
Abstract
Induced pluripotent stem cell (iPSC) technology has great promise in regenerative medicine and disease modeling. In this study, we show that human placenta-derived cell conditioned medium stimulates chemokine (C-X-C motif) receptor 2 (CXCR2) in human somatic cells ectopically expressing the pluripotency-associated transcription factors Oct4, Sox2, Klf4, and cMyc (OSKM), leading to mechanistic target of rapamycin (mTOR) activation. This causes an increase in endogenous cMYC levels and a decrease in autophagy, thereby enhancing the reprogramming efficiency of human somatic cells into iPSCs. These findings were reproduced when human somatic cells after OSKM transduction were cultured in a widely used reprogramming medium (mTeSR) supplemented with CXCR2 ligands interleukin-8 and growth-related oncogene α or an mTOR activator (MHY1485). To our knowledge, this is the first report demonstrating that mTOR activation in human somatic cells with ectopic OSKM expression significantly enhances the production of iPSCs. Our results support the development of convenient protocols for iPSC generation and further our understanding of somatic cell reprogramming.
Introduction
Human induced pluripotent stem cells (iPSCs) are reprogrammed from human somatic cells by the ectopic expression of the pluripotency-associated transcription factors Oct4, Sox2, Klf4, and cMyc (collectively referred to as OSKM).
They can also be used for clinical translation [1,2]. Thus, diverse reprogramming modulators have been investigated to improve the efficiency of reprogramming somatic cells into iPSCs. However, when combined with the expression of OSKM, many of these modulators do not enhance the overall reprogramming efficiency significantly, raising concerns regarding the convenience of these reprogramming procedures [3 –5]. Alternatively, researchers have been manipulating intracellular factors to enhance the reprogramming efficiency of human somatic cells. One of these factors is the mechanistic target of rapamycin (mTOR), a serine/threonine protein kinase that regulates cell growth, cell proliferation, cell motility, cell survival, protein synthesis, autophagy, and transcription [6]. Until now, most studies have focused on the inhibition of mTOR pathways, where reprogramming efficiency was shown to increase [7 –10]. However, the reported effects are diverse, and the reprogramming mechanism has not been elucidated. Recently, it was shown that a short treatment with rapamycin enhances reprogramming, a longer treatment decreases reprogramming, and an mTOR knockout has a negative impact on reprogramming [11]. Moreover, it was demonstrated that mTOR inhibition induces a paused pluripotent state in mouse blastocysts with a remarkable global suppression of transcription [12]. These findings highlight the complex role of mTOR in the reprogramming of somatic cells to iPSCs and how the mechanism underlying its role in reprogramming remains unknown. In this study, we used an experimental strategy that explored endogenous factors to enhance the reprogramming of human somatic cells, focusing on mTOR as a target. To develop an efficient protocol for reprogramming human somatic cells through mTOR manipulation, we used the human placenta-derived cell conditioned medium (hPCCM), which supports human pluripotent stem cells (hPSCs) through chemokine (C-X-C motif) receptor 2 (CXCR2)- and mTOR-dependent mechanisms [13,14]. This kind of approach has rarely been reported in reprogramming studies.
Materials and Methods
Cell culture
Human dermal fibroblasts (HDFs) and human umbilical vein endothelial cells (HUVECs) were obtained from the American Type Culture Collection (ATCC, Manassas, VA), and human embryonic cells (H1) were purchased from the WiCell Research Institute (Madison, WI). All cells were handled according to the supplier instructions. Human placental cells (HPCs) and hPCCM were collected as previously described. Manipulations and cell culture were performed in a clean germ-free facility at the stem cell laboratory of the Korea University Medical Center (Seoul, South Korea). The Institutional Review Board of Anam Hospital of the Korea University Medical Center approved the experimental design and procedures conducted in this study (2014AN0128).
Gene silencing using short hairpin RNA lentivirus
Lentiviral particles containing short hairpin RNA (shRNA) targeting CXCR2 (sc-40028-V), mTOR (sc-35409), β-catenin (sc-29209-V), and control shRNA (sc-108080; shControl) were purchased from Santa Cruz Biotechnology (Dallas, TX). For viral infection, HUVECs were seeded into a 24-well plate and cultured overnight. The next day, cells were treated with 6 μg/mL polybrene for 15 min and then infected with the viral particles (multiplicity of infection [MOI] = 10). After 24 h, the infection medium was replaced with fresh medium without polybrene, and stable cell lines expressing shRNA were generated by selection with puromycin (2 μg/mL). Silencing was confirmed by quantitative polymerase chain reaction (qPCR) and western blotting.
Gene overexpression using lentiviral activation particles
For CXCR2 activation experiments, we used lentiviral particles (sc-401404-LAC) and control lentiviral activation particles (sc-437282), both of which were purchased from Santa Cruz Biotechnology. HDFs were seeded in 24-well plates and cultured overnight. Cells were then treated with 6 μg/mL polybrene and infected with the viral particles (MOI = 5). Thereafter, cells were cultured in a medium containing 2 μg/mL puromycin. Overexpression was confirmed by qPCR and western blotting.
Cell reprogramming
Human somatic cells were reprogrammed using the CytoTune-iPS 2.0 Sendai Reprogramming Kit (Life Technologies, Carlsbad, CA) according to manufacturer's instructions. Cells were seeded (1 × 105 cells/well) into six-well plates. Two days later, the cells were transduced with OSKM-overexpressing Sendai viruses at MOI = 5. After 7 days, the cells were reseeded onto 0.1% gelatin-coated dishes containing hPCCM or cultured on Matrigel-coated dishes containing the mTeSR medium. Daily media change was performed thereafter. To confirm pluripotency, cells were live stained with a TRA1–60 antibody (Stemgent, Cambridge, MA).
Live staining
The StainAlive TRA-1-60 antibody was diluted 1:100 in Dulbecco's modified Eagle's medium (DMEM)/F-12 media and directly applied onto embryonic-like cell cultures. All staining procedures were conducted according to product specifications. After visualization or manual selection, the staining medium was replaced with normal culture medium to continue reprogramming.
Detection of alkaline phosphatase activity
Alkaline phosphatase (ALP) activity was detected using the ES Cell Characterization Kit (MilliporeSigma, Burlington, MA) according to manufacturer's instructions. The stained cells were examined and imaged using the IX71 microscope (Olympus, Tokyo, Japan).
