel drug targets for clinical therapy [4]. iPSC-derived cardiomyocytes have further been shown to highlight potential drug side-effects in specific patients and predict drug response [5]. In addition, the consistency and ability to control for other factors make iPSC disease models a rapid drug discovery system [6]. Recent discoveries in gene editing mean that iPSC lines can be mutated, providing a powerful tool for analyzing disease-causing mutations while controlling for genetic background [7].
iPSCs were first generated from mouse cells and subsequently from human cells [3,8,9]. Since then research focused on optimizing human iPSC generation and culture has allowed the whole process to be xeno-free and footprint-free (without inserted genetic sequences), providing a platform for clinical research [10]. Studies deriving iPSCs from other species have shown that the process is applicable across a wide range of animals. For example, multiple groups have derived iPSCs from dogs, horses, sheep, pigs, goats, and other wild species [11 –18]. With respect to felids, there are two studies describing the generation of iPSCs from wild species [19,20]. Currently no studies have described the generation of iPSCs from domestic cats, an important veterinary species. Cats also provide a spontaneous large animal model for important human diseases, including diabetes, cardiac disease, as well as a wide range of genetic conditions, allowing a one health approach to disease [21 –24]. For example, HCM in cats closely resembles human HCM at the genetic, clinical, and pathological levels [25]. Interestingly a causative mutation identified in cats is also present in a human family [26]. HCM has a high prevalence in the feline population, affecting around 15% of all cats and increasing to almost 30% in those older than 9 years of age [27]. Given the importance and similarities of disease in both humans and cats, discovering novel drug targets would be beneficial to the health of both species. Drug discovery utilizing feline iPSCs (fiPSCs) could be translated to the feline clinic, where benefits can be assessed more rapidly due to the high prevalence, rapid disease progression, and reduced regulation compared to human clinical trials.
iPSCs derived from species other than mouse or human remain cultured under conditions that are not fully optimized or defined. Many still rely on original methods of retroviral insertion of human genes, use of undefined animal-derived media and maintenance on inactivated mouse embryonic fibroblasts (iMEFs) [15,28]. Much of what is known about pluripotent stem cells from diverse species is based on knowledge of embryonic stem cell culture. In cats, there are no studies deriving bona fide embryonic stem cells. There are two reports of embryonic stem-like cells, however, these could not be maintained under the culture conditions indefinitely, raising the possibility that either the cells were not true ESCs or that the culture conditions were not able to sustain a pluripotent state [29,30].
The development of a method to derive iPSCs from feline somatic cells would advance feline genetic and embryological research, which is currently hampered also by the lack of ESCs. These methods would provide researchers with a platform to further optimize iPSC culture from a novel species and realize the same potential as for human iPSCs.
Materials and Methods
Fibroblast cell culture
Feline adipose tissue-derived fibroblasts, isolated as described elsewhere [31], had been cryopreserved in the liquid phase of nitrogen at passage 0 (P0) to P1 before commencement of this project as approved by the Ethics and Welfare Committee (Approval No. URN 2012 1192) at the Royal Veterinary College (RVC). Cells were thawed rapidly in a 37°C water bath, then resuspended in media consisting of Dulbecco's modified Eagle's medium (DMEM; Thermo Fisher), 10% v/v fetal bovine serum (FBS; Thermo Fisher), 1% v/v
Feeder cell preparation
Proliferating MEFs were purchased at P3 (EmbryoMax; Sigma-Aldrich). The cells were cultured in the same media as for feline fibroblasts but without bFGF. MEFs were thawed onto 0.2% gelatin (Sigma-Aldrich)-coated 75 cm2 flasks at a density of 1.3 × 104 cells cm−2 and passaged 1:3 at 70%–80% confluence. At passage 5, MEFs were mitotically inactivated with 10 μg mL−1 mitomycin C (Stem Cell Technologies) for 2 h and cryopreserved in Cell Banker 2 (AMS Bio) at a concentration of 1 × 106 cells mL−1.
Retroviral production and transfection
Moloney murine leukemia virus (MMLV)-based retroviral plasmids (pMXs) containing the coding sequences for human Oct4 (17217; Addgene), Sox2 (17218; Addgene), Klf4 (17219; Addgene), c-Myc (17220; Addgene), and Nanog (18115; Addgene) were purchased from Addgene. A GFP containing plasmid (Cell Biolabs) was used to assess for transfection efficiency. For production of MMLV virus, platinum-A packaging cells (Cambridge Bioscience), which express the gag, pol, and env genes, were plated 24 h before transfection at a density of 1 × 105 cells cm−2 in 60 mm cell culture dishes, one dish per retroviral vector plasmid. Next, 6 μg of each of the vectors was transfected to platinum-A cells separately using Lipofectamine 2000 (Thermo Fisher) according to the manufacturers' instruction. To optimize transfection efficiency, feline fibroblasts were transfected with GFP vector at a range of seeding densities (500–1 × 104 cells cm−2), cultured for 24, 48, or 72 h, and in media with and without bFGF 24 h. Following this optimization, fibroblasts were plated 24 h before transfection at a density of 500 cells cm−2 in 100 mm cell culture dishes, in media containing bFGF. Supernatants containing virus particles were collected from the packaging cells after 48 h, filtered through a 0.45 μm filter, and supplemented with polybrene (8 μg mL−1, Sigma-Aldrich) and 10 ng mL−1 bFGF. The five transcription factor virus supernatants were pooled in equal volumes and added to the fibroblasts. We did not quantify viral titer before infection since storage of MMLV leads to reduced viral titer making this impractical. Instead, we used a parallel GFP experiment to monitor that viral production and cell infection were successful. The procedure was repeated after removal of the previous viral supernatant 24 h later for a total of two rounds of transfection.
