
Abstract
Tripartite motif (TRIM) proteins participate in numerous biological processes. They are the key players in immune system and are involved in the oncogenesis. Moreover, TRIMs are the highly conserved regulators of developmental pathways in both vertebrates and invertebrates. In particular, numerous data point to the participation of TRIMs in the determination of stem cell fate, as well as in the neurogenesis. TRIMs apply various mechanisms to perform their functions. Their common feature is the ability to ubiquitinate proteins mediated by the Really Interesting New Gene (RING) domain. Different C-terminal domains of TRIMs are involved in DNA and RNA binding, protein/protein interactions, and chromatin-mediated transcriptional regulation. Mutations and alterations of TRIM expression cause significant disturbances in the stem cells' self-renewal and neurogenesis, which result in the various pathologies of the nervous system (neurodegeneration, neuroinflammation, and malignant transformation). This review discusses the diverse molecular mechanisms of participation of TRIMs in stem cell maintenance and self-renewal as well as in neural differentiation processes and neuropathology.
Introduction
Tripartite motif (TRIM) proteins usually contain three common motifs at the N-terminus: the Really Interesting New Gene (RING) domain, one or two B-box domains, and the coiled-coil (CC) domain [1,2] (Fig. 1). The RING domain is composed of 40–60 amino acids and is responsible for E3 ubiquitin ligase activity, which can mediate protein conjugation either with the ubiquitin [2], or with the small ubiquitin-like molecule modifier (SUMO) [3], or with the neural precursor cell (NPC)-expressed developmentally downregulated 8 (NEDD8) ubiquitin-like protein [4]. The B-boxes promote TRIM self-association, as well as interactions with other proteins [2]. The CC domain is a hyperhelical structure, which is responsible for dimerization of TRIM proteins and higher-order TRIM oligomerization [2].
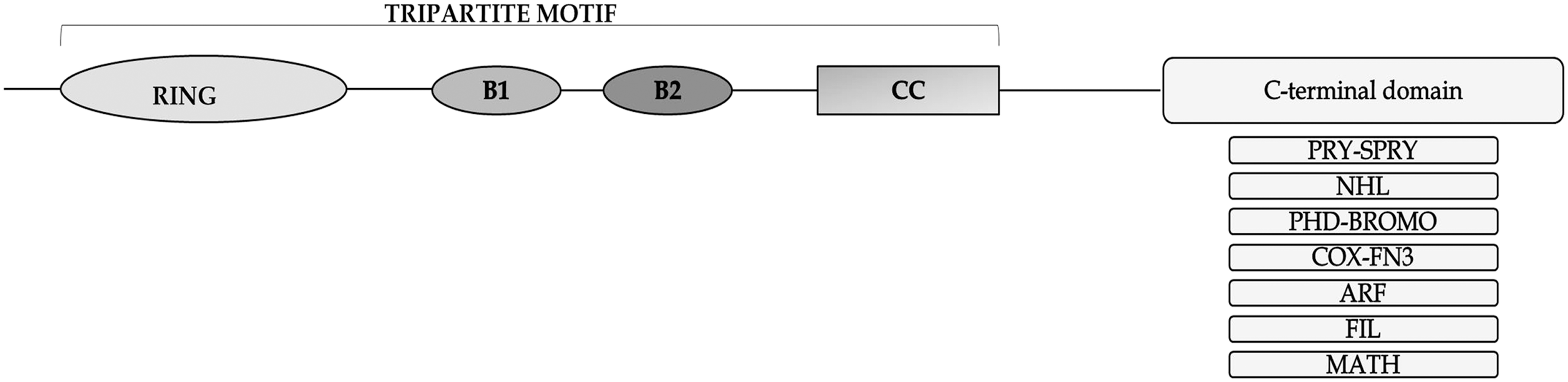
Schematic representation of the TRIM family protein domain structure. RING domain; B1, B-box 1; B2, B-box 2; CC, coiled-coil region. The most frequent C-terminal domains are also shown on the right side of the scheme. RING, Really Interesting New Gene; TRIM, tripartite motif.
The C-terminal part of TRIMs contains various domains, which are not conserved between all members of the TRIM family. For example, the PRYSPRY domain promotes protein/protein interactions [5]. The C-terminal subgroup one signature (COS) motif interacts with microtubules [6]. The fibronectin type III motif (FN3) contains b-sandwich structure and is detected in cell surface proteins, all of which are involved in a molecular recognition function [7]. The Plant Homeodomain (PHD)-Bromo domain is associated with chromatin-based transcriptional regulation [8].