Immunofluorescence
For immunofluorescence staining, iPSCs were cultured and fixed in eight-well slide chambers (BD Biosciences, Franklin Lakes, NJ) with 4% (w/v) paraformaldehyde, permeabilized with 0.1% (v/v) Triton X-100, and blocked for 1 h with 3% (v/v) normal horse serum (Gibco; Thermo Fisher Scientific, Waltham, MA) in phosphate-buffered saline (PBS) containing 0.1% (v/v) Tween 20 (Sigma-Aldrich, St. Louis, MO). Subsequently, the cells were incubated with a primary antibody overnight at 4°C and then with a secondary antibody for 1 h at 20–25°C. Between incubations, the cells were washed three to five times with 0.1% (v/v) Tween 20 in PBS. Before mounting the slides, the cells were incubated with 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes, Invitrogen, Carlsbad, CA) for 5 min in the dark. Fluorescence was preserved by an antifade mounting medium (Vector Labs, Burlingame, CA), and the cells were observed under a fluorescence microscope (Olympus). The primary antibodies used were as follows: SSEA-4 (MilliporeSigma); OCT-4 and p-mTOR (Cell Signaling Technology, Danvers, MA); CXCL1, CXCR2, and Nestin (Abcam, Cambridge, UK); β-catenin (Invitrogen); Nanog, AFP, and Desmin (Santa Cruz Biotechnology); and TUj1 (Covance, Princeton, NJ). Secondary antibodies (anti-rabbit IgG or anti-mouse IgG Alexa Fluor 488- or 594-conjugated) were purchased from Invitrogen.
Senescence-associated β-galactosidase staining
Senescence-associated β-galactosidase staining was performed on confluent somatic cells in 24-well plates using the Senescence Cells Histochemical Staining Kit (CS0030; Sigma-Aldrich) according to manufacturer's instructions. Briefly, cells were fixed in 1 × fixation buffer for 6 min at 20°C and stained overnight at 37°C outside the CO2 incubator. The next day, the cells were analyzed under a phase-contrast microscope. Blue-stained cells and the total number of cells were counted, after which the percentage of cells expressing β-galactosidase (senescent cells) was calculated.
Reverse transcription qPCR
Total RNA was isolated from cells using the Qiagen RNeasy Kit (Qiagen, Hilden, Germany), and the extracted RNA was quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific). cDNA was synthesized by adding 2 μg of total RNA to a 20-μL reaction mixture containing oligo (dT) primers and Superscript II reverse transcriptase (Gibco), according to manufacturer's instructions. Synthesized cDNA was amplified using a Bio-Rad iCycler iQ system with iQ SYBR Green qPCR Master Mix (Bio-Rad Laboratories, Hercules, CA). Primers used for qPCR are listed in Supplementary Table S1. The cycle threshold values of genes of interest were normalized to those of glyceraldehyde-3-phosphate dehydrogenase (GAPDH).
Flow cytometry
Cells were dissociated into single-cell suspensions in cold fluorescence-associated cell sorting (FACS) buffer (0.1% [v/v] bovine serum albumin [BSA] in phosphate buffered saline [PBS]). These cells were then incubated with nonconjugated primary antibodies against SSEA-4, SSEA-1, OCT-4, and SOX2 (from R&D Systems, Minneapolis, MN) and TRA-1-60 and TRA-1-81 (from MilliporeSigma) for 1 h on ice in the dark and then washed thrice with cold FACS buffer. Control cells were incubated with immunoglobulins (IgG and IgM) and then with the secondary antibodies as described above. Data were acquired using the BD FACSCanto II (Becton Dickinson, Franklin Lakes, NJ) and analyzed using FlowJo software (FlowJo LLC., Ashland, OR).
Western blotting
Cells were washed with cold PBS and lysed in lysis buffer (20 mM KCl, 150 mM NaCl, 1% NP-40, 50 mM NaF, 1 mM dtt dl-dithiothreitol (DTT), 1 mM ethylenediaminetetraacetic (EGTA) disodium salt, 1 × protease inhibitor, 10% glycerol, and 50 mM Tris-HCl, pH 7.5) for 30 min on ice. The lysate was then centrifuged at 13,400 × g for 15 min at 4°C. The protein concentration in the supernatant was determined by the Bradford assay (Bio-Rad Laboratories). The protein samples (30 μg) were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto polyvinylidene fluoride membranes. After blocking the nonspecific antibody-binding sites with skim milk for 1 h, the membranes were probed with primary antibodies (diluted 1:1,000) at 4°C overnight, followed by incubation with horseradish peroxidase-conjugated secondary antibodies (diluted 1:2,000) for 1 h. The primary antibodies used were as follows: CXCR2 (Abcam); OCT4, mTOR, p-mTOR, and β-catenin (Cell Signaling Technology); and Nanog and β-actin (Santa Cruz Biotechnology). The secondary antibodies anti-rabbit IgG and anti-mouse IgG were obtained from Cell Signaling Technology and Bio-Rad Laboratories, respectively. Chemiluminescent signals were developed using an ECL reagent (GE Healthcare, Chicago, IL) and a ChemiDoc Imaging System (Bio-Rad Laboratories).
Human cytokine array
The human cytokine array C3 (RayBiotech, Norcross, GA) was used according to manufacturer's instructions. Briefly, the cytokine array membranes were incubated overnight at 4°C in the conditioned media obtained from the basal medium of each somatic cell line. The next day, membranes were incubated with primary antibodies, and signals were detected using the ChemiDoc Imaging System.
Embryoid body differentiation
iPSCs were transferred to low-attachment surface plates and allowed to spontaneously differentiate through embryoid body (EB) formation in DMEM-F12 medium supplemented with 20% knockout serum replacement, 1% nonessential amino acids, and 0.1 mM β-mercaptoethanol (all from Gibco); the medium was changed every 2–3 days. After 1 week in suspension, the EBs were transferred to gelatin-coated dishes and cultured for 1 week. The embryoid bodies were then fixed and stained with antibodies against the markers of each embryonic germ layer (endoderm, AFP; mesoderm, desmin; ectoderm, TUJ1, and Nestin) and analyzed for immunofluorescence.
Teratoma formation
Teratomas were obtained by the subcutaneous implantation of ∼5–10 × 106 iPSCs into 5-week-old immune-deficient nonobese diabetic/severe combined immunodeficient mice. Mice were maintained under specific pathogen-free conditions. Teratoma growth was determined by palpation every week, and the mice were sacrificed 8–9 weeks after implantation. Teratomas were then fixed, and the sections were stained with hematoxylin and eosin. All animal experiments were approved by the Ethics Committee of the Korea University Medical School (KOREA-2016-0155-C1).