Generation and culture of iPSCs
At 24 h following the second retroviral infection, iMEFs were added to the plate of feline transfection fibroblasts at a density of 2 × 104 cm−2 in fibroblast media. The following day, the media was changed to fiPSC media, which consisted of DMEM-GlutaMAX, 20% v/v ESC-qualified FBS (Thermo Fisher), 1% v/v nonessential amino acids (Thermo Fisher), 0.1 mM 2-mercaptoethanol (Thermo Fisher), and 10 ng mL−1 feline leukemia inhibitory factor (fLIF; Kingfisher Biotech). Colonies were manually picked based on ESC-like morphology at 7–10 days using a dissecting microscope and P-20 pipette tip. Colonies were placed into a 96-well plate containing 20 μL of 0.25% trypsin-EDTA (Thermo Fisher), incubated at room temperature for 5 min and then mixed by pipetting to obtain single cells. This cell suspension was then transferred to one well of a 24-well plate, which had been seeded with iMEFs 24 h previously. Individual colonies were treated separately to obtain clonal cell lines. Hereafter, fiPSCs were passaged as single cells using 0.25% trypsin-EDTA at a 1:6–1:10 ratio every 2–3 days. Cells for cryopreservation were resuspended in CellBanker 2 media, transferred to −80°C in a Mr Frosty freezing container (Nalgene), then transferred to liquid nitrogen storage after 24 h.
Embryoid body formation and differentiation
To form embryoid bodies (EBs) fiPSCs were treated with 0.25% trypsin-EDTA for 5 min and cells were transferred to CELLSTAR cell repellent surface 24-well plates (Greiner Bio-One) using a 1:1 split ratio from a confluent 6-well plate. Media was changed to iPSC media without fLIF. After 4–5 days, EBs were collected and redistributed onto 0.2% gelatin-coated 48-well plates and allowed to differentiate for 3 weeks before antibody staining.
Alkaline phosphatase staining and immunocytochemistry
Alkaline phosphatase activity of fiPSC at passages 2, 15, and 22 was determined using VECTOR Red Alkaline Phosphatase (Red AP) Substrate Kit (Vector Laboratories). Cells for immunocytochemistry (ICC) at passages 15–22 were plated in 48-well plates for all staining procedures and allowed to adhere for at least 24 h. Immunostaining procedures were performed at room temperature following removal of culture media. Cells were washed once with Dulbecco's phosphate-buffered saline (DPBS) and fixed in 4% paraformaldehyde (Sigma-Aldrich) for 15 min. Fixative was then removed, and the cells were permeabilized using 0.4% Triton X-100 (Sigma-Aldrich) for 15 min. Cells were washed with 0.05% Tween 20/DPBS (Sigma-Aldrich) and blocked with 10% goat serum (Abcam) for 1 h. Cells were washed again and incubated with the primary antibody for 1.5 h. Following three washes cells were incubated with the secondary antibody for 1 h. Cells were then washed again three times and nuclei counterstained using NucBlue ReadyProbe according to the manufacturers' instructions (Thermo Fisher). The primary and secondary antibodies used for ICC are listed in Table 1. Images were captured using an EVOS FL color imaging system (Thermo Fisher).
A List of the Primary and Secondary Antibodies Used in This Study
Flow cytometry
To determine the GFP-positive percentage, cells were detached from culture vessels using 0.25% trypsin-EDTA 48 h second posttransfection. Cells were pelleted (400 g for 5 min) and resuspended in FACSFlow (BD Biosciences). Samples were acquired in polystyrene FACS tubes on a BD FACS Calibur flow cytometer (BD Biosciences). The instrument was calibrated using CaliBRITE 3 color FACS CompBeads (BD Biosciences) before acquiring and analyzing each set of samples using CellQuest Pro software (BD Biosciences). Noninfected cells were acquired to set the forward and side scatter parameters to center the cell population on the scatter plot. Fluorescence intensity was adjusted to set the noninfected cells within 100–101 on the log scale axis. Cells were then acquired with an event count set to a total of 1 × 104 events. Data were analyzed using FlowJo software (version 10; FlowJo, LLC).
Reverse transcription and quantitative real-time polymerase chain reaction
RNA was extracted from noninfected fibroblasts, infected fibroblasts, and iPSCs at passages 1–22 using the GenElute™ Mammalian Total RNA Miniprep Kit (Sigma-Aldrich). Genomic DNA contamination was removed using the TURBO DNA-free kit (Thermo Fisher). Then, 1 μg of RNA was reverse transcribed using the SensiFAST complementary DNA (cDNA) Synthesis Kit (Bioline). Real-time quantitative polymerase chain reaction (RT-qPCR) was performed using 2 μL of cDNA template, nonreverse transcribed control or water using the SensiMix SYBR Green No-ROX Kit (Bioline) and run on a CFX96 RT-PCR Detection System (Bio-Rad) with the following conditions: 95°C for 10 min, then 44 cycles of 95°C for 15 s, 60°C for 15 s, and 72°C for 15 s. A final ramp of 60°C–90°C at 0.5°C increments was used for melt curve analysis. Five housekeeping genes were first analyzed to ascertain stability across different cell types (noninfected fibroblasts, pooled infected fibroblasts, and three iPSC lines). RPS7 was scored as the most stable using RefFinder, which compares analyses from 4 computational programs (Delta CT, BestKeeper, NormFinder, and geNorm; Supplementary Fig. S1). This was then used to normalize qPCR data for other genes analyzed. The primer sequences are given in Table 2.