The ADP-ribosylation factor-like (ARF) domain mediates GTP hydrolysis activity [9]. The NHL domain is involved in protein and RNA binding, while the filamin domain possessing an immunoglobulin-like structure takes part in messenger RNA (mRNA) regulation in TRIM-NHL proteins [10]. Finally, the meprin and tumor-necrosis factor receptor-associated factor homology (MATH) domain takes part in receptor binding and oligomerization [11] (Fig. 1). Depending on the presence of different C-terminal domains, TRIM proteins are divided into 11 groups [12].
The majority of TRIMs possess the ubiquitin ligase activity due to their RING domain, which is critical for many cellular functions, including the signal transduction and regulation of innate immunity [13]. Although TRIM family is characterized by TRIM, some TRIM-like members lack one or more of these domains (eg, the RING domain is missed in TRIM14 [14] and in TRIM16 [15]), but possess the corresponding C-terminal domains and can be assigned to one of the 11 TRIM groups [12].
Over the last few years, increasing evidence has suggested that members of the TRIM family play an important role in the stem cell differentiation and pluripotency, apoptosis and autophagy, regulation of transcription and cell cycle progression, signaling pathways, chromatin remodeling, as well as neurogenesis, and oncogenesis [16,17]. However, the particular interest to those proteins has been attracted due to their involvement in innate immune system [16,18 –20]. More than 70% of TRIMs are directly implicated in the innate and intrinsic immune response [21]. The antiviral strategies employed by TRIMs are divided into three broad categories: the modulation of innate immune sensing or signaling, direct restriction of viruses, and regulation of autophagy-mediated antiviral defense. In recent years, an increasing number of studies have indicated that many antiviral signaling pathways are TRIM regulated [20,22].
At the same time, TRIMs are also involved in many other key cellular processes. In particular, they play an essential role in stem cell functioning and differentiation, as well as in neurogenesis (Fig. 2). The latter topics that until now were paid less attention will be considered in the current review.
The established functions of TRIM family members.
Functions of TRIMs in Stem Cells
The participation of TRIMs in stem cell regulation just begins to attract the attention of researchers. Currently, at least 10 TRIMs are known to be involved in the maintenance of stem cells' pluripotency and in regulation of their differentiation (Table 1). Proteins belonging to the C-VI (TRIM24, TRIM28, and TRIM33, containing PHD-Bromo domain), C-VII (TRIM-NHL proteins: TRIM32 and TRIM71), and to the most representative C-IV (TRIM6, TRIM14, TRIM25, and TRIM26, containing a PRYSPRY domain) subfamilies [12] are actively involved in stem cell regulation.
Tripartite Motif Proteins in Stem Cells
CDKN1A, Cyclin-Dependent Kinase Inhibitor 1A; ERK, extracellular signal-regulated kinase; ERVs, endogenous retroviruses; ESC, embryonic stem cell; FGF, Fibroblast growth factor; HP1, heterochromatin protein 1; HSP, heat shock protein; iPSC, induced pluripotent stem cell; KRAB-ZNF, Krüppel-associated box domain zinc finger; mESC, mouse embryonic stem cell; miRNA, microRNA; PHF, PHD Finger Protein; SETDB1, SET domain bifurcated 1; SHCBP1, SHC SH2 domain-binding protein 1; STAT, Signal Transducer and Activator of Transcription; TRIM, tripartite motif.
TRIM proteins, carrying PRYSPRY domain (C-IV subfamily)
C-IV subfamily consists of thirty-eight members, most of which are implicated in the antiviral response. At the same time, some data demonstrate the active involvement of several TRIMs of this subfamily in the maintenance of pluripotency and stem cells' self-renewal. The RING domain, common to most TRIM proteins, also takes part in the realization of these functions. For example, in mouse embryonic stem cells (mESCs), TRIM6 ubiquitinates c-MYC and weakens its transcriptional activity. However, overexpression of TRIM6 leads to increase of the expression of NANOG, the marker of the pluripotency of ESCs. Knockdown of TRIM6 induces differentiation of ESCs, suggesting that TRIM6 participates in maintenance of the pluripotency upstream of NANOG [23] (Table 1).
Another member of the C-IV subfamily, TRIM14, lacks the RING domain, but is able to promote the differentiation of mESCs into the embryoid bodies [24]. In addition, TRIM14 overexpression in mESCs does not influence endodermal, but increases mesodermal, and decreases ectodermal differentiation in these cells [25].
Analyses of mESCs have revealed several RNA-binding proteins, which are expressed significantly stronger in ESCs as compared with differentiated cells, implying their role in the maintenance of stem cells. Among them are TRIM25 and TRIM71 [26]. Nevertheless, unlike in TRIM71, where NHL domain supports RNA-binding, in TRIM25 the PRYSPRY domain is responsible for binding to RNA [27]. TRIM25 takes part in the degradation of precursor of let-7 microRNA (miRNA), the regulator of cell differentiation, as the RNA-specific cofactor for Lin28s/TuT4-mediated uridylation [28].