Karyotype analysis
The karyotyping of each cell line was carried out by G-banding as previously described [13]. Briefly, iPSCs were incubated in 0.075 M KC1 for 20 min at 37°C. After fixation with a solution of 3:1 methanol/acetic acid, the karyotype of iPSCs was determined at the 300-band level of resolution.
Short tandem repeat genotyping
Genomic DNA was extracted from somatic cells and iPSCs using a QIAamp® DNA Micro Kit (Qiagen) according to manufacturer's instructions. Sixteen different genetic loci were amplified from the extracted DNA using the PowerPlex 16 System Kit (Promega, Madison, WI) or the AmpFLSTR® Identifiler® PCR Amplification Kit (Applied Biosystems, Foster City, CA), after which capillary electrophoresis was carried out using a 3130xl Genetic Analyzer (Applied Biosystems).
Statistical analysis
Statistical significance was determined using two-tailed Student's t-tests or two-way analysis of variance (ANOVA) followed by Tukey's test for multiple comparisons. All experiments were performed in triplicate. P values <0.05 were considered statistically significant.
Results
hPCCM promotes reprogramming of human somatic cells through CXCR2 signaling
We assessed reprogramming efficiency using three different somatic cell types—HUVECs, HPCs, and HDFs. Cells were infected with Sendai viruses encoding Oct4, Sox2, Klf4, and cMyc. After 7 days, the transduced cells were transferred to hPCCM (Fig. 1A). iPSC colonies appeared within 24 h in hPCCM, and their characteristics were confirmed using TRA-1-60 live staining (Fig. 1B). The reprogramming efficiency of HUVECs was significantly higher compared with the other cell lines at 7 days after exposure to hPCCM (P < 0.05; Fig. 1C, D). We determined the constitutive expression levels of CXCR2 in each somatic cell line before transduction and found that it was strongly expressed by HUVECs at both mRNA and protein levels (P < 0.05; Supplementary Fig. S1A, B). Moreover, the iPSC colonies expressed pluripotency markers (OCT4, Nanog, Rex-1, and SSEA4) and were able to differentiate into the three germ layer derivatives in vitro and in vivo (Supplementary Fig. S1C–F). The hPCCM-induced iPSC lines presented normal karyotypes (Supplementary Fig. S2A), and short tandem repeat (STR) analyses showed that they correspond to the karyotypes of their origin cells (Supplementary Fig. S2B). To assess the expression of CXCR2 signaling during reprogramming, we analyzed protein expression at different time points after OSKM transduction (Fig. 1E, Supplementary Fig. S2C, D). High levels of phosphorylated mTOR and β-catenin, together with increased CXCR2 levels, were detected 1 day after HUVECs were exposed to hPCCM. Moreover, the expression levels of the pluripotency-associated markers Nanog and OCT4 increased 3 days after the medium was changed to hPCCM (Fig. 1E, F). These data indicate that CXCR2, mTOR, and β-catenin signaling occurs during reprogramming, while hPSC characteristics are maintained [13,14]. Subsequently, we compared the efficiency of the hPCCM protocol with that of the ordinarily recognized reprogramming protocols (E8 or mTeSR) using ALP staining. Cells cultured in hPCCM had significantly higher reprogramming efficiency than those cultured in E8 or mTeSR at 14 days following OSKM transduction (P < 0.01 and P < 0.001, respectively; Fig. 1G, H). To further investigate whether hPCCM can facilitate reprogramming, we introduced hPCCM 1 day after OSKM transduction. We found that the reprogramming duration was markedly shortened, resulting in increased reprogramming (hPCCM, 14 days vs. E8, 21 days; Fig. 1G and Supplementary Fig. S3A–D). Based on our previous findings [13,14], we hypothesized that the CXCR2 ligands in hPCCM stimulate CXCR2 in human somatic cells and enhance reprogramming.
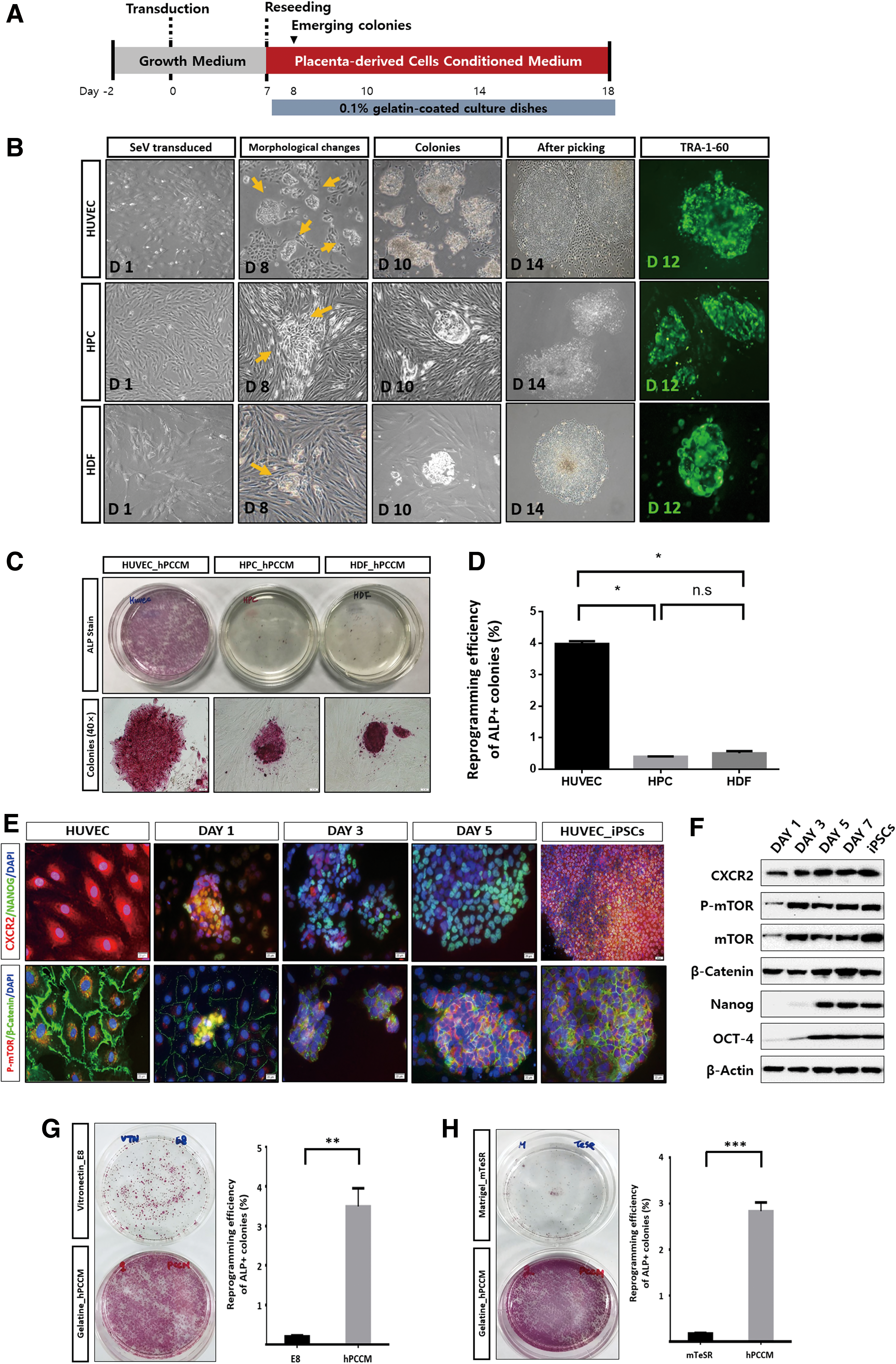
Generation of iPSCs in hPCCM through CXCR2 signaling.