Primer Sequences Used in This Study
f, feline gene; h, human gene.
Karyotyping
fiPSCs were incubated for 18 h at 37°C in standard cell culture conditions in media containing 0.13 μg mL−1 colcemid (KaryoMAX; Thermo Fisher). Next cells were dissociated using 0.25% trypsin-EDTA, pelleted (300 g for 3 min), and resuspended by adding 4 mL of 0.075 M KCL dropwise to the cell pellet and then incubated at 37°C for 25 min. Then, 10 drops of fixative (3:1 methanol/acetic acid) were added, and cells were mixed by inversion and incubated for 10 min at room temperature. Cells were then pelleted as before, resuspended in 4 mL fixative and incubated for 30 min at room temperature. Following a final centrifugation, cells were resuspended in 2 mL of fixative and chromosomes visualized using DAPI staining. At least 20 metaphase spreads were counted, with a haploid number of 38 considered normal.
Statistical analysis
The SPSS software package (version 23 for Mac; IBM) was used for statistical analysis. Data were assessed for normality using histogram analysis. All data are presented as the mean ± standard error of the mean of three individual clonal iPSC lines derived from the same reprogramming experiment, with samples run at least in triplicate. Fibroblast data are obtained from three independent analyses of the same cell line or transfected pool that generated the iPSC lines. Comparisons between two independent samples were performed using Student's two-tailed t-test and between three or more groups using one-way analysis of variance with post hoc Tukey's analysis. A P value of <0.05 was considered significant.
Results
Amphotropic retrovirus infects feline cells with high efficiency
Transduction efficiency of feline fibroblasts was assessed by transfection with a GFP containing amphotropic MMLV. After a number of optimizations, the transfection efficiency could be increased from an initial 10%–20% to >99% (Fig. 1a, b). Seeding the cells at low density (500 cells cm−2) in media containing bFGF 24 h before transfection led to the highest GFP expression.

Efficiency of amphotropic MMLV-retrovirus transfection of feline fibroblasts. Fluorescence microscopy showed that feline fibroblasts transfected with viral supernatant with a vector containing GFP show high expression at 48 h posttransfection
Infected feline fibroblasts express human pluripotent genes
Once we confirmed efficient transduction, feline fibroblasts were transduced with pooled viral supernatant containing coding sequences for five human pluripotency associated genes (OSKMN). Immunocytochemistry demonstrated expression of the pluripotent factors at the protein level with a nuclear localization as expected (Fig. 2a). RT-qPCR confirmed that the feline cells had high gene expression levels of integrated human pluripotent factors (Fig. 2b).

Expression of viral transgenes in transfected feline fibroblasts. Immunocytochemistry shows positive nuclear staining for Oct4, Sox2, Klf4, cMyc, and Nanog in infected fibroblasts versus noninfected fibroblasts
fiPSCs require fLIF for maintenance
Following a number of different derivation conditions, we ascertained a protocol for fiPSC induction. A timeline for this is shown in Fig. 3a. Feline fibroblasts were seeded 24 h before transduction at low density in media containing bFGF. Following two rounds of retroviral infection 24 h apart, iMEFs are added directly to the plate of infected feline fibroblasts. Colonies are then observed from days 3 to 7 (Fig. 3b–e). The fiPSC colonies were observed to be initially relatively two dimensional, peripherally well demarcated, and with cells exhibiting a high nuclear to cytoplasm ratio, more typical of human embryonic stem cell morphology (Fig. 3c). Following the first passage, colonies formed a domed morphology with a phase bright surface and refractile edges more typical of mouse ESC morphology (Fig. 3d, e). iPSCs were able to be propagated using a 1:6–1:10 split ratio every 2–3 days for over 20 passages.