In 2018, Zhao et al. [29] described a new mechanism of resolving replication stress in mESCs. The mESC-specific Filia-Floped complex overcomes stalled replication forks by recruiting TRIM25. Next, TRIM25 binds and ubiquitinates the Bloom syndrome helicase (BLM), a key regulator of stalled replication forks. In this way, TRIM25 ensures genomic stability of mESCs [29]. Another member of this subfamily, TRIM26, is required to generate induced pluripotent stem cells (iPSCs) during somatic cell reprogramming. This protein ubiquitinates by its RING domain the PHD Finger Protein (PHF)20, promoting degradation of PHF20, and prevents the activation of Oct4 expression thus leading to not fully reprogrammed iPSCs [30].
C-V subfamily of TRIM proteins
The C-V subfamily of TRIMs does not contain the known domains at the C-terminus. TRIM8 is the only member of C-V subfamily that was found to participate in stem cell maintenance (Table 1). TRIM8 is detected in undifferentiated mESCs, whereas differentiated cells do not express this protein [31]. Okumura et al. [31] demonstrated that TRIM8 binds to heat shock protein (HSP) 90β, which, in turn, regulates the translocation of phosphorylated Signal Transducer and Activator of Transcription (STAT)3 into the nucleus. These processes lead to the downregulation of transcription of NANOG in ESCs. Interestingly, RING domain is not involved in these functions of TRIM8 [31].
TRIM proteins, containing PHD-Bromo domain (C-VI subfamily)
TRIM24, TRIM28, and TRIM33 participate in the regulation of the balance between maintenance of pluripotency and differentiation of the stem cells [32 –37]. It was shown that PHD and Bromodomain of TRIM24 jointly recognize both the unmethylated lysine 4 and acetylated lysine 23 or 18 of histone H3 [8]. TRIM24 binds to multiple enhancers of genes encoding OCT4, SOX2, and NANOG (the transcription factors essential to maintain the pluripotent embryonic stem cell phenotype), activates them and thus maintains the pluripotency of mESCs. At the same time, TRIM24 decreases the expression of developmental genes, but activates the cell cycle genes in mESCs [32].
In addition, TRIM24 binds to P53, ubiqutinates it by the RING domain, which promotes P53 degradation and thus prevents spontaneous ESCs apoptosis [38], as well as differentiation [33]. It was shown that TRIM24 and HDM2 [also known as Mouse Double-Minute 2 Homolog (MDM2)], both possessing E3 ubiquitin ligase activity, are the negative regulators of P53 in human ESCs (hESCs). They promote P53 degradation, thereby preventing differentiation. In hESCs, P53 is deacetylated and maintained at the low level in the nucleus. Being acetylated at lysine 373, P53 is released from the HDM2 and TRIM24 and interacts with Cyclin-Dependent Kinase Inhibitor 1A (CDKN1A; P21), thereby promoting the G1 phase of cell cycle and preventing cell death. Additionally, P53 can stimulate expression of miR-34a and miR-145, which suppresses the expression of major stem cell factors OCT4, Kruppel-Like Factor 4 (KLF4), Lin-28 Homolog A (LIN28A), and SOX2 in hESCs, thus preventing their self-renewal [33].
PHD domain of TRIM28, another member of the C-VI subfamily, was shown to direct SUMO conjugation with its own bromodomain, and this sumoylation is necessary for the Kruppel-associated box domain zinc finger (KRAB-ZNF) protein TRIM28-mediated transcriptional repression [39]. TRIM28 interacts with the KRAB-ZNF protein and recruits various histone-modifying complexes [NuRD complex, SET domain bifurcated 1 (SETDB1)—H3K9-specific histone methyltransferase, and heterochromatin protein 1 (HP1)—the H3K9me2/3 reader] to the specific histone marks. As a result, TRIM28/KRAB-ZNF promotes transcription silencing of genes [34].
TRIM28, along with zinc finger protein ZFP809 and SETDB1 (ESET) factors, is responsible for H3K9me3-mediated silencing of exogenous proviruses and endogenous retroviruses (ERVs) in ESCs [40]. TRIM28 was also shown to silence human-specific endogenous retroelements in hESCs [41]. The phosphorylated TRIM28 variant forms complex with the transcription factor Oct4 and induces expression of ESC-specific genes thereby maintaining their undifferentiated state [35].