Degree of CXCR2 stimulation affects reprogramming efficiency to iPSCs
We next investigated the role of CXCR2 in somatic cell reprogramming by altering its levels and assessing reprogramming efficiency. To knockdown CXCR2 expression, HUVECs showing high CXCR2 expression were infected with lentiviruses containing shRNA targeting CXCR2. CXCR2 levels decreased by 70%–90% compared with those of shControl, both at the protein (Fig. 2A) and mRNA (Fig. 2B) levels. We then measured ALP activity in the CXCR2-silenced HUVECs transduced with OSKM and grown in hPCCM. The reprogrammed cells were subjected to ALP+ colony staining at 14 days following OSKM transduction, and quantification showed that reprogramming efficiency was significantly reduced after knocking down CXCR2 expression (P < 0.0001; Fig. 2C, D). Moreover, we observed that CXCR2 knockdown attenuated the increase in mTOR phosphorylation and β-catenin protein levels, and this effect was more pronounced for mTOR than for β-catenin (Fig. 2E). The expression of pluripotency-associated markers in CXCR2-silenced cells transduced with OSKM and cultured in hPCCM was considerably lower than in nonsilenced cells. To investigate the effects of CXCR2 overexpression, we selected the HDFs and HPCs with relatively low CXCR2 expression rather than HUVECs with high CXCR2 expression (Supplementary Fig. S1A, B). We used a lentivirus to transduce the cells with a synergistic activation mediator transcription activation system designed to specifically upregulate CXCR2 expression [15]. qPCR showed that CXCR2 mRNA was significantly increased after CXCR2 activation (Fig. 2G), and immunofluorescence staining confirmed higher CXCR2 expression levels compared with the basal level (Fig. 2H). Similarly, western blot analysis showed that CXCR2, mTOR, and β-catenin expression levels were markedly increased (Fig. 2F). To examine whether CXCR2 can enhance cell reprogramming, the CXCR2-overexpressing HDFs were transduced with OSKM. ALP staining of these cells showed that the number of ALP+ colonies significantly increased in the CXCR2-activated group compared with that in the control group after 14 days (P < 0.05; Fig. 2I, J). We also performed the same experiment using mTeSR (a commercial iPSC growth medium that does not include CXCR2 ligands) but observed no increase in ALP+ colonies. We obtained similar findings for CXCR2-activated HPCs using the same protocol (Supplementary Fig. S3E–H). Next, we investigated whether the mTOR and β-catenin pathways were activated by CXCR2 during reprogramming to iPSCs. We observed high expression levels of mTOR, β-catenin, and pluripotency markers upon CXCR2 activation in cells cultured in hPCCM 14 days after OSKM transduction (Fig. 2K, Supplementary Fig. S3I). However, the CXCR2-activated cells were rarely reprogrammed in mTeSR, which lacks CXCR2 ligands.

Regulatory role of CXCR2 in reprogramming efficiency.
Inhibiting mTOR before OSKM transduction and activating mTOR by CXCR2 stimulation after transduction enhance reprogramming
To further investigate the role of CXCR2 in iPSC generation, we examined its downstream signaling pathways during reprogramming. Specifically, we evaluated the effects of mTOR and β-catenin knockdown on the generation of iPSCs. To this end, HUVECs were infected with lentiviral particles overexpressing shRNA targeting mTOR and β-catenin, and the successfully transduced cells were selected with puromycin. Western blot assays showed successful silencing of mTOR and β-catenin (Fig. 3A). Thereafter, we generated iPSCs from HUVECs with downregulated mTOR or β-catenin expression. mTOR knockdown resulted in an ∼twofold increase in the reprogramming efficiency of HUVECs at 14 days following OSKM transduction. In contrast, β-catenin knockdown decreased the efficiency of iPSC generation (Fig. 3B, C). We next examined the expression of CXCR2 and Nanog in iPSCs generated from mTOR-silenced cells cultured in hPCCM and found that their expression levels after OSKM transduction were similar to those observed in human embryonic stem cells (H1or hESCs). Moreover, we compared expression levels of CXCR2, mTOR, and Nanog in mTOR-silenced cells cultured in hPCCM after OSKM transduction to those in the mTOR-active control cells. We observed that the fluorescence levels corresponding to CXCR2 and Nanog expression were similarly increased in a time-dependent manner (Fig. 3D–F). In addition, we analyzed CXCR2 and Nanog expression by measuring immunofluorescence in control cells (expressing β-catenin) and β-catenin-silenced cells cultured in hPCCM after OSKM transduction. We found that CXCR2 and Nanog expression gradually increased in control cells, but not in the β-catenin-silenced cells (Supplementary Fig. S4A–C). Next, we asked whether mTOR activation in human somatic cells cultured in hPCCM after OSKM transduction was due to the presence of CXCR2 ligands in hPCCM or the ectopic expression of OSKM. We thus cultured mTOR-silenced cells in mTeSR, which lacks CXCR2 ligands, and hPCCM and assessed their efficiency of reprogramming iPSCs under the same conditions at 14 days following OSKM transduction. We found that mTeSR did not activate CXCR2 and that reprogramming efficiency was very low (Fig. 3G–J).
