
Abstract
Bone marrow mesenchymal stem cells (MSCs) are a rare subset of nonhematopoietic progenitor cells and are appealing biomaterial for multiple tissue damage repairs. Transplantation of MSCs is proved to improve heart function after myocardial ischemia. However, the limitations of MSC injection approaches are equally obvious. As a multiple-function cell, platelets (PLTs) are also known playing important roles in cardiac recovery after myocardial infarction. In this study, we analyzed circulating MSC–PLT aggregate numbers in acute myocardial infarction (AMI) patients by flow cytometry. We found more MSC–PLT aggregates in patients with AMI than in healthy controls, and the patients with higher MSC–PLT aggregates had better prognosis. When stromal cell-derived factor 1 (SDF-1) binds to its receptor CXC chemokine receptor 4 (CXCR4), they play an important role in MSC migration and engraftment. We explored SDF-1 and CXCR4 expression on PLT surface by flow cytometry and found relative mean fluorescence intensity of PLT CXCR4 and the number of MSC–PLT aggregates showed a significant correlation. Meanwhile, in vitro experiments demonstrated that SDF-1/CXCR4 was crucial in MSC–PLT aggregate formation, which might suggest a novel mechanism that SDF-1/CXCR4 is involved in MSCs homing and myocardial repair after AMI. There may be another strategy to encourage myocardial repair in AMI patients by increasing the expression of SDF-1 on MSCs and promoting the formation of MSC–PLT aggregates.
Introduction
Bone marrow mesenchymal stem cells (BM-MSCs) are a rare subset of nonhematopoietic progenitor cells. MSCs have the potential of multidirectional differentiation and can differentiate into muscle, fat, bone, cartilage, and other tissues. Therefore, MSCs are appealing biomaterial for multiple tissue damage repairs, such as myocardial repair [1], wound healing [2], cartilage regeneration [3], bone repair, and reconstruction [4]. Postmyocardial infarction (MI) repair has always been one of the important topics in the study of tissue repair of MSCs. In addition to multipotency of differentiation, MSCs have the abilities of extensive paracrine secretome [5] and provision of support to other tissue cells, anti-inflammatory and immunomodulatory characteristics, and the propensity for genetic engineering to augment reparative effectiveness.
Strategies of MSC therapy for cardiac repair are mainly delivery of autologous or allogeneic MSCs by intramyocardial or intravenous injection [6]. These MSCs can be categorized as unmodified [7], engineered [8,9], combined with other cells [10], and antigenically selected [11]. A flurry of clinical trials has demonstrated that transplantation of MSCs did improve heart function [6,12 –16]. However, the limitations of these MSC injection approaches are equally obvious, including poor retentions of the transplanted cells, significant entrapments of MSCs in the lungs when delivered intravenously, and low storage/shipping stabilities of live cells [17].
Meanwhile, as the second most abundant cell in the peripheral blood, platelets (PLTs) have multiple functions, including traditional roles in physiological hemostasis and pathological thrombosis and critical roles in inflammation and tissue repair and regeneration through the release of bioactive cargos and the cell–cell interactions. However, roles of PLT in cardiac recovery after MI are paradoxically beneficial [18].
Stromal cell-derived factor 1 (SDF-1), also known as CXCL12, is expressed and critically functional in cardiac myocytes. When it binds to its receptor, CXC chemokine receptor 4 (CXCR4), SDF-1 stimulates and enhances the cellular signal, which attracts potentially beneficial stem cells for tissue repair within the ischemic myocardium.
In this study, we found more MSC–PLT aggregates in peripheral blood of patients with acute myocardial infarction (AMI) than in healthy controls, and the more the MSC–PLT aggregates were in circulation, the longer the patients would survive. In addition, we demonstrated that adhesion molecules CXCR4 and SDF-1 might play an important role in this phenomenon and might suggest a novel mechanism that SDF-1/CXCR4 is involved in MSCs homing and myocardial repair after AMI.
Materials and Methods
Study approval
The protocol for this study was approved by the Ethics Committee of West China Hospital, Sichuan University. We also obtained written informed consent from all the participants.
Patients
One hundred thirty-two patients (88 males and 44 females) with AMI for the first time were included in the AMI group according to the universal definition of AMI in the European Society of Cardiology (ESC) Guidelines [19,20]. One hundred twenty-four unrelated, sex- and age-matched healthy volunteers (82 males and 42 females) who were free of MI, stroke, transient ischemic attack, venous thromboembolism, and cancer history were included as the healthy control group. All subjects, AMI patients, and healthy controls were recruited by advertisement at West China Hospital, Sichuan University. After percutaneous coronary intervention, two antiplatelet drugs, aspirin and clopidogrel or ticagrelor, were used for 1 year or 6 months according to the ESC guidelines. Then, only one antiplatelet drug was used for life according to a cardiologist assessment. However, patients were not analyzed for antiplatelet activity-related genes for aspirin and clopidogrel.
Two milliliters of peripheral blood was collected from healthy controls and AMI patients within 4–7 days after the onset of AMI, using heparin as an anticoagulant. Clinical data, such as medical history, electrocardiogram, coronary angiography, blood pressure, blood sugar, blood lipids, troponin, and other biochemical indicators, were recorded.
Cytometric analysis
Cytometric analysis was performed in a BD FACSCanto II Flow Cytometer (Becton Dickinson) using BD FACSDiva Software Systems (Becton Dickinson).
MSC–PLT aggregates analysis
For MSC–PLT aggregates analysis, monoclonal antibodies against CD105 (fluorescein isothiocyanate-conjugated, clone 266), CD90 [phycoerythrin (PE)-conjugated, clone 5E10], CD45 [peridinin–chlorophyll–protein complex (PerCP)-conjugated, clone 2D1], CD34 [(PE Cy7)-conjugated, clone 581], CD41a [allophycocyanin (APC)-conjugated, clone HIP8], and corresponding isotype controls were purchased from Becton Dickinson (San Jose, CA). A total of 250 μL of whole blood was added to 10 μL of each antibody and then incubated for 30 min at room temperature (RT) in the dark. One milliliter of lysis solution was added (BD FACS Lyse, Lysing Solution; Becton Dickinson, Germany) for erythrocyte lysis and leukocyte fixation. MSC–PLT aggregates (CD105+CD90+CD34−CD45−CD41a+cells) and total MSCs (CD105+CD90+CD34−CD45−cells) were evaluated by flow cytometry. At least 200,000 cells were analyzed at a time.