TRIM28 plays a crucial role in somatic cell reprogramming into the iPSCs [36,42,43]. Oleksiewicz et al. [36] have demonstrated that upon reprogramming into iPSCs, the interaction of three KRAB-ZNF factors (ZNF114, ZNF483, and ZNF589) with TRIM28 is necessary for the maintenance of the pluripotent state and for DNA methylation of the major differentiation genes. Klimczak et al. [43] have received similar data. At the same time, Miles et al. [44] have shown that TRIM28 represses the transcription of ERVs and the genes located in the surrounding genome regions in somatic cells. These regions are enriched in the H3K9me3 marks, and knockdown of TRIM28 leads to more decondensed chromatin state promoting reprogramming of somatic cells. Thus, TRIM28 may be a key regulator of gene expression in the repressive chromatin regions upon reprogramming.
Another member of the subfamily, TRIM33 is able to form complex with Mothers against decapentaplegic homolog (Smad)2/3 through its PHD and Bromo domains, which mediates the signal transduction from the receptors of transforming growth factor beta (TGFβ) superfamily. The complex replaces HP1γ at the promoters of mesendoderm regulators Gsc and Mixl1 marked by H3K9me3 in stem cells. Binding of TRIM33-Smad2/3 complex makes these promoters available for the Smad4-Smad2/3 complex, which recruits histone acetyltransferases, switches the master regulators Gsc and Mixl1 from poised to activated state, and thereby promotes subsequent mesendoderm differentiation of mESCs [37]. Xia et al. [45] have shown that TRIM33 can regulate Wnt-mediated response of Mixl1 in mESCs. Besides, TRIM33 is necessary for the normal maturation of embryoid bodies in mESCs [46].
TRIM-NHL proteins (C-VII subfamily)
TRIM71 is an important regulator of the pluripotency in mESCs [26]. One of the known functions of NHL domain is RNA binding. TRIM71 maintains stem cell identity, contributes to the G1-S transition and promotes ESCs self-renewal. To perform these functions, it binds to Argonaute2 and microRNAs and inhibits the expression of CDKN1A [47]. However, Mitschka et al. [48] have shown that the loss of TRIM71 in mESCs does not affect the expression of such regulators of pluripotency as NANOG and OCT4. Recently, it was shown that TRIM71 stabilizes SHC SH2 domain-binding protein 1 (SHCBP1) and promotes fibroblast growth factors (FGF)/extracellular signal-regulated kinase (ERK) signaling, which controls self-renewal and differentiation in ESCs. The authors described new long noncoding RNA Trincr1 (TRIM71 interacting noncoding RNA1) that represses TRIM71 and its ability to stabilize SHCBP1 [49].
Besides, TRIM71 can ubiquitinate P53 and counteract P53-dependent apoptotic and differentiation signaling pathways in TRIM71-inducible mESCs [50]. TRIM71 is negatively regulated by let-7 miRNA, which is the inhibitor of stem cells' self-renewal [51,52]. let-7 inhibition and subsequent activation of TRIM71 promotes reprogramming of differentiated cells into iPSCs [53]. Unlike TRIM71, TRIM32 does not maintain stem cell pluripotency. It inhibits reprogramming of mouse embryonic fibroblasts into iPSCs, through ubiquitination by RING domain and subsequent proteasomal degradation of c-MYC and OCT4 [54]. These data demonstrate that, despite the structural similarity, TRIMs can either favor or prevent stem cell differentiation (Table 1).
Thus, TRIMs play the key role in various processes in stem cells (Table 1). Their functions may be linked to either the ability to ubiquitinate proteins leading to their degradation, or to the different functionalities mediated by various C-terminal domains. Interestingly, some properties of TRIMs, such as RNA binding, can be performed by distinct C-terminal domains (NHL or PRYSPRY). In conclusion, this combination of various domains is responsible for a wide variety of properties of TRIM proteins in stem cells.
Functions of TRIM Proteins in Embryogenesis
As could be expected, besides involvement in the stem cells' regulation, TRIMs actively participate in the early stages of embryogenesis [55 –64] (Table 2). For example, in Xenopus laevis oocytes and unfertilized eggs, Trim36 (subfamily C-I) ubiquitination activity is necessary for microtubule assembly. It attenuates the dynamics of plus-end microtubule growth and controls its orientation [55,56]. At the same time, TRIM28 (subfamily C-VI) was shown to participate in embryonic epigenetic reprogramming. The absence of maternal TRIM28 leads to the hypomethylation of Rbmy1a1 gene promoter. Its activation causes developmental arrest and male-specific early embryonic lethality [57].
Tripartite Motif proteins in Embryogenesis
PAX, Paired Box; TGFβ, transforming growth factor beta.