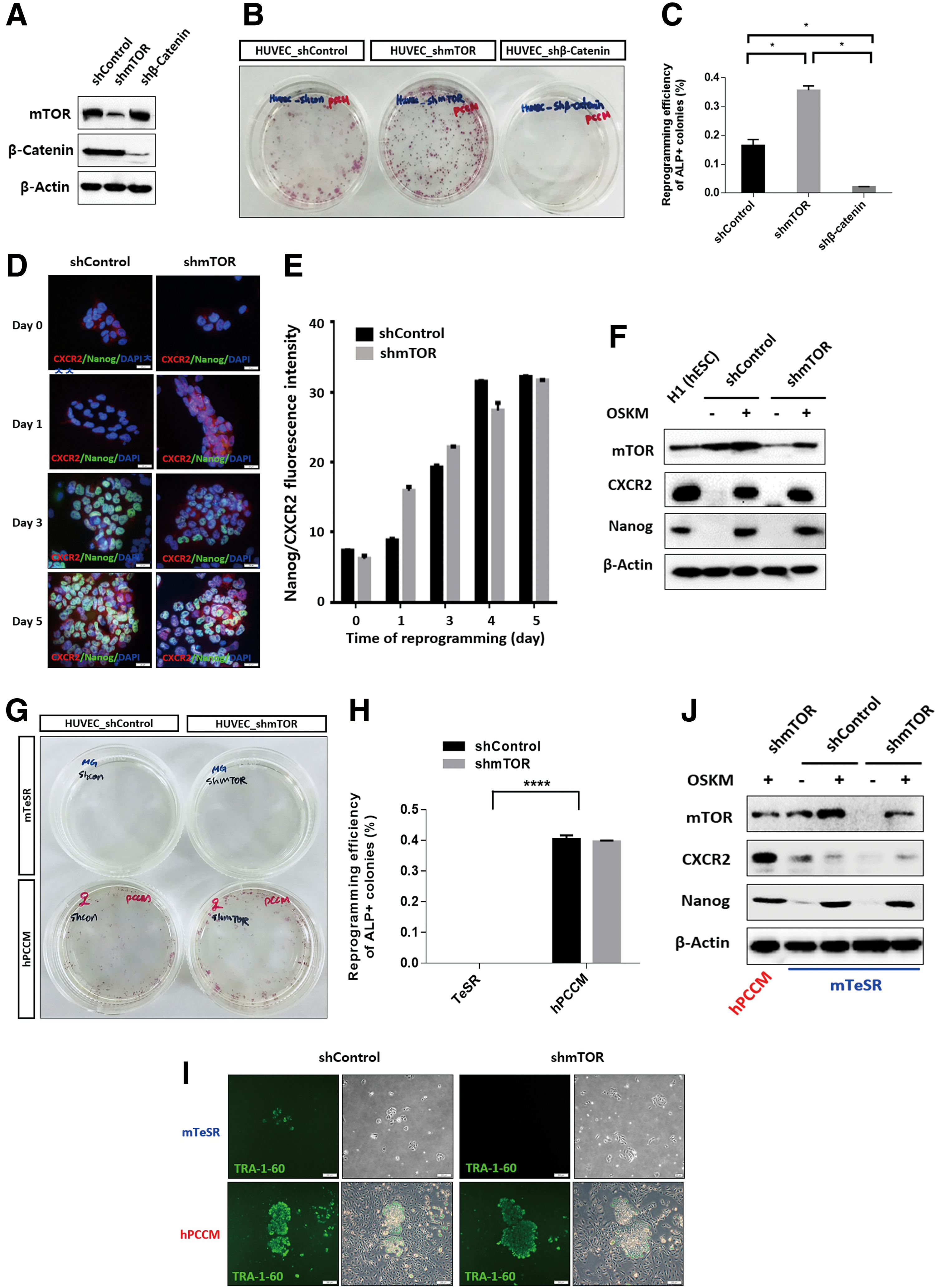
Effect of suppressed mTOR and β-catenin expression on reprogramming.
Endogenous cMYC induction and autophagy suppression by mTOR after OSKM transduction can promote reprogramming
We assumed that mTOR may induce the endogenous production of pluripotency-associated factors by creating a synergic effect with CXCR2 on reprogramming. To investigate the role of endogenous OSKM expression in reprogramming, we cultured human somatic cells in hPCCM and growth medium for 24 h and measured the mRNA levels of OSKM, CXCR2, and mTOR. Cells cultured in hPCCM had significantly higher expression of endogenous cMYC, CXCR2, and mTOR than those cultured in growth media (P < 0.05, P < 0.01, and P < 0.0001, respectively; Fig. 4A, B). In contrast, endogenous cMYC, CXCR2, and mTOR expression was markedly decreased in CXCR2-silenced HUVECs cultured in hPCCM compared with HUVECs normally expressing CXCR2 under the same conditions (P < 0.0001; Fig. 4C). To examine the role of endogenous cMYC and mTOR in reprogramming, we assessed the expression of endogenous OSKM in mTOR-silenced cells or mTOR-active control cells cultured in hPCCM. Endogenous cMYC expression in mTOR-active control cells was significantly higher than in mTOR-silenced cells (P < 0.0001; Fig. 4D). Furthermore, CXCR2, mTOR, and β-catenin protein levels in mTOR-silenced cells cultured in hPCCM after OSKM transduction were higher compared with cells cultured in growth medium, and the LC3α/β protein levels suggested that the degree of autophagy was considerably decreased in response to CXCR2 stimulation (Fig. 4E, F). Culturing control mTOR-active HUVECs (expressing high levels of endogenous cMYC) reprogrammed by ectopic OSK expression (not including cMyc) for 7 days and then exposing them to hPCCM for 7 days resulted in an acceptable reprogramming efficiency (∼0.1%); however, the same was not observed in mTOR-silenced HUVECs. Interestingly, culturing mTOR-silenced HUVECs ectopically expressing OSKM in hPCCM resulted in significantly enhanced reprogramming efficiency compared with that of mTOR-active control cells under the same conditions (twofold increase, P < 0.0001). Even though suppression of mTOR before OSKM transduction enhances reprogramming, reprogramming efficiency can be improved by CXCR2 ligand-mediated mTOR activation after transduction (Fig. 4G–I).