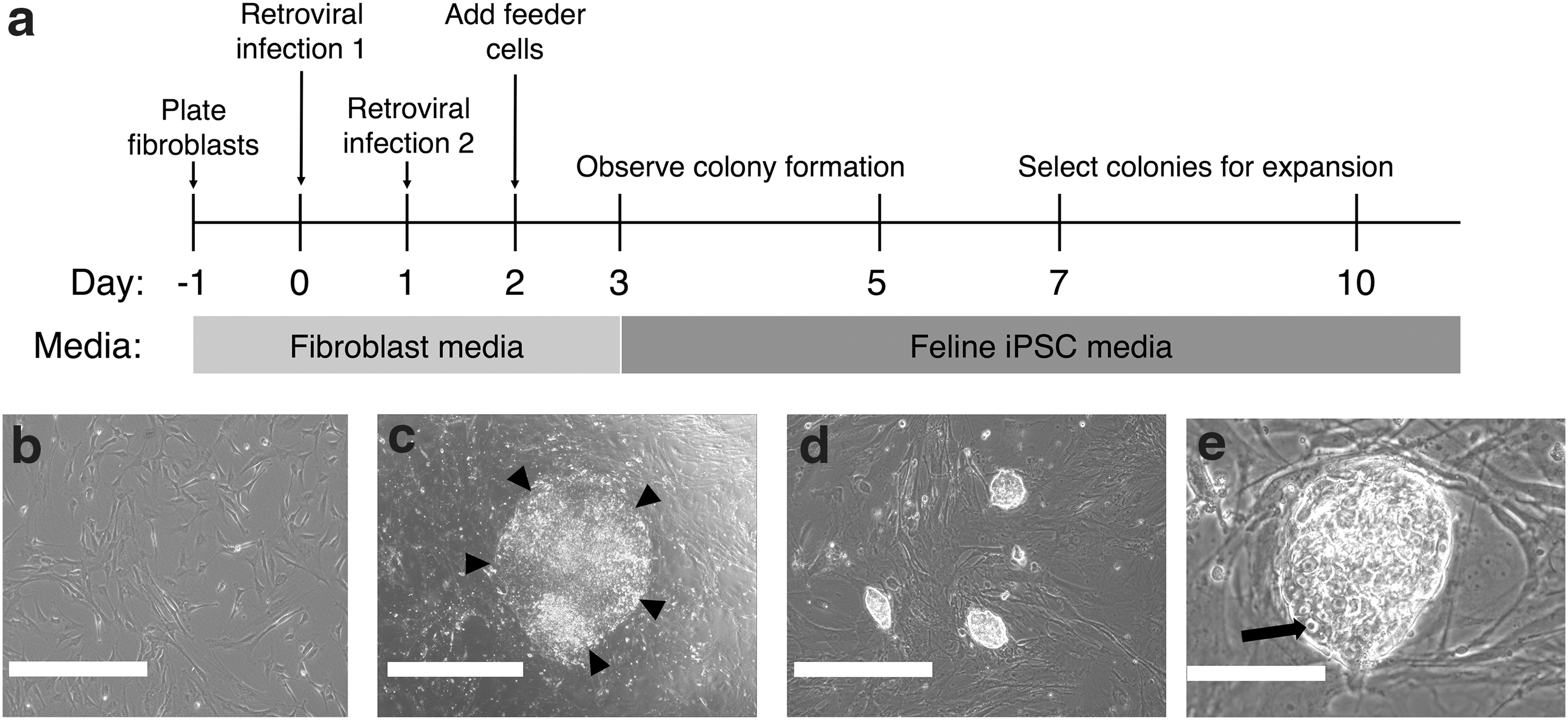
fiPSC generation and timeline of events during iPSC generation
fiPSC were further observed to be dependent on FBS and fLIF for maintenance. Culture of fiPSC in media containing knock-out serum replacer (KSR) or removing fLIF led to loss of colony morphology within 3–5 days (data not shown). Media containing bFGF with or without leukemia inhibitory factor (LIF) allowed small colonies to form, but they did not progress beyond 10–20 cells and could not be expanded (data not shown). In addition, murine LIF was unable to maintain alkaline phosphatase expression of fiPSCs (Fig. 4a, b).
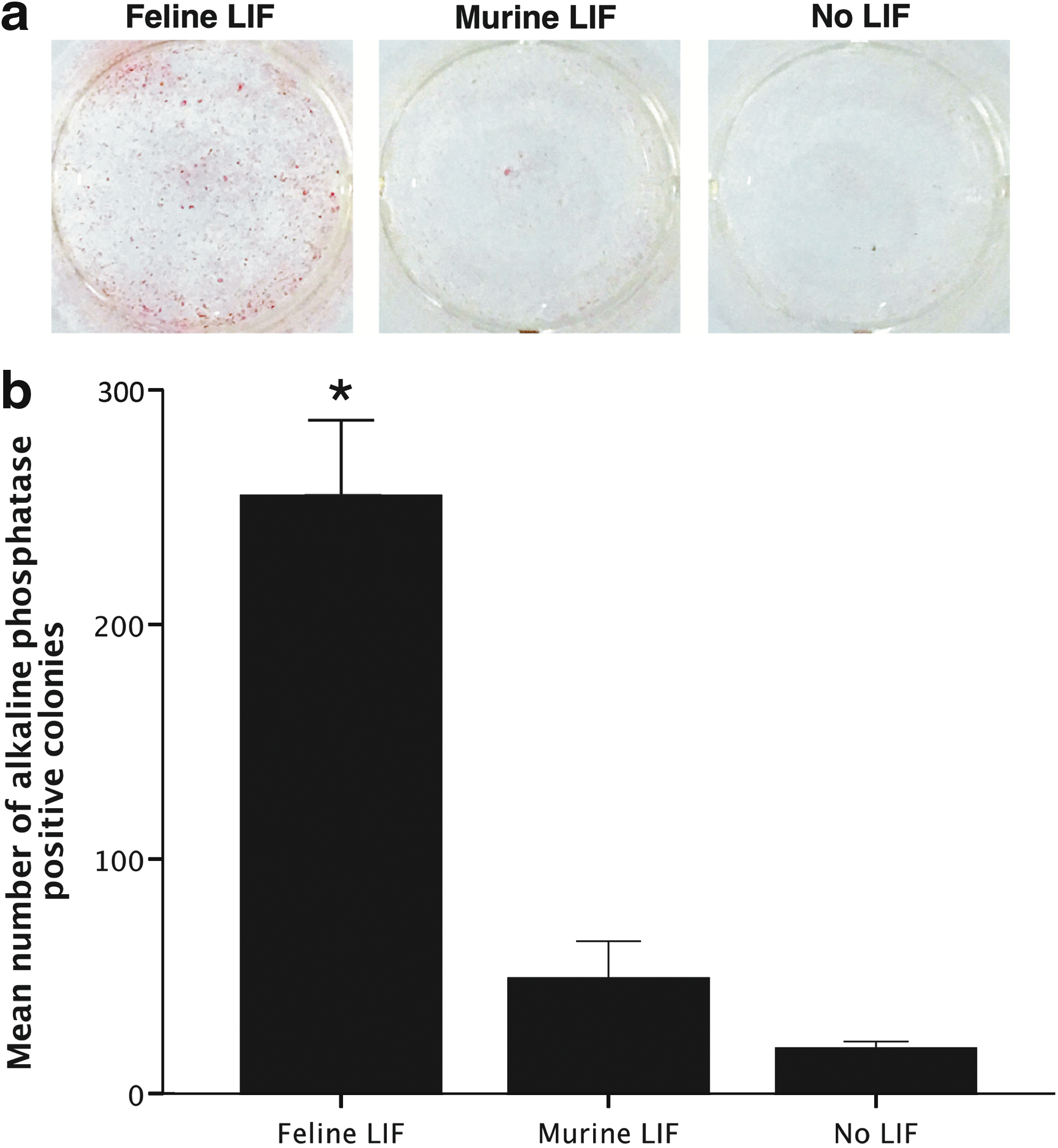
Dependency of fiPSCs on fLIF. When cells are exposed to fLIF, they are positive for alkaline phosphatase staining, but lose expression when fLIF is changed to murine LIF or no LIF
fiPSCs express key pluripotent markers and reactivate Nanog expression