Another TRIM protein from subfamily C-VI, TRIM33, is required for the restriction of Nodal Growth Differentiation Factor (Nodal)/TGFβ signaling during early vertebrate embryogenesis. TRIM33 forms the complex with Smad4, which participates in the intracellular signal transduction in response to the Nodal signaling, ubiqutinates it, and thereby can negatively regulate Nodal/TGFβ signaling. In the trophoblast, TRIM33 modulates Nodal activity to balance stem cells' self-renewal and differentiation. In epiblast, TRIM33-mediated regulation of Smad4 is responsible for the correct formation of anterior primitive streak derivatives [58].
TRIM59 (subfamily C-XI) is shown to be important in early embryo development (from the blastocyst stage to the gastrula) [59]. Depletion of TRIM59 downregulates the expression of primary germ layer formation-associated genes (Brachyury, lefty2, Cer1, Otx2, Wnt3, and Bmp4), disrupts the formation of primary germ layers, and leads to the embryonic lethality. TRIM59 also ubiquitinates F-actin and promotes its polymerization during gastrulation [59]. At the same time, TRIM71 mutants (truncated protein without NHL domain and 3’UTR, the site of the let-7 miRNA binding) manifest the embryonic lethality, thus indicating the important role of the TRIM71 in embryogenesis [60,61].
Yoshigai et al. [62] detected Trim36 (subfamily C-I) expression during early embryogenesis of X. laevis, mainly in the nervous system and in a section of the posterior somite. Knockdown of Trim36 inhibits somite formation from mesodermal tissue [62]. The expression of the protein of C-IV group, trim69 (the homologous gene of human TRIM69), was revealed in zebrafish embryo brain at different stages of early development [63]. TRIM11 (subfamily C-IV) ubiquitinates Paired Box (PAX)6, which is an important developmental regulator, and promotes its degradation, thereby inhibiting PAX6 transcriptional activity [64].
Thus, the TRIMs can participate in the embryogenesis from the very beginning. At later stages, they are involved in the processes of maturation of the nervous system.
TRIM-Mediated Regulation of the Nervous System Development and Functioning
Most TRIMs, linked to the stem cells' regulation, are reported to be also involved in the development and functioning of the nervous system (Table 3). Several TRIMs affect the early stages of neurogenesis, as well as the development of the nervous system. Below, we will focus on the existing data concerning the functions of TRIMs in the normal and pathological processes in the nervous system.
Tripartite Motif Proteins Associated with Neurogenesis
Bim, Bcl-2-interacting mediator of cell death; CNS, central nervous system; MCL, Myeloid Cell Leukemia; NFATc, Nuclear Factor of Activated T Cells; NPC, neural precursor cell; NSC, neural stem cell; PHOX, Paired Mesoderm Homeobox Protein; PIAS3, protein inhibitor of activated STAT3; PNS, peripheral nervous system; Pum1, Pumilio 1; SNAP25, Synaptosome-Associated Protein 25; SUMO, small ubiquitin-like molecule modifier; VASP, Vasodilator-Stimulated Phosphoprotein.
C-I subfamily TRIM proteins
The C-I TRIMs possess several C-domains, including a COS-box motif, a FN3 domain, and the PRYSPRY domain. Brain-specific E3 ubiquitin ligase TRIM9 actively takes part in the functioning of mouse central nervous system (CNS). During mouse embryogenesis (from E13.5), it is expressed mainly in the CNS, in particular in developing neocortex, dorsal thalamus, midbrain, basal area of the hindbrain, and in the spinal cord [65]. In adult brain, this protein is mainly found in Purkinje cells of the cerebellum, as well as in the pyramidal cells of the hippocampus, and in the external layers of the cortex [65]. Moreover, TRIM9 interacts and ubiquitinates netrin-1 receptor DCC and thereby decreases dendritic and axonal branching of adult-born hippocampal neurons as well as cortical neurons [66 –68].
The Trim9 deletion in mice causes the impairment of hippocampal-dependent spatial learning and memory in the Morris Water Maze [66]. Besides, Menon et al. [69] have shown that TRIM9 localizes to neuronal growth cone filopodia, binds to and ubiquitinates polymerase vasodilator-stimulated phosphoprotein (VASP), and modulates the filopodia density and stability in response to netrin in cortical neurons [69]. Moreover, TRIM9 directly interacts with Synaptosome-Associated Protein 25 (SNAP25), and thereby inhibits N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex formation located on the plasma membrane, which plays a key role in exocytosis in axon branching [67]. Abundant axonal arborization and unwanted innervation may cause neurodevelopmental disorders. Therefore, TRIM9 is an important regulator of the correct axon branching.