PLT surface CXCR4 and SDF-1 analysis by whole blood flow cytometry
PLTs in whole blood were analyzed for the surface expression of CXCR4 and SDF-1 gating for the PLT-specific marker GPIIb. The anticoagulated peripheral blood samples were diluted, and the final PLT concentration was 1 × 1010/L ± 10%. One hundred microliters of blood samples was added to monoclonal antibodies against CXCR4 [(APC)-conjugated, clone 12G5; BD Bioscience] 20 μL, SDF-1 (PE-conjugated, clone 79018; R&D System, Bio-Techne, MN), and CD41a (PerCP-conjugated, clone MEM-06; Abcam, Cambridge, United Kingdom) 10 μL each and then was incubated for 30 min at RT in the dark. One milliliter of lysis solution was added for erythrocyte lysis. The relative mean fluorescence intensity (RMFI; RMFI = PLT MFI/RBC MFI) of CXCR4 and SDF-1 expressed on CD41+ PLTs were evaluated by flow cytometry. At least 5,000 PLTs were analyzed at a time.
Patient follow-up
The total follow-up time was 36 months. The patients' clinical data, treatment status, and final outcome were collected. Patients were divided into high- and low-MSC–PLT aggregates groups according to the best cutoff value from receiver operating characteristic (ROC) analysis, and the survival time between the two groups was compared. The survival curves were drawn with the primary end point as the composite of death, nonfatal MI, stroke, and hospitalization for worsening heart failure.
Bone marrow MSCs
Human MSCs from bone marrow were isolated from healthy donors and characterized as previously described [21]. MSCs (passages 2–5) were cultured in proliferation medium, containing Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Sigma), 2 mM
Preparation of engineered SDF-1 overexpression MSCs
When the confluence of proliferating MSCs reached or surpassed 70% between the second and fifth passages, the cells were transduced with recombinant adenovirus vectors carrying SDF-1 gene or blank virus at a multiplicity of infection (viruses/cells) of 1,500. The medium was changed after 2 h. After 36–48 h, further experiments were carried out according to GFP expression using a Nikon TE2000-U inverted fluorescent microscope.
Cocultures of SDF-1 overexpression MSCs and PLTs from AMI patients
Sterile glass coverslips were placed in the bottom of the six-well plates. Blood collected from healthy controls and AMI patients in ACD buffer was centrifuged at 200 g for 20 min at RT to obtain platelet-rich plasma (PRP). Using MSCs transduced with blank virus as control, 106 MSCs transduced with SDF-1 and 1 × 109 PLTs from AMI patients and healthy controls were cocultured in DMEM containing 10% fetal bovine serum at 37°C in a humidified atmosphere of 5% CO2 for 24 h.
Immunofluorescence staining
The culture medium was aspirated from each well of the six-well plates, and the cells grown on the sterile glass coverslips were gently rinsed twice in phosphate-buffered saline (PBS) at RT. Then, the cells were fixed with 4% paraformaldehyde PBS for 20 min at RT, rinsed three times with PBS for 10 min, blocked with 10% normal goat serum in PBS for 1 h, labeled with PLT with mouse monoclonal anti-CD41a-PE (PE-conjugated, clone HIP8; BD Bioscience), and incubated at RT for 2 h in the dark. Then, the fluorescently labeled antibody was removed. The coverslips were permeabilized with 0.3% Triton X-100 and 4′,6-diamidino-2-phenylindole (DAPI) staining solution was added at RT for 15 min or more. The cells were then washed three times with PBS containing 0.1% Tween-20 for 10 min at RT. After washing, the coverslips containing the immunolabeled cells were mounted with an antifade mounting medium (Biomeda Gel mount; Electron Microscopy Sciences, Foster City, CA) and observed under a confocal Leica TCS SP5 microscope (Leica Microsystems).
Cocultures of unmodified MSCs and PLTs from AMI patients with or without CXCR4 inhibitor and immunofluorescence staining
Sterile glass coverslips were placed in the bottom of the six-well plates. 106 MSCs and 1 × 109 PLTs from AMI patients were cocultured in DMEM containing 10% fetal bovine serum, with or without AMD3100 (final concentration 5 μg/mL; Sigma), at 37°C in a humidified atmosphere of 5% CO2 for 24 h. The fluorescence staining method was the same as that described above using mouse monoclonal anti-CD41a-PE and DAPI, and the immunolabeled cells were observed under a confocal Leica TCS SP5 microscope.
Real-time quantitative polymerase chain reaction
After coculturing with PLTs, MSCs with and without PLTs (from AMI patients and healthy controls) were collected. Total RNA of the cells was extracted using a TRIzol Extraction Kit (Invitrogen) according to the manufacturer's instructions. cDNA was produced from the total RNA using a QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). real-time polymerase chain reaction (RT-PCR) was performed with SYBR FAST qPCR Kit Master Mix (2 × ) Universal (KAPA) to estimate the expression of CXCR4 and SDF-1 with β-actin as the internal control. Primers are listed in Supplementary Table S1. Genes were amplified in triplicate. The relative expression amount of SDF-1 and CXCR4 mRNA was analyzed using the 2−ΔΔCt method.
Western blotting
The total protein was extracted using modified radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% SDS, 1% NP-40, 2 mM DTT, 1 mM EDTA, 1 mM PMSF, 1 mM NaF, 0.25% Na-deoxycholate, 1 mg/mL aprotinin, 1 mg/mL leupeptin, 1 mg/mL pepstatin). The total protein content in the supernatants was quantified by Bio-Rad protein assay (Bio-Rad Laboratories, Milan, Italy) following the manufacturer's instructions. Afterward, 100 μg of protein was resolved by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and electroblotted onto a polyvinylidene difluoride membrane (Millipore, Billerica, MA). Immunodetection using antibodies against CXCR4, SDF-1, or β-tubulin (all from Abcam, MA) was carried out for 1 h at RT or overnight at 4°C. Antibody–antigen complexes were detected using the enhanced chemiluminescence system (Amersham Bioscience, Piscataway, NJ). β-Tubulin served as a loading control.