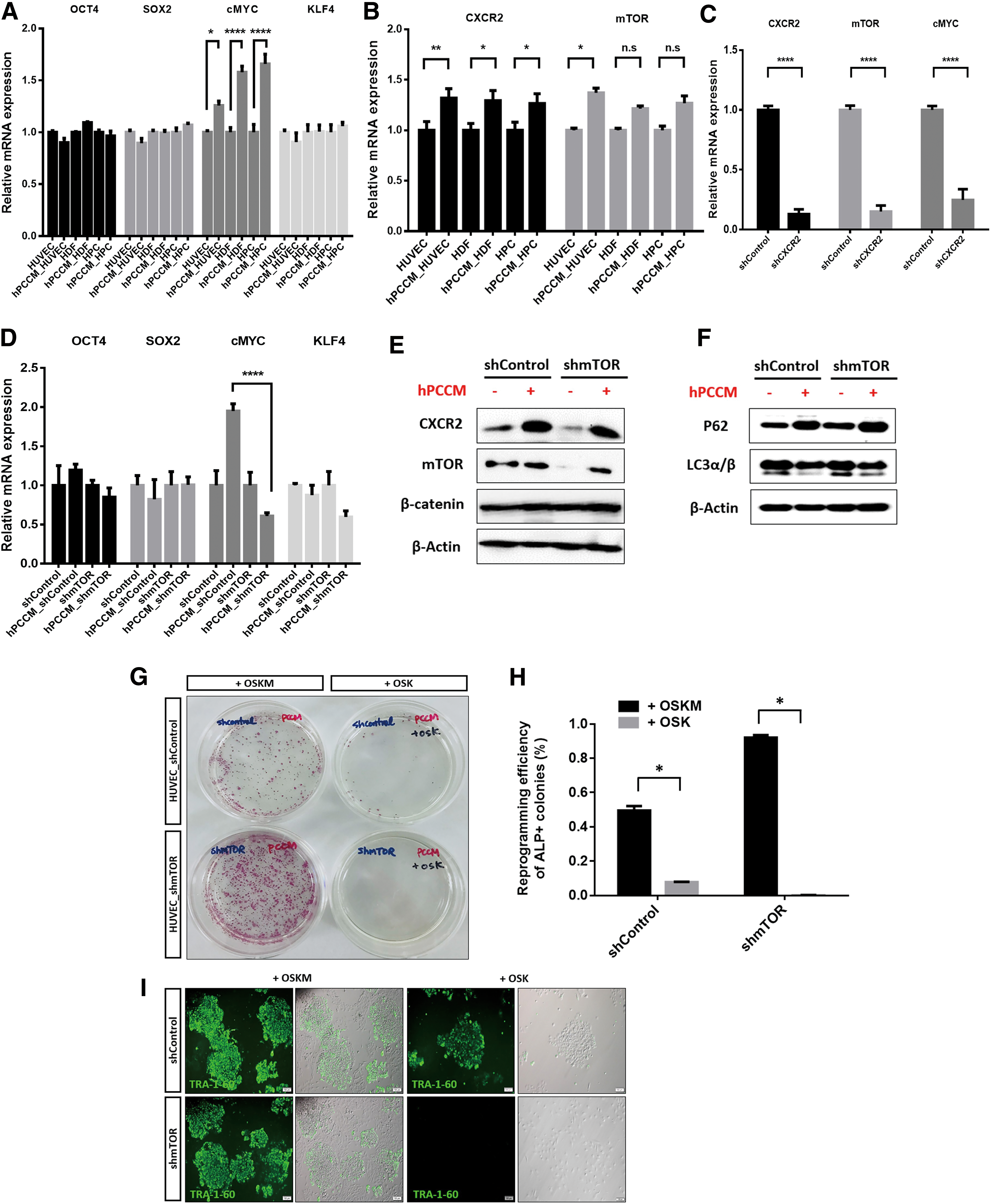
Enhanced reprogramming efficiency through CXCR2 and mTOR stimulation in human somatic cells after OSKM transduction is associated with upregulated cMYC levels and decreased autophagy.
Proof of concept using established reprogramming media
Considering that hPCCM has several disadvantages, one of which is its undetermined composition, we designed a standardized experiment with a widely used reprogramming protocol that includes growth media, mTeSR, and Matrigel. In our previous study, we quantitatively measured the levels of major cytokines in hPCCM and found high levels of CXCR2-related ligands [13]. First, we confirmed that growth-related oncogene α (GROα), a CXCR2-specific ligand, increased mTOR and cMYC expression in somatic cells (Fig. 5A, B). Next, GROα or another CXCR2 ligand, interleukin (IL)-8, was added 2 days before or 1 day after OSKM transduction. The media (growth media or hPCCM) were then changed every 2days, and cells were seeded on Matrigel-coated dishes with mTeSR on the seventh day (Fig. 5C). The administration of CXCR2 ligands to human somatic cells before OSKM transduction resulted in senescence without reprogramming (Supplementary Fig. S5A–C). In contrast, exposure of human somatic cells to CXCR2 ligands 1 day after OSKM transduction significantly enhanced reprogramming at 21 days (P < 0.01 and P < 0.0001; Fig. 5D–G, Supplementary Fig. S6A, B). Of note, the media containing GROα showed a remarkable reprogramming efficiency comparable to that of hPCCM (Fig. 5E). Finally, we assessed reprogramming in cells treated with MHY1485, an mTOR activator, under the same conditions as the CXCR2 ligand experiments. We evaluated the effect of MHY1485 on CXCR2, mTOR, and cMYC expression and autophagic flux and found that CXCR2, mTOR, and cMYC protein levels increased and LC3α/β expression levels decreased without p62 degradation, indicating reduced autophagic flux (Fig. 6A). Considering that CXCR2 expression was used to determine mTOR activity in previous experiments (Fig. 2E, F), it is noteworthy that MHY1485 enhanced CXCR2 expression. Next, we treated human somatic cells with rapamycin, a classic mTOR inhibitor, to induce autophagy and found that the cells previously treated with MHY1485 showed no signs of increased autophagy (Fig. 6B). After MHY1485 was administered to human somatic cells cultured using the same protocol after OSKM transduction, we obtained significantly improved reprogramming efficiency at 21 days compared with that of untreated cells (P < 0.05; Fig. 6C, D, Supplementary Fig. S6C). We observed that the reprogramming efficiencies of the widely used reprogramming protocols using either CXCR2 ligands (GROα or IL-8) or an mTOR activator (MHY1485) were similar compared to those of hPCCM alone (Supplementary Fig. S6D–F).