Another member of this subfamily, TRIM46, controls the initial polarization of neuronal cells and microtubule organization during axon formation. These properties are mediated by the C-terminal COS domain, which is responsible for interaction with microtubules [70]. Mouse TRIM67 inhibits cell proliferation and induces neural differentiation, including neuritogenesis. This protein interacts with Brain-Specific Phosphatidic Acid Phosphatase-Like Protein 1 (PRG-1) and Protein Kinase C Substrate 80K-H, ubiquitinates and promotes further degradation of 80K-H, and negatively regulates RAS signaling in the neuroblastoma cell line N1E-115 followed by growth arrest and neuritogenesis in NPCs [71]. Besides, Boyer et al. [72] have shown that TRIM67 is essential for the correct morphology of several brain regions as well as for cognitive abilities and social behavior of mice [72,73].
C-IV subfamily TRIM proteins
TRIM11, through its C-terminal SPRY domain, directly interacts with the homeodomain-containing transcription factor, Paired Mesoderm Homeobox Protein (PHOX) 2B, which plays an essential role during the development of noradrenergic neurons in both the CNS and the peripheral nervous system [74]. TRIM11 inhibits PAX6-dependent corticogenesis. Depletion of endogenous TRIM11 in the mouse cortex leads to an accumulation of inclusion bodies, containing PAX6, and to apoptosis [64]. TRIM16 participates in the differentiation of ganglia cells. It affects cell cycle progression through Cyclin D1 and p27. Known domains do not take part in these functions suggesting the presence of other important functional sites [15].
TRIM17 promotes neuronal apoptosis through ubiquitination and subsequent degradation of phosphorylated form of BCL2-like antiapoptotic protein Myeloid Cell Leukemia (MCL)-1 [75]. In addition, TRIM17 interacts with the SUMOylated form of Nuclear Factor of Activated T Cells (NFATc)3, which is a proapoptotic factor of cerebellar granule neurons. This SUMOylation prevents nuclear localization of NFATc3 [76]. The knockdown of trim69 in zebrafish causes significant defects in embryo brain and attenuates neuronal differentiation markers. TRIM69 mediates these effects through interaction with c-JUN and subsequent negative regulation of its expression [63].
C-V subfamily TRIM proteins
TRIM8 and TRIM44, belonging to the C-V subfamily of TRIMs, were also shown to participate in early neurogenesis. RNA-seq analysis of TRIM8-expressing primary mouse embryonic neural stem cells (NSCs) [77] resulted in the identification of CNS-related pathways that were especially enriched in comparison to the control cells. TRIM8 interacts with STAT3 and increases its transcriptional activity by direct binding with the STAT3-inducible element (SIE) sequences [77]. In addition, TRIM8 can ubiquitinate and promote proteasomal degradation of protein inhibitor of activated STAT3 (PIAS3) and thereby activates STAT3. The overexpression of TRIM8 increases the expression of p-STAT3, c-MYC, SOX2, NESTIN, and CD133 (Prominin 1) and enhances glioma stem cell self-renewal [78]. TRIM44 is expressed in neuroblasts and in developing neurons in adult mice brain and in embryonic brain from day 14 [79].
C-VI subfamily TRIM proteins
TRIM28 and TRIM33 belonging to C-VI subfamily and playing the key role in self-renewal or differentiation of the pluripotent stem cells were also shown to regulate NSCs proliferation or differentiation [80 –83]. TRIM28 represses ERVs and the nearby protein-coding genes by maintaining a localized heterochromatin regions in NPCs in the same manner as in pluripotent stem cells [80 –82]. Recently, Pavlaki et al. [84] have demonstrated that TRIM28 itself is regulated by long noncoding RNA Paupar, which forms a ribonucleoprotein complex with TRIM28 and transcription factor PAX6 on chromatin region enriched for regulators of neural proliferation and differentiation. This complex is important for controlling of olfactory bulb neurogenesis. Interestingly, knockout of TRIM28 in mice leads to changes in exploratory activity, spatial learning, and memory suggesting a role of TRIM28 in the hippocampus [85].
Another protein of this subfamily, TRIM33, in complex with SMAD4, regulates the TGFβ pathway and thereby controls proliferation, differentiation, and cell cycle exit of NSCs in the midbrain [83].
C-VII subfamily TRIM proteins
The C-VII subfamily of TRIM-NHL proteins participates in neurogenesis, employing their E3 ubiquitin ligase activity or functions of NHL domain (Table 2). TRIM2 promotes specification of axons in cultured mouse hippocampal neurons [86]. It ubiquitinates the neurofilament light chain, mediates its degradation, and thereby prevents axonopathy [87]. TRIM2 also ubiquitinates and promotes the degradation of proapoptotic factor Bcl-2-interacting mediator of cell death (Bim) in neurons following brief ischemia and thus contributes to rapid ischemic tolerance neuroprotection [88].