fiPSCs were examined for markers of pluripotent stem cells. Colonies were positive for alkaline phosphatase (Fig. 5a). In three individual clonal fiPSC lines, there was high expression of feline Nanog, which was not detectable in the original fibroblasts (Fig. 5b). In addition, cells were positive for the surface marker SSEA1 (Fig. 5c–e), negative for SSEA4 (Fig. 5f–h), and positive for nuclear transcription factors Oct4 (Fig. 5i–k), Sox2 (Fig. 5l–n) and Nanog (Fig. 5o–q). fiPSC could be passaged >20 times without losing proliferative potential, maintained the above phenotype, and had a normal karyotype (Fig. 6).
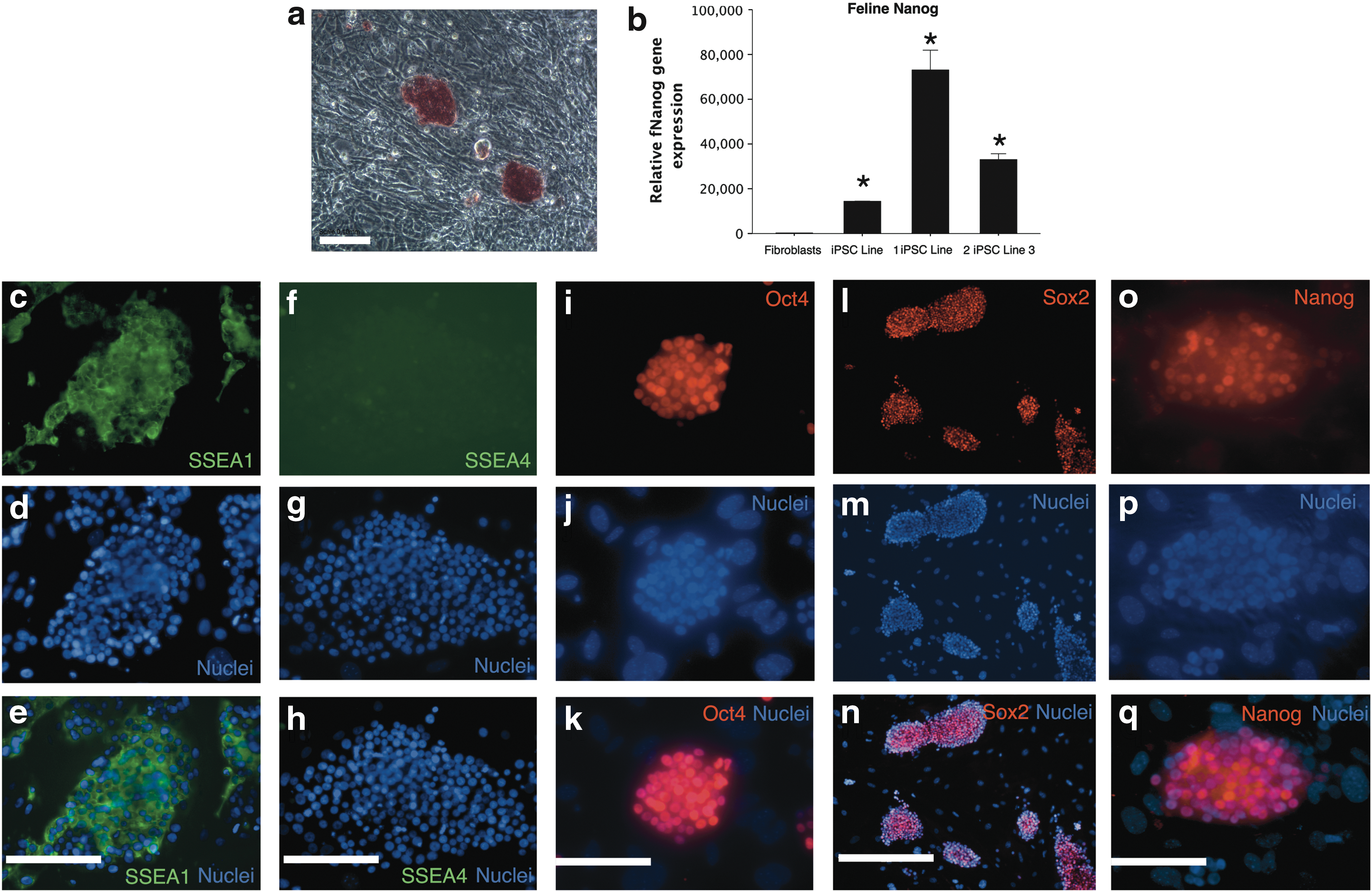
Characterization of fiPSCs. fiPSC colonies express alkaline phosphatase (red staining)

Karyotype analysis of fiPSCs at passage 22 reveals a normal haploid chromosome number of 38. Results are representative of 20 metaphase spreads. Scale bar = 100 μm. Color images are available online.