Statistical analyses
All statistical analyses were performed using SPSS version 11.0 (SPSS, Inc., Chicago, IL) and GraphPad Prism 7 (GraphPad, La Jolla, CA). Continuous variables are expressed as mean ± standard deviation for variables with a normal distribution. Normally distributed data were compared using independent Student's t-test. Correlations between RMFIs of PLT CXCR4 and the number of MSC–PLT aggregates were assessed by Spearman's rank correlation coefficient (r). ROC analysis and relative area under the curve (AUC) statistics were used to optimize the cutoff value of the number of MSC–PLT aggregates. Those patients who were lost to follow-up were excluded. The best cutoff value was selected with the maximum Youden's index (sensitivity+specificity −1). Survival analysis was carried out with the Kaplan–Meier method. P < 0.05 was considered statistically significant.
Results
Patient characteristics
A total of 132 AMI patients and 124 healthy controls were enrolled in this study based on the inclusion and exclusion criteria. Patients' characteristics (age, gender, heart rate, blood pressure, and blood lipids) of this study are available in Table 1. Compared with healthy controls, AMI patients had higher diastolic blood pressure, blood sugar, and triglycerides and lower high-density lipoprotein cholesterol, low-density lipoprotein cholesterol, and cholesterol.
Clinical Characteristics of the Healthy Control and Acute Myocardial Infarction Groups
P < 0.05, data are presented as mean value ± SD.
AMI, acute myocardial infarction; HDL-c, high-density lipoprotein cholesterol; LDL-c, low-density lipoprotein cholesterol; MSC, mesenchymal stem cell; PLT, platelet; TG, triglycerides; WBC, white blood cell.
MSC–PLT aggregates analysis
To see whether AMI patients have more MSC–PLT aggregates in their peripheral blood, we analyzed microparticles expressing both MSC and PLT markers by flow cytometry (Fig. 1A–E) and we found that AMI patients had more MSC–PLT aggregates than healthy controls (11.61 ± 3.52 cells vs. 7.48 ± 2.06 cells/200,000 nucleated cells, P < 0.001) (Fig. 1F).

Circulating MSC–PLT aggregates analysis.
SDF-1 and CXCR4 expression on PLT surface
We compared SDF-1 and CXCR4 expression on PLT surface between healthy controls and AMI patients by flow cytometry using red blood cells as control to calculate RMFI (Fig. 2A–C). In AMI patients, RMFI of CXCR4 on PLT surfaces was much higher than that in healthy controls (12.18 ± 3.87 vs. 8.53 ± 2.60, P < 0.0001) (Fig. 2E), but no difference was found in RMFI of SDF-1 on PLT surfaces between AMI patients and healthy controls (0.42 ± 0.18 vs. 0.38 ± 0.20) (Fig. 2D).

SDF-1 and CXCR4 expression on PLT surface.
Correlation between PLT CXCR4 RMFI and the number of MSC–PLT aggregates
RMFI of PLT CXCR4 and the number of MSC–PLT aggregates showed a significant correlation in AMI patients (n = 132, r 2 = 0.6527, P < 0.0001) (Fig. 3A), healthy controls (n = 124, r 2 = 0.4838, P < 0.0001) (Fig. 3B), and all the individuals involved in this study (n = 256, r 2 = 0.6934, P < 0.0001) (Fig. 3C).
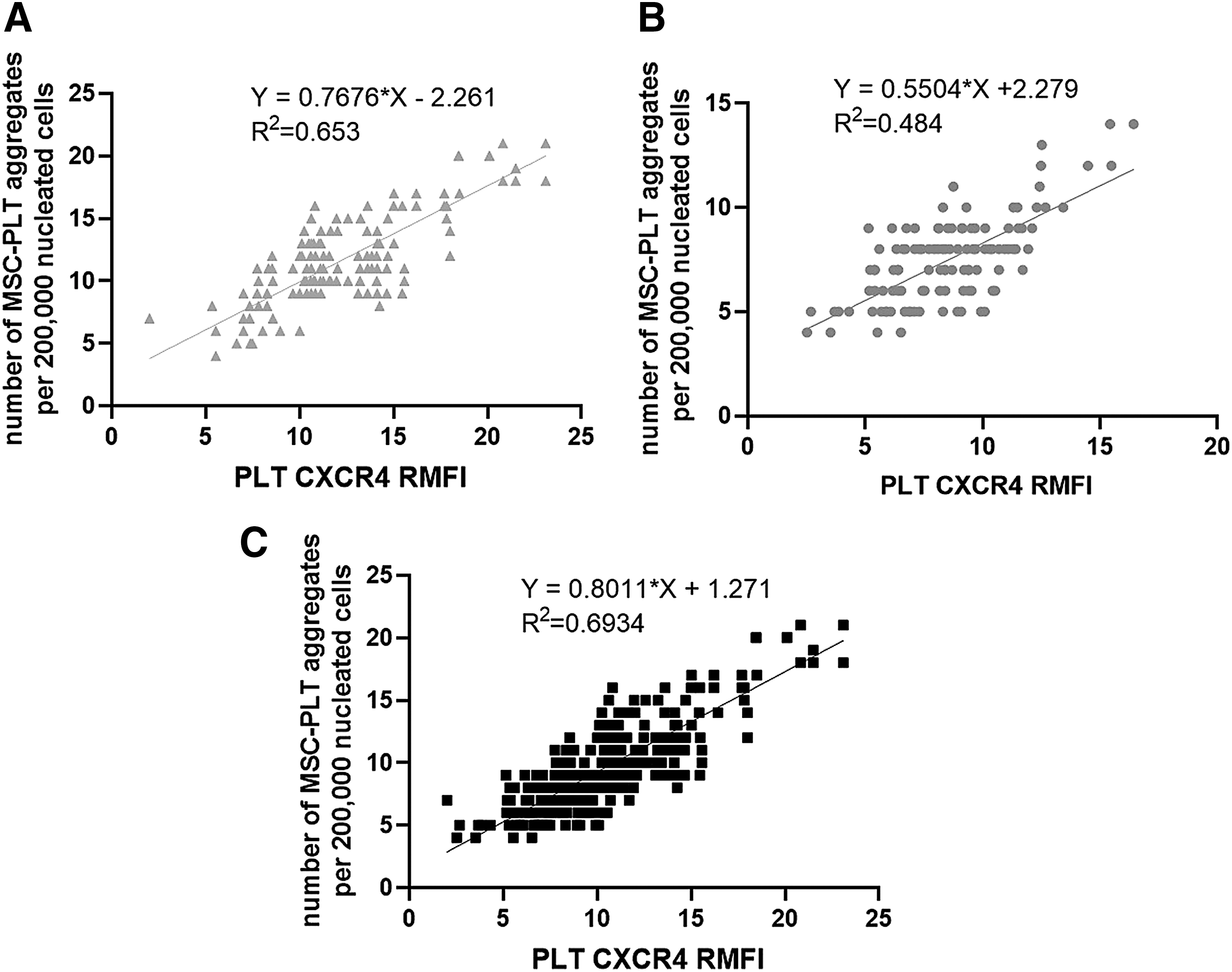
Correlation between RMFI of PLT CXCR4 and the number of MSC–PLT aggregates.