TRIM3 regulates NSC proliferation and differentiation by influencing on the Notch signaling pathway [89]. TRIM3 increases the degradation of postsynaptic density proteins, Discs Large Homolog-Associated Protein 1 (DLGAP1/GKAP/SAPAP) by ubiquitination and supposedly decreases the interaction between GKAP and the other scaffold protein SHANK1, thereby inhibiting growth of dendritic spine heads in rat hippocampal neurons [90]. However, knockout of TRIM3 in mice does not influence on the GKAP or SHANK1 levels. TRIM3 can polyubiquitinate γ-actin and regulates the timing of hippocampal plasticity, learning, and memory [91]. Moreover, the expression of TRIM3 is regulated in p53-dependent manner after the treatment with antagonist of Gamma-Aminobutyric Acid GABA(A) receptor [GABA(A)R] signaling. In turn, TRIM3 regulates the presence of GABA(A)Rs with γ2-subunit at the cell surface and thereby can mediate susceptibility to seizure in mice model of epilepsy [92].
TRIM32 determines cell fate in NSCs and affects the equilibrium between stem cell maintenance and neuronal differentiation, as well as controls the production of olfactory bulb neurons in adults [93 –96]. Loss of TRIM32 alters the behavior in the open field test, but does not influence exploratory and anxiety-related behavior as well as spatial learning [97]. TRIM32 is asymmetrically distributed in dividing NPCs, promotes the degradation of c-MYC by ubiquitination, enhances neuronal differentiation, and suppresses self-renewal [95].
In addition, TRIM32 forms complex with Argonaute-1 (Ago1) and microRNAs by NHL domain. It was shown that TRIM32 expression activates Let-7a as well as some other miRNAs [95]. Also, TRIM32 binds to the RNA helicase DEAD Box Polypeptide (DDX)6, Ago2, and RNA-binding protein Pumilio 1 (Pum1) in vivo. This complex is supposedly involved in let-7a miRNA activation during NSC differentiation [93]. TRIM32 can promote the enhancement of neural differentiation by interacting with retinoic acid receptor α and by increasing its downstream activity in the presence of retinoic acid [94].
TRIM71 modulates the neural differentiation, but does not regulate pluripotency. The loss of TRIM71 in Trim71(−/−) mESCs promotes the expression of neural markers and upregulation of several miRNA (let-7e, mir-132, mir24–2, mir-200) and directs mESCs toward neural differentiation [46]. In addition, mouse TRIM71 is negatively regulated by let-7 miRNA resulting in the correct neural tube closure during embryogenesis [48]. TRIM71 is likely not essential for adult neurogenesis and is found only in the ependymal cells lining the walls of the four ventricles [65].
In summary, the existing data show the important role of TRIMs in the CNS processes both in NSCs and in different types of terminally differentiated neurons. Their functions in CNS may be mediated either by the RING or by the C-terminal domains.
TRIMs in Neuropathology
Compelling evidences point to the essential role of many TRIMs both in immune system and in the development and functioning of CNS. It is therefore not surprising that there is a relationship between alterations of expression of these genes and various neuropathologies. Several data indicate that an expression of some TRIMs in brain is affected by viral infection. For example, rabies virus infection in mice results in the downregulation of TRIM9, as well as in the accumulation of synaptic vesicles in the presynaptic neurons [98]. Japanese encephalitis virus infection increases the expression of TRIM21 in human microglial (CHME3) cells [99].
During the last few years, a lot of data have emerged that indicate that innate immunity is an active player in the neurodegeneration processes [100]. Several genetic alterations associated with neurodegeneration were shown to increase the innate immunity response [101]. It becomes clear that chronic neuroinflammation as an attribute of innate immunity plays an important role in the processes of different neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD) [102 –105]. Neurodegenerative diseases are manifested by the degeneration and death of the specific neurons in the brain, and the inflammation may greatly contribute to their degeneration. Consistent with known neuronal functions, including protective ones, defects in TRIM proteins or alterations of their expression are associated with different neurological pathologies (Table 4). Below, we consider in more detail the existing evidence of the involvement of TRIM proteins in the PD, AD, and HD.
Tripartite Motif Proteins Associated with Neuropathology
AD, Alzheimer's disease; DUSP6, dual specificity phosphatase 6; HD, Huntington's disease; hESC, human embryonic stem cell; LRRK, Leucine-Rich Repeat Kinase; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; PD, Parkinson's disease.