fiPSCs are capable of in vitro differentiation into derivatives of the three germ layers
We next sought to assess whether the fiPSC had the potential to differentiate into cells representative of the 3 germ layers. fiPSCs seeded onto a low attachment surface in the absence of fLIF formed EBs (Fig. 7a). Outgrowth cells from EBs attached to gelatin-coated plates displayed multiple morphologies (Fig. 7b). Immunostaining revealed that subsets of these cells expressed the mesodermal markers smooth muscle actin (Fig. 7d) and α-sarcomeric actin (Fig. 7f), ectodermal marker βIII-tubulin (Fig. 7h), and endodermal marker SOX17 (Fig. 7j). None of these markers was expressed in the undifferentiated iPSCs (Fig. 7c, e, g, i). Together these results indicated the in vitro pluripotency of fiPSCs.
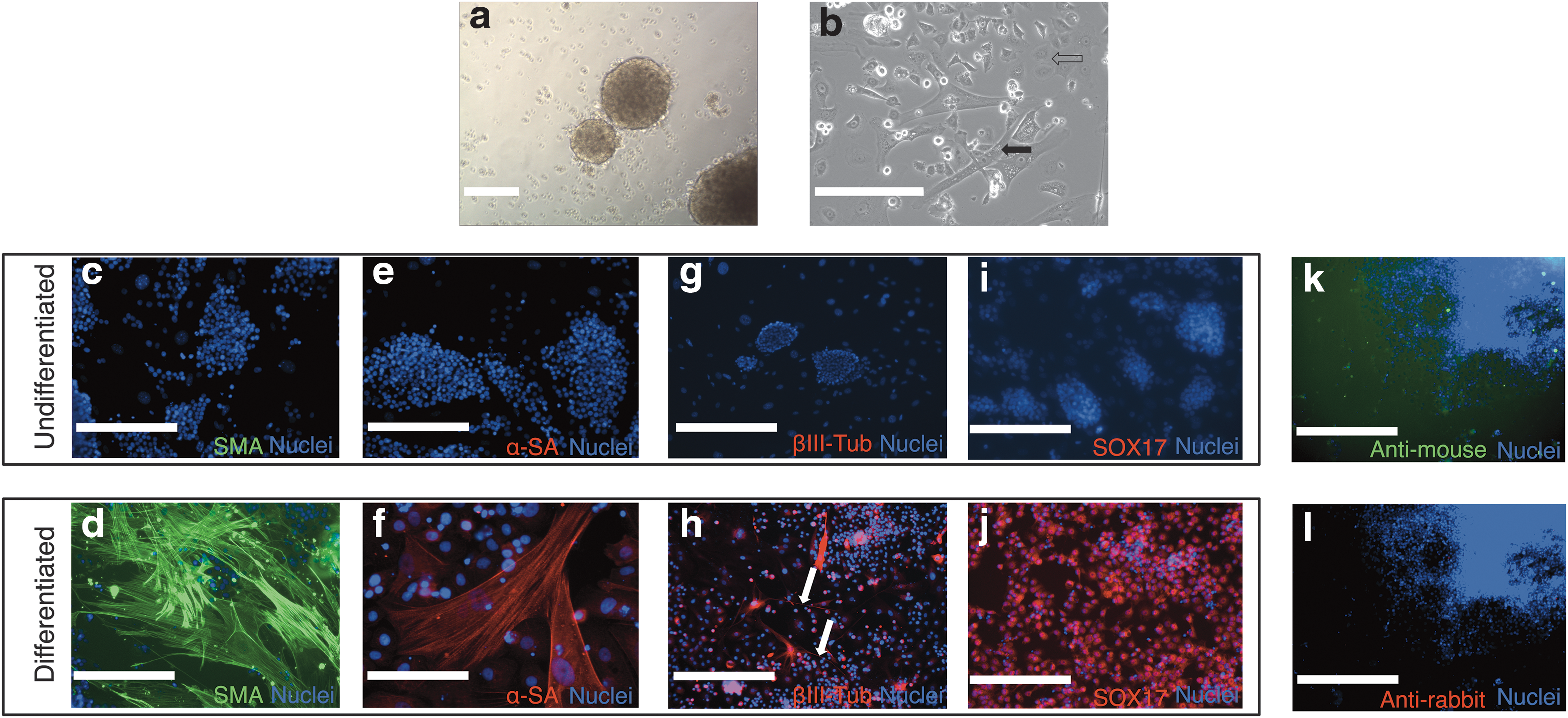
Multilineage differentiation of fiPSCs. When grown in suspension culture plates, iPSCs form embryoid bodies
fiPSCs continue to express viral transgenes
fiPSCs showed expression of the viral transgenes in the infected fibroblasts and in three iPSC lines at passage 7 and passage 22 (Fig. 8). Expression levels for Oct4 and Sox2 were higher in the iPSC lines compared to the infected fibroblasts (P < 0.05). Expression of cMyc was higher in passage 7 iPSCs compared to infected fibroblasts (P < 0.05). Nanog expression was lower in iPSCs compared with the infected pool, however, this difference was not significant (P = 0.252 for passage 7 and P = 0.720 for passage 22). There was no difference in expression levels between passage 7 and passage 22 iPSCs for any of the transgenes (P = 0.993 for Oct4, P = 0.997 for Sox2, P = 0.223 for Klf4, P = 1.00 for cMyc, and P = 0.881 for Nanog).