Patient follow-up
As MSCs and PLTs are related to myocardial repair mechanisms, we performed follow-up of 132 AMI patients for 36 months to assess a possible influence of the number of MSC–PLT aggregates on patients' prognosis after AMI. A total of 27 patients who were lost to follow-up were excluded. To perform ROC analysis, the number of MSC–PLT aggregates was plotted against the survival status for each patient. The AUC was 0.711, and the best cutoff value was selected as 10.5 (Fig. 4A). The patients were then dichotomized into a high- (≥11, n = 62/105) and low-MSC–PLT aggregates (<11, n = 43/105) group. For the whole cohort, the mean duration of follow-up was 19.8 months. The mean survival time was slightly higher for the high-MSC–PLT aggregates group than that for the low-MSC–PLT aggregates groups (20.61 ± 11.19 months vs. 18.51 ± 10.32 months), but no significant difference was found between the two groups (Fig. 4B). However, the percentage of patients at the primary end point was lower in the high-MSC–PLT aggregates group than in the low-MSC–PLT aggregates group (3% vs. 16%), and the survival curves demonstrated significant difference between the high- and low-MSC–PLT aggregates groups (Fig. 4C, P < 0.001). No differences were found in white blood cells and PLT count between low- and high-MSC–PLT groups in healthy controls, AMI patients, and all individual groups, but the number of total MSCs was much higher in the high MSC–PLT group than in the low MSC–PLT group in all the three groups. We found no differences in the expression of SDF-1 and CXCR4 between the low- and high-MSC–PLT groups (Table 2). There was no significant difference in drug use between the two groups.
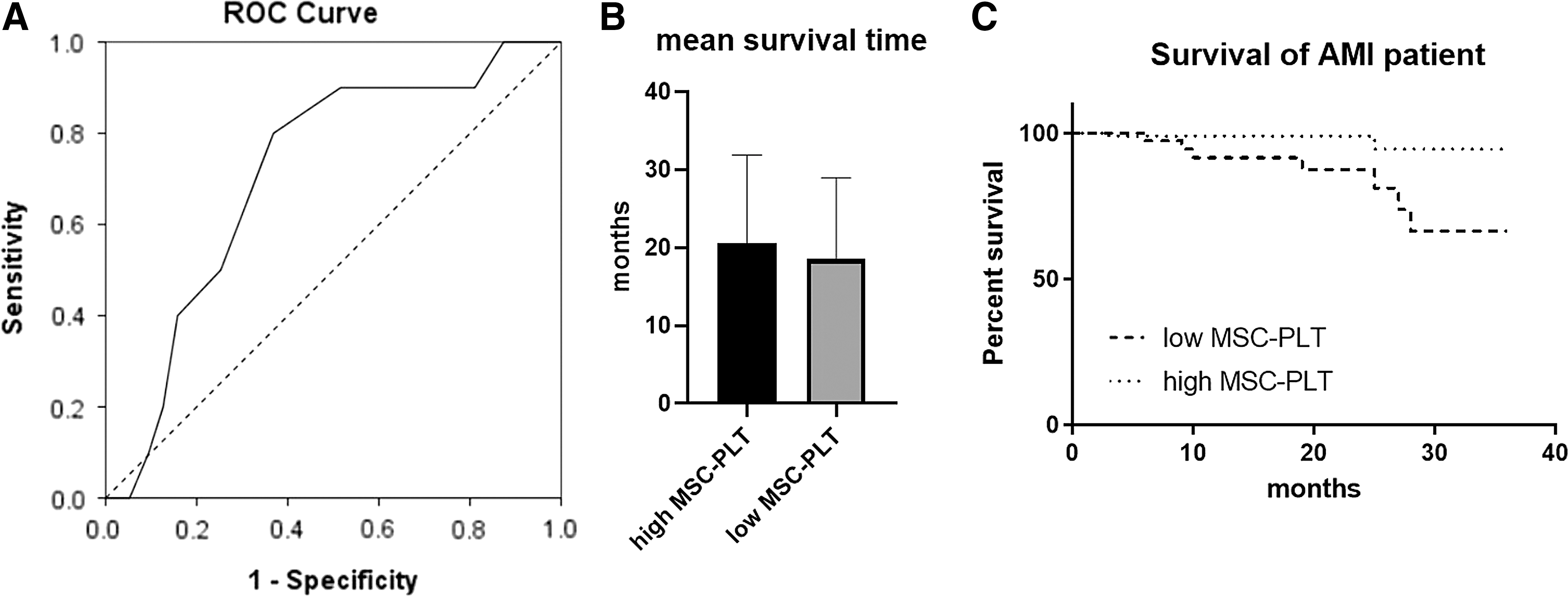
Survival analysis.
Comparison of White Blood Cells, Platelets, and Total Mesenchymal Stem Cells and Expression of Stromal Cell-Derived Factor 1 and CXC Chemokine Receptor 4 on Platelets in Low- and High-Mesenchymal Stem Cell–Platelet Groups
P < 0.05, data are presented as mean value ± SD.