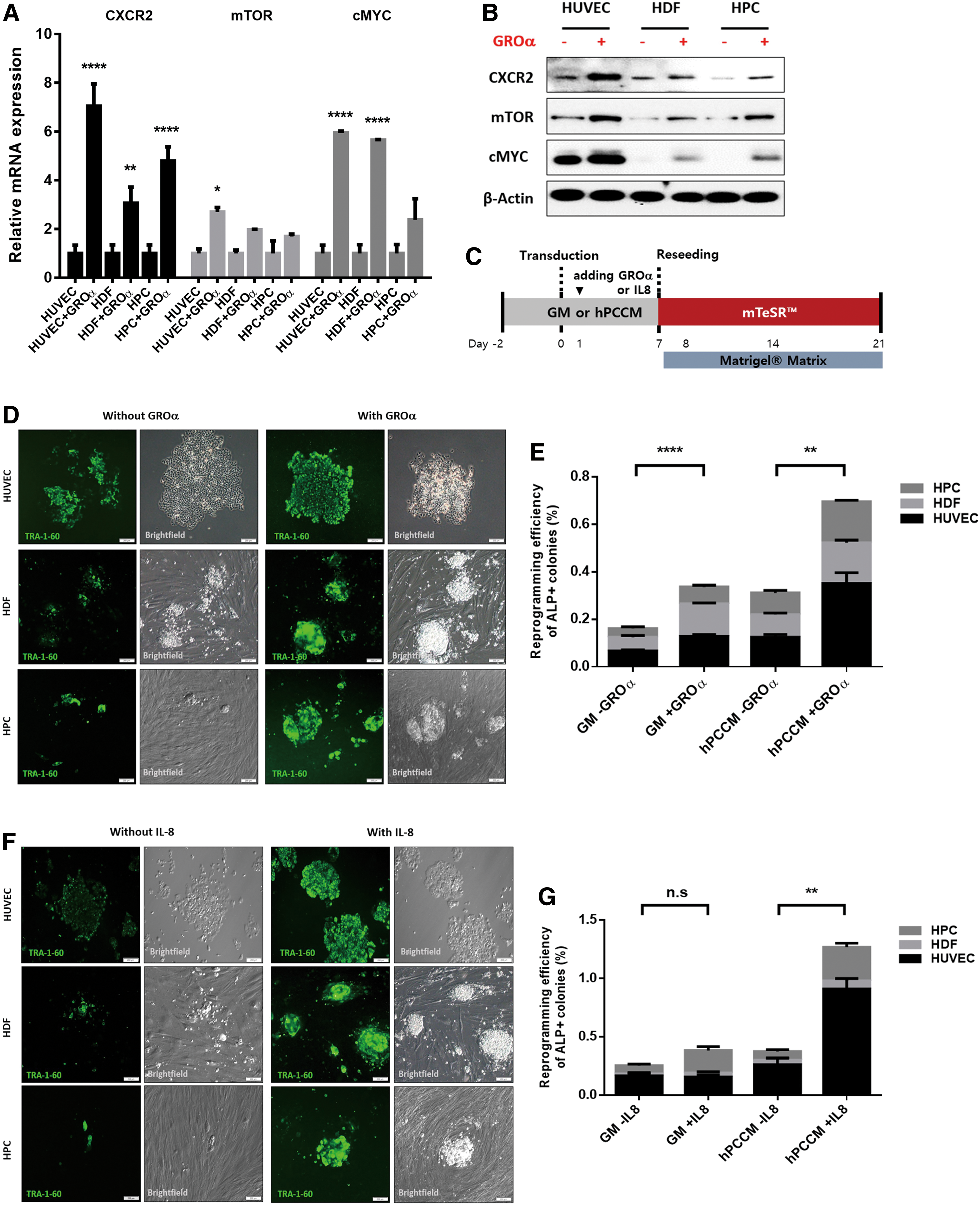
Proof of concept using commonly used reprogramming media supplemented with CXCR2 ligands. The CXCR2-specific ligand GROα directly activates CXCR2, mTOR, and cMYC. Somatic cells were cultured in growth medium or treated with GROα (1 μg/mL) in growth medium for 24 h.

Proof of concept using commonly used reprogramming media supplemented with MHY1485, an mTOR activator. mTOR activation directly activates CXCR2 and cMYC and inactivates autophagy. Somatic cells were cultured in growth medium or treated with MHY1485 (2 μg/mL) for 24 h.
Discussion
In this study, we developed a highly optimized, relatively easy, and cost-effective culture protocol that allows efficient reprogramming of human somatic cells ectopically expressing pluripotency-associated factors into iPSCs. This approach provides an opportunity to shorten the time needed to generate iPSCs from human somatic cells in a feeder-free system with significantly higher reprogramming efficiency compared with the commonly used protocols (hPCCM ∼4% vs. E8 ∼ 0.1%). To date, studies aiming to improve the reprogramming efficiency of human somatic cells by targeting mTOR pathways have focused only on suppressing these pathways [7,8,10,11]. Previously, we reported on the role of CXCR2 in supporting the nature of hPSCs cultured in hPCCM and considered its possible interaction with mTOR and β-catenin [13,14]. Considering that the CXCR2-mTOR-β-catenin axis is involved in the maintenance of hPSC characteristics, we designed experiments to test the role of this axis and elucidate its mechanism in the reprogramming process. We observed that suppression of CXCR2 and β-catenin expression impaired the reprogramming of human somatic cells, indicating a role for these components in iPSC generation. In the case of β-catenin, it was previously reported that Wnt/β-catenin signaling promotes self-renewal and inhibits primed state transition in naive hESCs [16]. Moreover, β-catenin was shown to promote porcine cell reprogramming by upregulating the expression of pluripotency-associated genes [17]. Accordingly, our results highlight the importance of β-catenin for iPSC production. Since our main objective was to assess the roles of CXCR2 and mTOR in reprogramming, we did not further explore the role of β-catenin. The transduction of OSKM genes into human somatic cells with downregulated (knockdown) mTOR expression promoted reprogramming in hPCCM. These suppressed mTOR levels became activated during reprogramming to iPSCs and autophagy decreased. To uncover why mTOR is activated from its repressed status, the same experiment was performed using the widely used reprogramming protocol that does not involve CXCR2 ligands, and we observed no mTOR activation and iPSC generation. Therefore, it is reasonable to consider that CXCR2 activates mTOR, resulting in enhanced reprogramming. This effect is similar to our previous results showing the role of CXCR2 and mTOR in the maintenance of hPSC features [13,14].