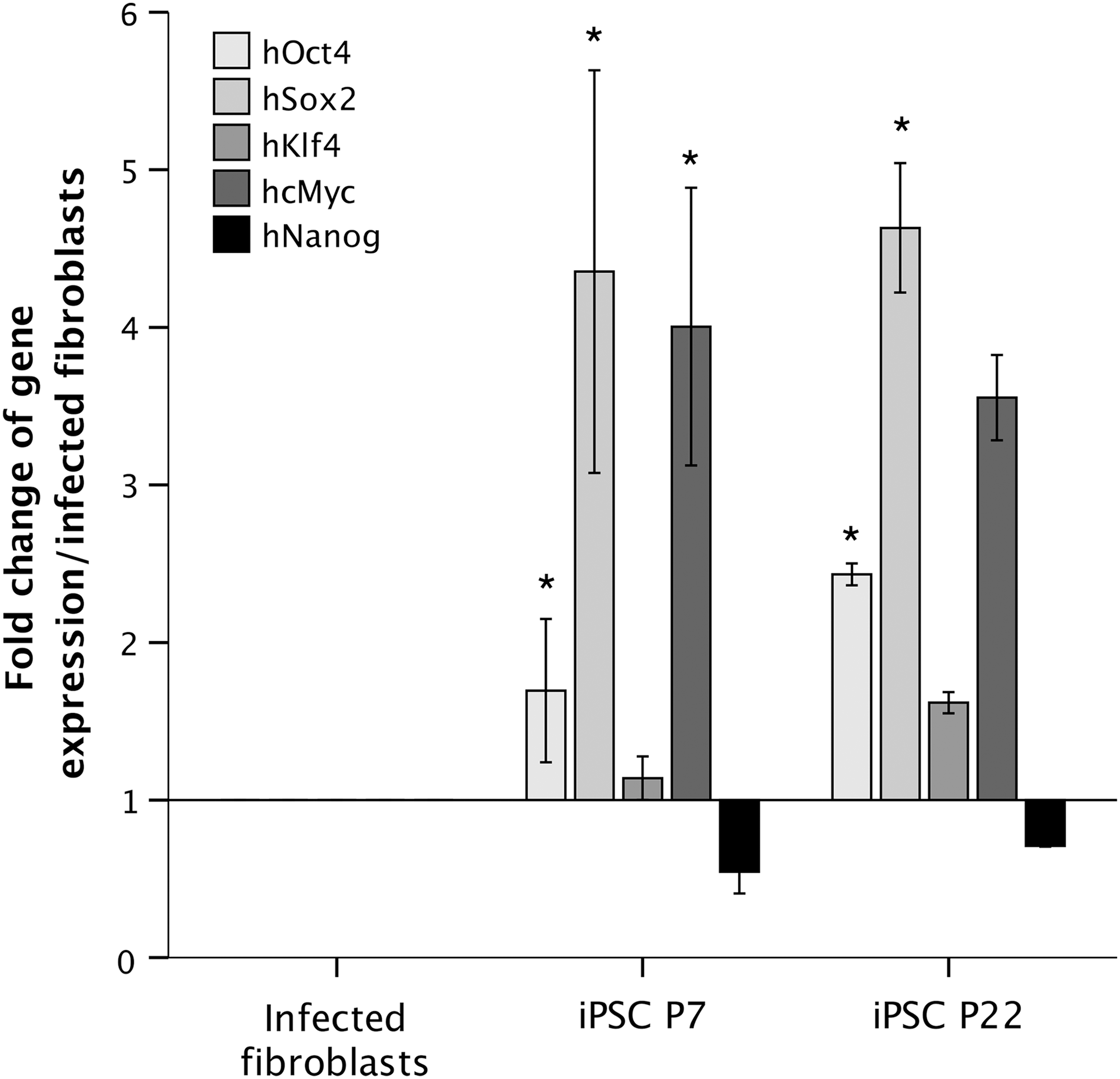
RT-qPCR analysis of transgene expression in infected fibroblasts and iPSCs at passage 7 and 22. Relative gene expression is shown as the fold change when compared with the pool of infected fibroblasts. Origin = 1. Expression levels for Oct4 and Sox2 were higher in the iPSC lines compared to the infected fibroblasts (P < 0.05). Expression of cMyc was higher in passage 7 iPSCs compared to infected fibroblasts (P < 0.05). There was no difference in expression levels between passage 7 and passage 22 iPSCs for any of the transgenes (P = 0.993 for Oct4, P = 0.997 for Sox2, P = 0.223 for Klf4, P = 1.00 for cMyc, and P = 0.881 for Nanog). Error bars represent the mean ± SEM of three independent experiments. *P < 0.05 in one-way ANOVA.