CXCR4, CXC chemokine receptor 4; RMFI, relative mean fluorescence intensity; SDF-1, stromal cell-derived factor 1.
Preparation of engineered SDF-1 overexpression MSCs
MSCs were induced to overexpress SDF-1 by transduction with an adenoviral vector encoding SDF-1 (Fig. 5A). MSCs expressed GFP with 75% and 100% of luminescence rates at 36 and 48 h, respectively, after transduction with recombinant Ad-SDF-1 at a multiplicity of infection (viruses/cells) of 1,500 (Fig. 5C, D).
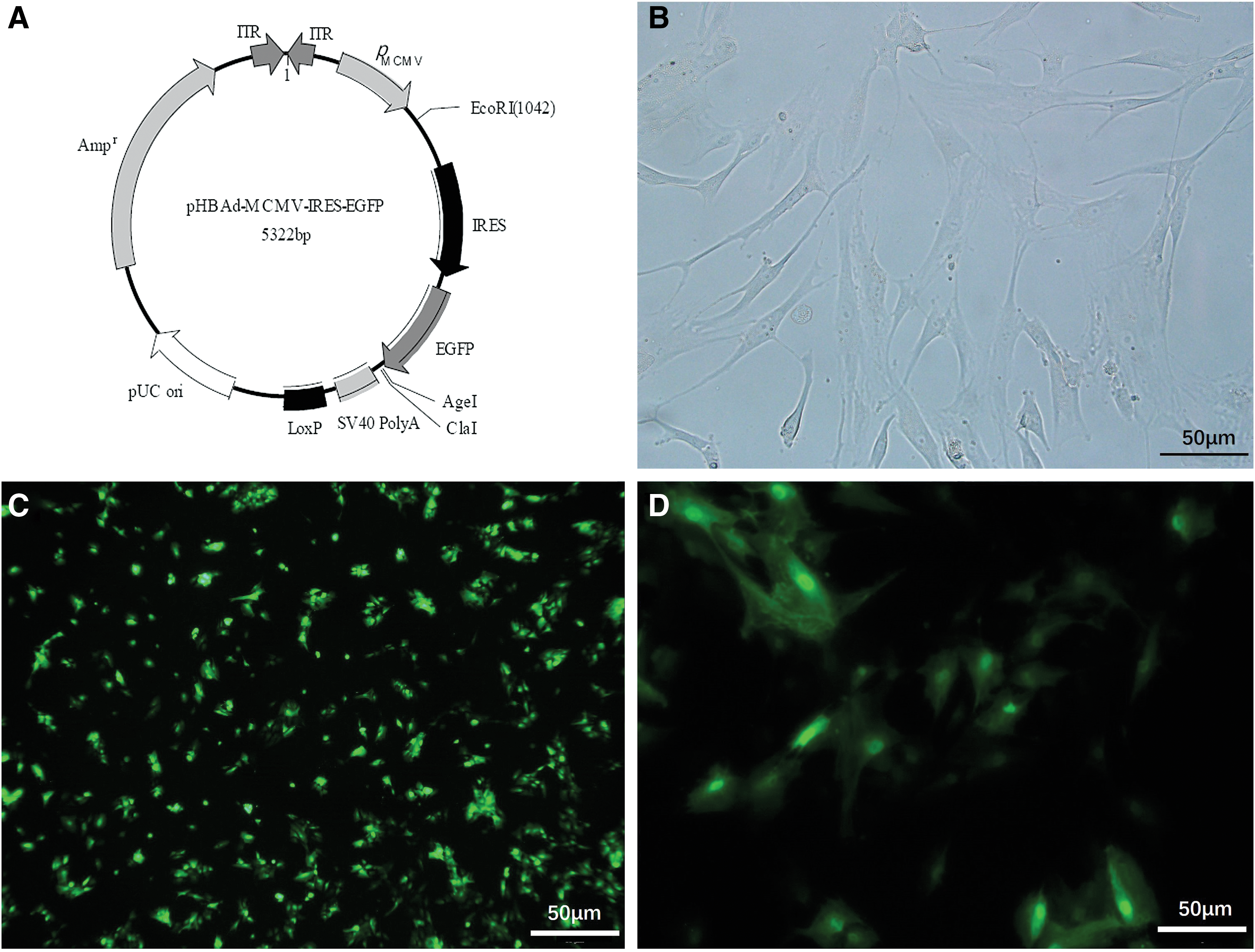
The phase-contrast and fluorescence images of transduced MSCs.
Cocultures of SDF-1 overexpression MSCs and PLTs from AMI patients
To analyze the role of SDF-1/CXCR4 axis in the formation of MSC–PLT aggregates, we constructed SDF-1 overexpression MSCs. They were cocultured with PLTs from AMI patients and healthy controls, and the number of PLTs on the MSCs and MSC–PLT aggregates/MSCs ratio was counted after immunofluorescence staining (Fig. 6A–D). The number of PLTs on each MSC and the number of MSC–PLT aggregates/MSCs were found increased in SDF-1 overexpression MSCs cocultured with AMI patient PLTs compared with those cocultured with healthy control PLTs and MSCs transduced with blank virus cocultured with both kinds of PLTs (Fig. 6E, F).