TRIMs' involvement in PD pathogenesis
PD results in the death of dopaminergic neurons in the substantia nigra. The immune system plays a significant role in the pathogenesis and progression of PD through inflammation and autoimmune reactions [140,141]. In particular, different TRIMs were found to be associated with PD (Table 4). The level of the TRIM9 is decreased in the affected brain areas upon PD and dementia with Lewy bodies [112]. In 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-generated cellular model of PD in PC12 cells, TRIM10 promotes dual specificity phosphatase 6 (DUSP6) ubiquitination and inhibits its expression. The knockdown of TRIM10 decreases MPTP-caused apoptosis and neurotoxicity in PC12 cells [115]. Also, TRIM24 expression was significantly changed in the blood of MPTP-treated monkeys [120]. In addition, TRIM24 was identified among genes with de novo mutations associated with PD pathogenesis [121].
TRIM32 regulates the transcription of one of the major risk genes of PD, encoding α-synuclein [129]. TRIM32 also suppresses the miRNA, which inhibits the Leucine-Rich Repeat Kinase (LRRK)2, one of the well-known factors associated with PD [130]. The expression of TRIM27 gene is increased in the patients with PD as well as in the substantia nigra of mice with MPTP-generated model of PD. At the same time, Trim27 knockout reduces the loss of dopaminergic neurons in MPTP-treated mice [124]. The E3-ubiquitin ligases, TRIM17 and TRIM41, modulate α-synuclein expression and thus may contribute to the pathogenesis of PD. In addition, infrequent allelic variants of the TRIM17 and TRIM41 genes were associated with some familial forms of PD [118].
A relatively new technology for studying molecular mechanisms of PD is the use of human iPSCs, allowing to compare cells from PD patients and from healthy donors. iPSCs derived from fibroblasts of patients with PD and the corresponding neural progenitors and neuronal cultures (enriched with dopaminergic neurons) allow to identify the dysfunctional pathways at the early stages of the disease [142,143]. In this way, the expression of 15 TRIM-encoding genes in the iPSCs derived from PD patients with hereditary and sporadic forms of disease was recently analyzed. The expression of more than half of these genes appears to be altered in PD patients, as compared with healthy individuals, upon reprogramming of fibroblasts into iPSCs and their subsequent neuronal differentiation [106]. These data support an association of multiple TRIM genes (TRIM1, TRIM2, TRIM6, TRIM9, TRIM14, TRIM16, TRIM24, TRIM32, and TRIM37) with PD at the early stages of disease (Table 4).
TRIMs' involvement in AD pathogenesis
Numerous data indicate the relationship of AD with immune system impairments [144]. Inflammation may contribute to AD pathogenesis and promote its progression. Upon AD, the oligomeric forms of beta-amyloid are accumulated in neurons. Their deposition in synapses seems to cause the memory loss in AD [145]. Current data suppose that microglia, which represents the innate immune cells in brain, becomes activated in response to the oligomeric forms of beta-amyloid. The resulting microgliosis is suggested to promote AD development [144].
Some TRIMs (TRIM15, TRIM35, TRIM32, and TRIM37) are supposedly associated with AD pathogenesis and progression [117,128,133] (Table 4). TRIM2 is one of the target genes of miR-9 and miR-181 miRNAs that are known to be downregulated upon AD [146]. TRIM11 promotes the degradation of humanin, a neuroprotective peptide, which was shown to suppress AD-related neurotoxicity [116]. The hypermethylation of TRIM59 potentially contributes to apoptosis upon AD [136] (Table 4). Yet, it is evident that the potential causative role of TRIMs in the AD and PD pathogenesis is not firmly established and awaits further studies.
TRIMs' involvement in HD pathogenesis
HD is a neurodegenerative disease caused by genetic defect in the gene encoding for huntingtin (HTT). The disorder is characterized by motor and cognitive dysfunction. Feyeux et al. [108] searched for genes associated with mutations in HTT gene using the hESC lines derived from embryos with a mutant-HTT allele and from healthy donors. TRIM4 was identified among genes, which were differentially expressed in HD cells (hESCs and corresponding NSCs) compared with wild-type cells. The expression of TRIM4 (at mRNA and protein levels) was shown to be upregulated [108].
Conclusion
Complex structural composition and diversity of functions result in many faces of TRIM proteins not only in the immune response to various pathogens but also in the stem cells' maintenance and differentiation, including neurogenesis. These common pathways brightly illustrate the intimate links between immune and nervous systems.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was funded by the Russian Foundation for Basic Research (grants no. 17-29-06008, 18-04-00733, 19-29-04080), Program for Molecular and Cellular Biology from Presidium of Russian Academy of Sciences (no. 01201356275), Russian Fundamental Scientific Research Program (no. 01201355481).