Discussion
In this study, we describe the first generation of iPSCs from the domestic cat. We present a number of optimization steps required for successful iPSC generation and anticipate that these results will advance feline medical research similar to the advances made in human medical research, particularly for genetic diseases [32,33]. These new technologies have been widely applied in human neurological and cardiac diseases, where patient-specific iPSC lines act as powerful tools as in vitro disease models [34 –36]. These include new gene therapy strategies for iPSC-derived cellular models of HCM [37]. Our work provides the initial steps to realizing these advancements for feline patients and enhances the potential of cats as large animal models for analogous human disease where prospective therapies can be rapidly translated from the feline to the human clinic.
A major barrier to inducing pluripotency is the efficient delivery and expression of pluripotent genes (Oct4, Sox2, Klf4, cMyc, and Nanog; OSKMN) [38]. There are few reports on viral delivery of genes in domestic cat cells [39,40]. Initially, we trialed fiPSC generation using a Sendai virus-based delivery of OSKM (Cytotune 2.0; Thermo Fisher), however, although the virus was able to infect feline cells, iPSC colonies never appeared despite several attempts with various modifications (data not shown). We then selected an MMLV-based retrovirus delivery system as this has been previously reported for the generation of iPSCs from wild feline species [19,20]. In addition, this allowed us to add Nanog into the reprogramming factors, as this is required for iPSC generation in wild felids [20]. This system yielded low transfection efficiencies when replicating conditions used for mouse or human cells. We hypothesized that this was due to slow growth of feline fibroblasts in DMEM/10% FBS media since MMLV retrovirus is only able to integrate into cells that are undergoing mitosis [41]. To increase the proliferation of the fibroblasts, media was supplemented with bFGF [42] together with seeding cells 24 h before transfection and at low density to increase the multiplicity of infection. This led to a maximum GFP expression of about 99%. This is in line with retroviral-based delivery methods for iPSC generation in other species [8,11,20]. We further demonstrated that feline cells are able to express the transduced human genes at the messenger RNA (mRNA) and protein level.
It was also necessary to optimize the conditions for iPSC colony emergence. We observed mesenchymal-to-epithelial transition (MET) of fibroblasts within 2–3 days of viral transfection, which is thought to be one of the earliest stages in the reprogramming process [43]. Under conditions used for human iPSC generation (including KSR and bFGF), these epithelial-shaped cells reverted back to a mesenchymal morphology. We also observed that if MEFs were added after day 5, no colonies emerged. Since MET was occurring within a few days, it appeared that reprogramming in feline cells was following a similar timeline as for mouse iPSCs [44], which form quicker than human iPSCs [45]. We therefore added iMEFs to the transfected cells and included fLIF as early as 1 day after virus was removed which yielded colonies with typical ESC and iPSC morphology [46].
These fiPSCs expressed Oct4, Sox2, Nanog, and SSEA1 but not SSEA4 similar to mouse ESCs and iPSCs [47]. Further characterization of the fiPSCs showed that they were able to differentiate into cells representing the three germ layers and had a normal karyotype after >20 passages. Nanog, a marker of cellular reprogramming [48], was also activated.
Culture conditions for iPSCs from different species show considerable variation between publications, which makes determining the optimal conditions for a novel species challenging [15]. This difference may be due to the developmental stages represented by iPSCs [49]. For example, mouse iPSCs represent a “naive” type pluripotent stem cell, and have a domed and phase-bright appearance [50]. However, human iPSCs represent a “primed” pluripotent cell, similar to mouse epiblast cells, with flattened colony morphology [51]. Both types can be converted into the other type by adjusting culture conditions [52,53]. The naive type relies on LIF for maintenance of pluripotency and self-renewal, whereas the primed type is dependent on bFGF [54]. It is less clear where iPSCs derived from other species fit along this spectrum, with iPSCs derived from monkeys, pigs, horses, cows, goats, and dogs fitting both definitions [11,13,14,16,18, and 55–57]. With respect to our fiPSCs, they closely resemble the naive type, which is substantiated by their morphology and surface expression of SSEA1 and not SSEA4, as is the case for mouse iPSCs. Specifically, feline cells relied on fLIF, with murine LIF unable to sustain alkaline phosphatase expression, and this is likely due to the differences between feline and murine LIF protein and receptors [58].
Although we describe conditions for fiPSCs, there is potential for the system to be further optimized. For example, the use of FBS and iMEFs led to inconsistent colony morphology and behavior. Further optimization may identify other growth factors or molecules that would enable a more stable phenotype and facilitate feeder cell removal. The suboptimal culture conditions in our study may have led to the persistent transgene expression that we observed in the fiPSCs [59]. In human and mouse studies, transgene expression is expected to cease at around passage 10–15, which was not the case for the iPSCs in this study [60]. This is common issue with iPSCs derived from domestic animals and where expression of inserted factors has been directly measured the majority of reports demonstrate persistent transgene expression [15], as is also the case for wild felid iPSCs [19,20]. Although these cells would be useful for in vitro drug discovery, continued transgene expression creates an issue for clinical application since implantation of these cells may cause tumorigenesis [6], therefore, transgene-free fiPSCs would be a useful next step, and this has been the process for human iPSCs.
In conclusion, we sought to establish a platform to generate the first iPSCs from domestic cats with the expectation that these will advance feline medicine. Outcomes from their application to the understanding and management of disease are not limited to the cat since similarities of genetic pathologies between cats and humans will provide invaluable information for human conditions where similar clinical translation is more difficult and outcomes take longer.
Footnotes
Author Disclosure Statement
The authors declare no competing financial interests.
Funding Information
This study was funded by grants from the Beryl Evetts and Robert Luff Animal Welfare Trust, Boehringer Ingelheim Vetmedica GmbH and the Winn Feline Foundation (project no. W17-008).
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.