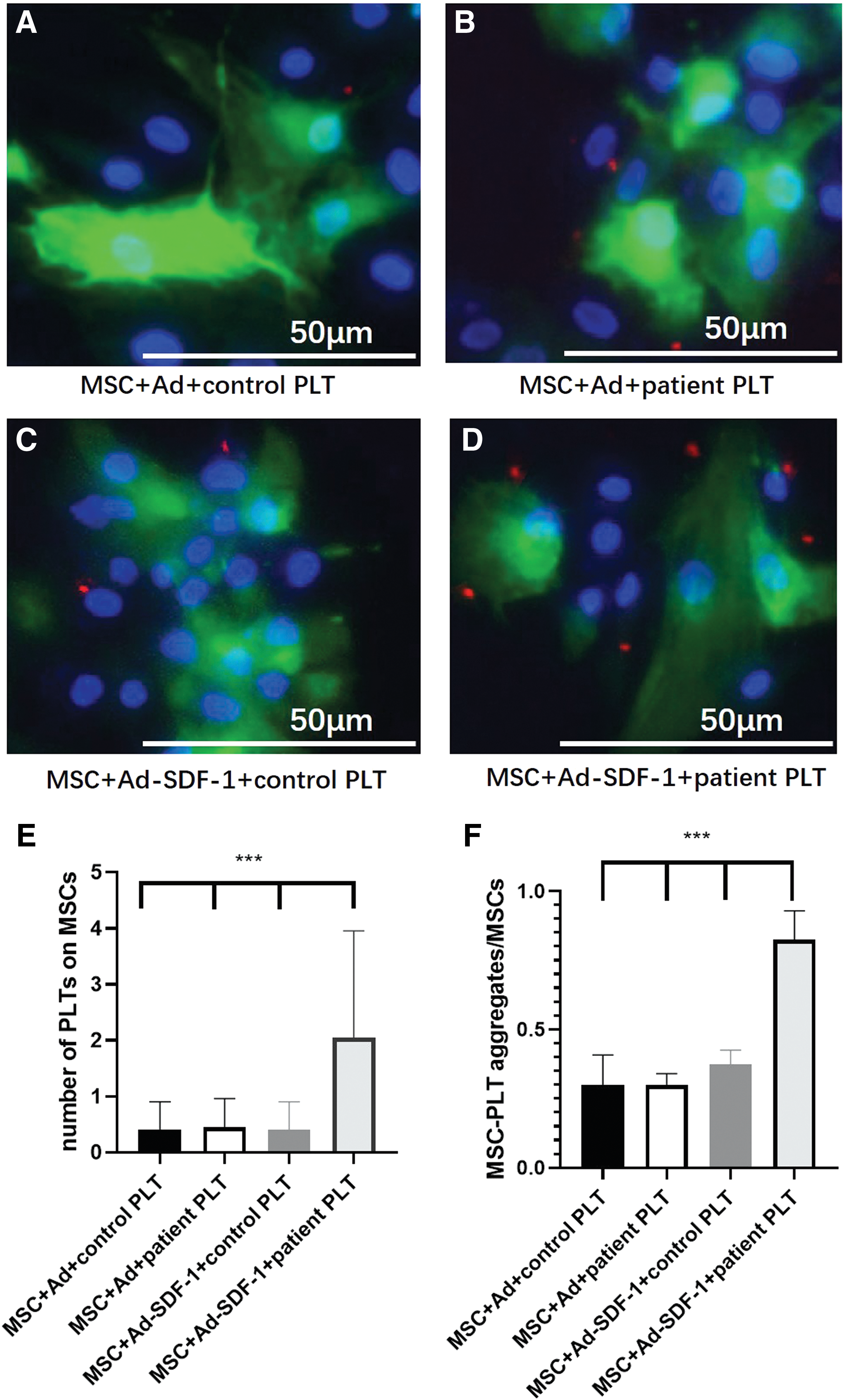
Combination of PLTs to SDF-1 overexpression MSCs to form aggregates.
Cocultures of unmodified MSCs and PLTs from AMI patients with or without CXCR4 inhibitor
AMD3100, a small bicyclam molecule, is a CXCR4 antagonist, which was utilized to assess whether the formation of MSC–PLT aggregate was induced through the binding of SDF-1 to its receptor. MSCs and PLTs from AMI patients were cocultured with or without AMD3100, using PLTs from healthy controls as the control (Fig. 7A–F). We found that the average PLT number combined with MSCs was much higher in MSCs cocultured with AMI patient PLT than in MSCs cocultured with healthy control PLT (3.94 ± 1.75 vs. 0.83 ± 0.87, P < 0.0001), and the combination was blocked by the AMD3100 (3.94 ± 1.75 vs. 1.53 ± 1.01, P < 0.0001) (Fig. 7G).
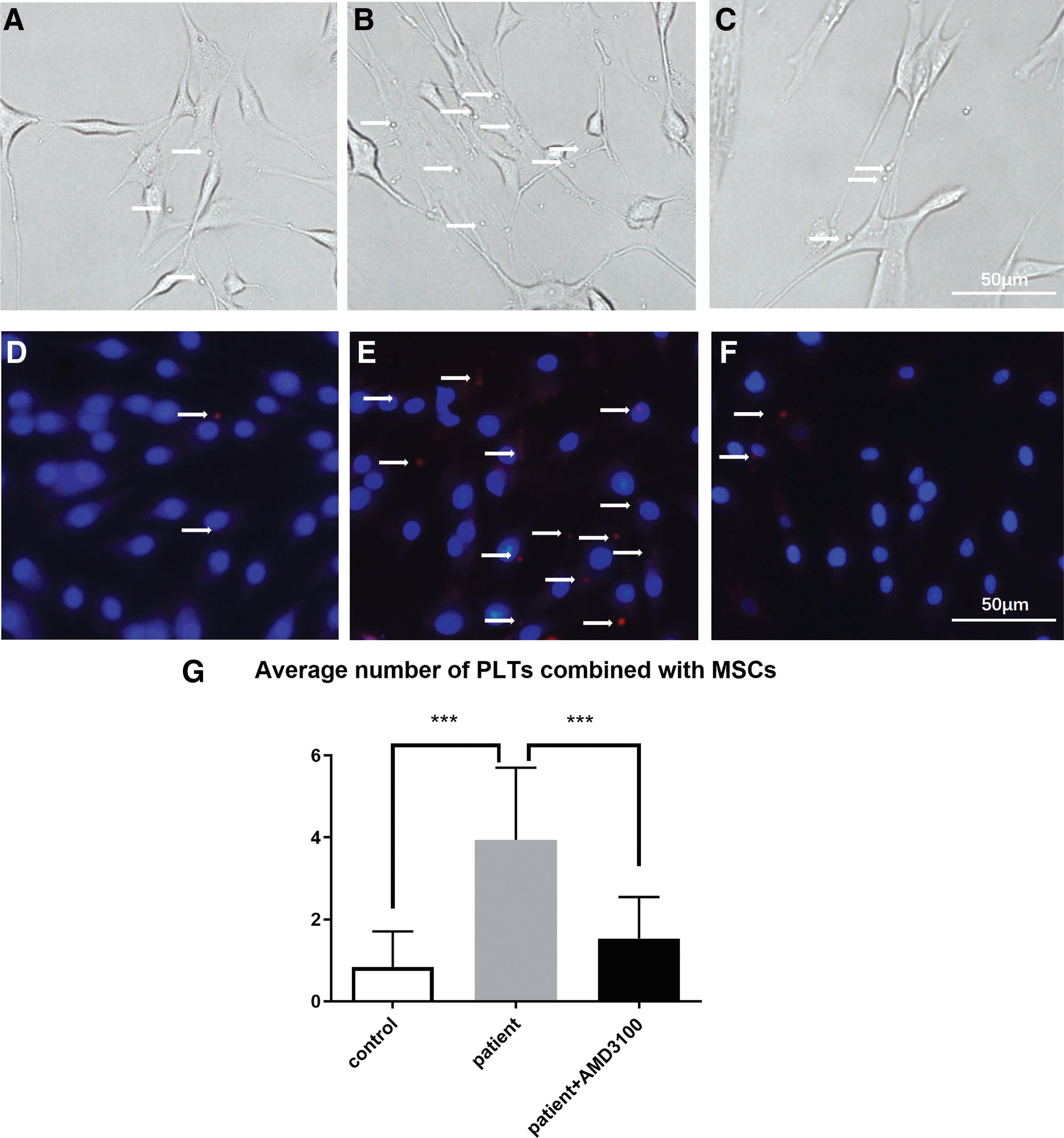
The phase-contrast and fluorescence images of PLTs (arrows) cocultured with MSCs.