We theorized that mTOR may induce endogenous production of pluripotency-associated factors, creating a synergistic effect with ectopic OSKM expression as a mechanism enhancing reprogramming after mTOR activation. Therefore, human somatic cells were cultured in hPCCM, and the changes in CXCR2, mTOR, and OSKM expression were analyzed. The result was significantly higher in intracellular cMYC expression with simultaneous activation of CXCR2 and mTOR in all human somatic cells cultured in hPCCM compared with those cultured in growth media without CXCR2 ligands. To further examine the role of endogenous cMYC overexpression in reprogramming, we reprogrammed HUVECs, which showed the highest constitutive cMYC expression levels, by ectopic expression of OCT4, SOX2, and KLF4 (OSK, without cMyc) in hPCCM and obtained successful reprogramming with an efficiency within the range mentioned in most published reports. Moreover, HUVECs with knocked down mTOR and cultured in hPCCM exhibited low expression of endogenous cMYC and could not be reprogrammed by ectopic OSK transduction alone. This indicated the important role of mTOR-induced endogenous cMYC. In contrast, cells transduced with OSKM showed enhanced reprogramming rates and decreased autophagy. An established reprogramming protocol without CXCR2 ligands could not reproduce these findings. Based on these results, we were able to confirm the role of CXCR2 in reprogramming enhancement through mTOR activation, induction of endogenous cMYC expression, and autophagy suppression. Therefore, we hypothesized that endogenous cMYC produced in response to mTOR activation can promote reprogramming and that the effect of autophagy suppression may be positive for reprogramming after OSKM transduction. The role of autophagy in somatic cells after OSKM transduction was not further explored herein. Nevertheless, our observation that reprogramming of HUVECs by ectopic OSK expression and endogenous (instead of ectopic) cMYC is feasible may provide a new perspective in the development of human iPSCs, eliminating the concern of using ectopic cMYC, which is known as a strong proto-oncogene [18,19]. Furthermore, we used hPCCM as a supporting medium to directly enhance the transduction of somatic cells with ectopic OSKM expression, assuming that hPCCM can facilitate reprogramming. As a result, reprogramming was accelerated, with significantly shorter reprogramming duration and higher efficiency than those of the commonly used protocols (up to 1% within 14 days).
Although hPCCM enhanced human somatic cell reprogramming and its mechanism was identified as mTOR activation by CXCR2 ligands, it was not clear whether CXCR2 ligands directly stimulate mTOR activation because unknown components in hPCCM may facilitate this process. To address this issue, we repeated the experiment using a commonly used reprogramming culture media and supplemented it with GROα 1 day after OSKM transduction. This resulted in significantly enhanced reprogramming efficiency. Similar results were obtained when another CXCR2 ligand (IL-8) was added to the medium. Moreover, the addition of an mTOR activator (MHY1485) had similar effects on the reprogramming of somatic cells. Notably, reprogramming efficiency in these experiments (up to 1%) was significantly higher compared with the commonly used reprogramming protocols that do not include CXCR2 ligands or mTOR activators. In contrast, administration of CXCR2 ligands or mTOR activator to human somatic cells 2 days before OSKM transduction resulted in senescence without reprogramming. These findings reveal a possible dual role of mTOR between reprogramming and senescence. It was recently suggested that the senescence-associated secretory phenotype (SASP) components, such as IL-6, can promote reprogramming, whereas senescence is detrimental for mTOR-mediated reprogramming [11]. Herein, CXCR2 ligands—also known as SASP components—such as GROα and IL-8, enhance reprogramming through mTOR signaling. The possible role of other components, such as IL-6, was not assessed because the main objective of our study was to determine the role of CXCR2 and mTOR in the reprogramming of human somatic cells to iPSCs. Moreover, the expression of IL-6 in hPCCM was shown to be much lower compared with the CXCR2 ligands in our previous study. In addition, the secretion of SASP components from CXCR2-activated cell lines with enhanced reprogramming compared to their original status was similar to the findings of our previous study using hPCCM [14]. Therefore, it is plausible to hypothesize that CXCR2 ligands are the potent SASP factors stimulating mTOR (Supplementary Fig. S5D).
Furthermore, to our knowledge, there are no studies reporting the association of mTOR and cMYC in reprogramming. In contrast, this association has been widely investigated in other medical contexts [20 –22]. For instance, it was shown that cMYC levels are correlated with mTORC1 activation, the mTOR pathway is induced in cMYC-driven liver tumor cells, and activated mTORC1 is required for cMYC-driven tumor development in vivo [20]. These findings are in agreement with the observed effect of mTOR on the production of endogenous cMYC in human somatic cells during reprogramming.
Taking into consideration our findings and those of other studies [7,8,10 –14,18,20], we propose an mTOR handling strategy to enhance the reprogramming efficiency of human somatic cells. Such a strategy includes mTOR inhibition before transduction of pluripotency-associated factors in human somatic cells followed by mTOR activation after transduction. Our results provide compelling evidence for the feasibility of this strategy. The optimal combination of CXCR2 ligands and mTOR activator in commonly used reprogramming protocols should be further investigated to maximize their efficiency. Other issues related to the proposed strategy may be expected based on the present study. First, although CXCR2 is expressed in all human somatic cells, its expression may differ among cell types and influence reprogramming efficiency. This problem can be solved using mTOR activators to directly affect mTOR instead of depending on CXCR2. Second, because many unknown factors that support the characteristics of human stem cells may still be present in hPCCM, further studies are needed to further explore the mechanisms of reprogramming enhancement by hPCCM.
In conclusion, hPCCM enhances the reprogramming efficiency of human somatic cells by activating CXCR2 and mTOR. The addition of CXCR2 ligands or an mTOR activator to a commonly used reprogramming medium also improves reprogramming efficiency. This indicates the central role of mTOR in enhancing reprogramming efficiency. This approach is not only simple but also convenient and cost-effective, because it only requires hPCCM on 0.1% gelatin-coated dishes without substratum for cell growth. Moreover, the significantly higher reprogramming efficiency of our protocol using hPCCM supplemented with CXCR2 ligands or an mTOR activator compared with using hPCCM alone indicates that the concentration of CXCR2 ligands or mTOR activators in the culture media can be adjusted to maximize reprogramming efficiency. To our knowledge, this is the first report demonstrating that mTOR activation enhances reprogramming of human somatic cells upon ectopic expression of pluripotency-associated factors.
Footnotes
Acknowledgments
This work was supported, in part, by Brain Korea 21 Plus Grant Program from the Ministry of Education, Republic of Korea.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.