Real-time quantitative polymerase chain reaction and western blotting
We quantified SDF-1 and CXCR4 mRNA levels in MSCs cocultured with PLTs from AMI patients and healthy controls, using MSCs without PLTs as control. Both SDF-1 and CXCR4 mRNA levels increased in MSCs cocultured with PLTs from AMI patients compared with MSCs cocultured with PLTs from healthy controls and MSCs alone (Fig. 8A). Western blotting was performed for measurement of SDF-1 and CXCR4 protein levels. Both SDF-1 and CXCR4 protein levels were significantly increased in MSCs cocultured with PLTs from AMI patients and were blocked by AMD3100.
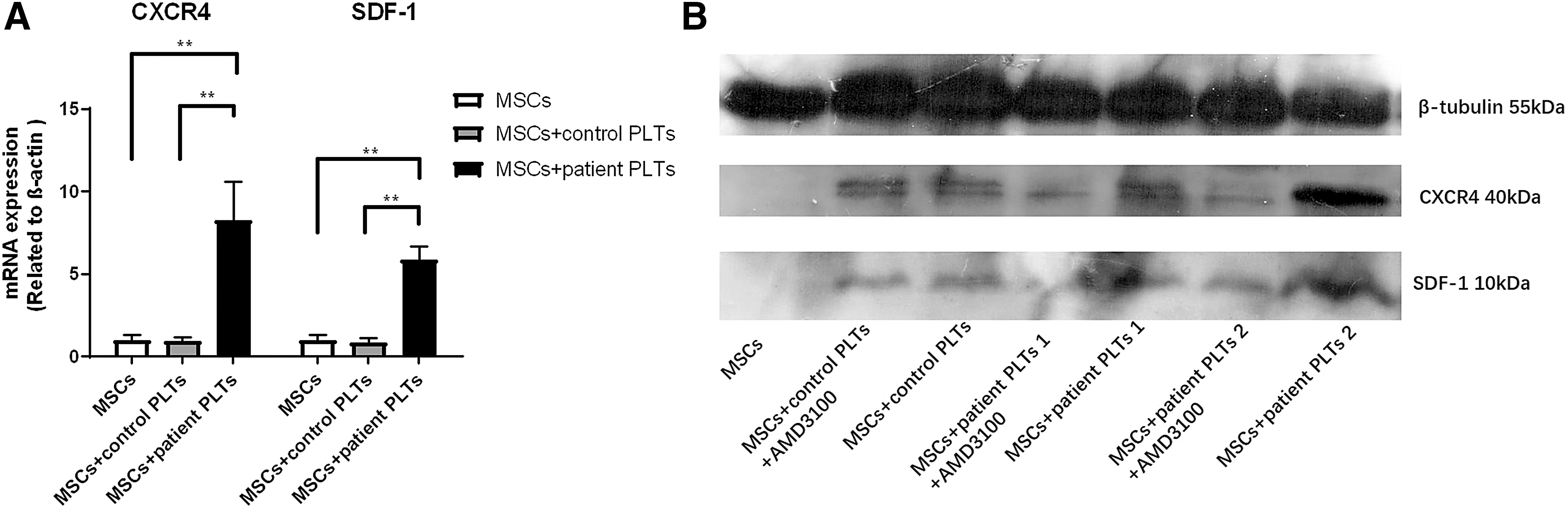
SDF-1/CXCR4 mRNA and protein levels analysis.
Discussion
In the present study, we found that AMI patients had more MSC–PLT aggregates than healthy controls. The percentages of patients at primary end point and the survival curves demonstrated significant difference between high- and low-MSC–PLT aggregates groups in AMI patients, which suggested that AMI patients with higher MSC–PLT aggregates had better prognosis. Meanwhile, we found that CXCR4 expression was increased on the PLT surface and was correlated with the number of MSC–PLT aggregates in AMI patients. To investigate the role of SDF-1/CXCR4 axis in MSC–PLT aggregate formation, we conducted a series of experiments, including overexpression SDF-1 in MSCs in vitro, coculture of overexpression SDF-1 MSCs and PLTs from AMI patient, and blocking CXCR4 in coculture of MSCs and PLTs. All these results illustrated that SDF-1/CXCR4 was crucial in MSC–PLT aggregate formation.
Transplantation of autologous or allogeneic MSCs into the ischemic myocardium has emerged as a promising therapy for myocardial repair [22]. MSCs transplantation after AMI dose has been proven by several clinical trials to improve cardiac function [6 –8]. The mechanisms by which MSCs improve cardiac function are controversial. Whether it is by regeneration of cardiac myocytes, modulation of remodeling, or preservation of injured tissue through paracrine function [5] remains unrevealed.
The study of PLT function has been traditionally focused on its roles in physiological hemostasis and pathological thrombosis. However, PLTs have more complex pathophysiological roles since they can release over 300 proteins (comprising growth factors, chemokines, and adhesive ligands), nucleotides, and neurotransmitters stored in 3 different classes of secretable granules (α−, dense, and lysosomal granules) [23]. Meanwhile, PLT microparticles/exosomes that account for the most abundant form of extracellular vesicles in circulation [24] add the complexity of PLT function by loaded with multiple bioactive molecules including proteins, lipids, and microRNAs [25]. PLTs are also considered the healer of damaged tissue because it has been recognized to be majorly involved in all the cellular events impacting tissue repair, indeed including cellular migration, extracellular matrix remodeling, cell proliferation, differentiation, and angiogenesis [26]. Although roles of PLTs in coronary thrombosis and subsequent cardiac damage are widely accepted, there is sufficient credible evidence to imply that PLTs have beneficial roles in cardiac repair processes by releasing bioactive cargos [18].
Several studies have demonstrated an increase of PLT–neutrophil complexes after MI [27,28]. PLT–neutrophil complexes and neutrophils accumulate in the myocardium, which is thought to further exacerbate the extent of myocardial damage [29]. In this study, we found that MSC–PLT aggregates increased in AMI patients' circulation, and the more the MSC–PLT aggregates were in peripheral blood, the longer the patients would survive. It is clear that increased MSC–PLT aggregates are beneficial to myocardial repair. We speculate that MSC–PLT aggregate formation may benefit myocardial repair processes by several means. First, PLT can serve as nutritional support for MSCs and is able to stimulate and proliferate their expansion. PLT represents a natural reservoir of soluble mediators, including PLT-derived growth factor-AB/BB, transforming growth factor-β isoform 1, insulin-like growth factor 1, epidermal growth factor, vascular endothelial growth factor, fibroblast growth factor-2, hepatocyte growth factor, and bone morphogenetic protein-2 [30]. As reported recently, human PLT lysate made from intercept-treated PLT concentrates can replace fetal bovine serum as xeno-free supplement for ex vivo expansion of MSCs [31]. Second, combination of PLT and MSC can play a greater role than a single cell. For instance, combined use of BM-MSCs and PRP can more effectively stimulate proliferation and differentiation of myoblasts in vitro than PRP alone in skeletal muscle regeneration [32]. In another study, combination of MSCs and PRP increases tenocytes and MSC migration speed, as well as extracellular matrix protein production compared with the use of MSCs alone on tenocyte artificial wound healing [33]. Third, PLT can act as a medium for adhesion of MSCs to other cells. MSC–PLT aggregate as a whole can adhere to myocardial injury sites, and these specific contact-based abilities MSC possessed can be in action. For example, direct contact of MSCs cultured atop fixed endothelial cells (EC) or its EC medium would coax the former to differentiate into EC [34], and the complicated mechanisms of PLT adhesion to other cells nowadays are gradually revealed. For instance, the main mechanisms of PLT adhesion to the activated endothelium involve PLT alphaIIbbeta3, endothelial alphavbeta3, and intercellular adhesion molecule 1 [35]. The specific mechanism involved in MSC–PLT aggregates beneficial to myocardial repair needs further experiments to verify.
SDF-1 is an important stem cell homing factor, and its expression is elevated in most tissues secondary to injury [36]. When SDF-1 binds to its receptor CXCR4, a G-protein coupled receptor, SDF-1 signaling is initiated [37,38]. Mobilization and migration of bone marrow-derived progenitors [39] and MSCs are mainly controlled by the SDF-1/CXCR4 axis [40], which has an instrumental role during cardiac development. In several studies using rodent model systems [36,41,42], it was demonstrated that the SDF-1/CXCR4 axis played an important role in impaired post-AMI cardiac repair and was a potential therapeutic target for ventricular remodeling after AMI. In this study, we found that SDF-1/CXCR4 was critical in MSC–PLT aggregate formation, which might indicate that SDF-1/CXCR4 axis acted in MSCs homing and cardiac repair after AMI.
It has been reported that activated PLTs secrete SDF-1, thus supporting further primary adhesion and migration of progenitor cells [43]. When analyzing the expression of SDF-1 and CXCR4 on the surface of PLTs, we did not add prostaglandin E1 to inhibit PLT activation when blood samples were collected. Therefore, SDF-1 expression did not increase on PLT surfaces in our study, probably because SDF-1 has already been released into the blood, which is not consistent with the results in previous research [44]. However, we did detect an increase in the relative fluorescence intensity of CXCR4 on PLT surface.
Our study had several limitations. First, we included a small number of participants (132 AMI patients and 124 healthy controls), which was due to the difficulty in recruiting volunteers. The high-MSC–PLT aggregate group had only 43 patients, and 3 patients reached the primary end point around the 24-month follow-up, 1 for stroke and 2 for hospitalization for worsening heart failure. The small number of cases might be the major cause of the sudden worsening of AMI patients. More cases need to be included to confirm this result. Second, we did not analyze the SDF-1/CXCR4 expression on MSC–PLT aggregates by flow cytometry because of the complexity of antibody combination in identifying MSCs. Third, in vivo experiments should be designed to validate roles of MSC–PLT aggregates in MSC homing and cardiac repair after AMI, which can be carried out in our future research.
In conclusion, we found increased MSC–PLT aggregates in AMI patients compared with healthy controls, and AMI patients with higher MSC–PLT aggregates had better prognosis. Meanwhile, RMFI of PLT CXCR4 and the number of MSC–PLT aggregates showed a significant correlation in AMI patients. In vitro experiments demonstrated that SDF-1/CXCR4 was crucial in MSC–PLT aggregate formation, which might suggest a novel mechanism that SDF-1/CXCR4 is involved in MSC homing and myocardial repair after AMI. Increasing the expression of SDF-1 on MSCs and promoting the formation of MSC–PLT aggregates may be another strategy to encourage myocardial repair through MSCs in AMI patients.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was funded by the National Natural Science Foundation of China (81301493).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.