
Abstract
A major objective of modern biomedical research aims to promote tissue self-regeneration after injury, obviating the need for whole organ transplantation and avoiding mortality due to organ failure. Identifying the population of cells capable of regeneration, alongside understanding the molecular mechanisms that activate that population to re-enter the cell cycle, are two important steps to advancing the field of endogenous tissue regeneration toward the clinic. In recent years, an emerging trend has been observed, whereby polyploidy of relevant parenchymal cells, arising from alternative cell cycles as part of a normal developmental process, is linked to restricted proliferative capacity of those cells. An accompanying hypothesis, therefore, is that a residual subpopulation of diploid parenchymal cells retains proliferative competence and is the major driver for any detected postnatal cell turnover. In this perspective review, we examine the emerging literature on residual diploid parenchymal cells and the possible link of this population to endogenous tissue regeneration, in the context of both heart and liver. We speculate on additional cell types that may play a similar role in their respective tissues and discuss outstanding questions for the field.
Introduction
Somatic polyploidy is defined as a single somatic cell having at least three copies of a haploid genome. Many cells in the mammalian body are polyploidy, including cardiomyocytes, hepatocytes, trophoblast giant cells of the placenta, various pancreatic cells, keratinocytes, retinal pigment epithelial cells, and skeletal muscle cells. There is substantial variability across species and tissue types in the frequency of polyploid cells within a tissue, and the age at which they can be found (Table 1). For example, mammalian cardiomyocytes are overwhelmingly polyploid. In humans, mice, rats, and other surveyed mammals, >90% of cardiomyocytes are polyploid, induction of which occurs at the time of, or soon after, birth [1,2]. Hepatocytes are also predominantly polyploid, with reports of ∼90% in certain strains of mice [3] and ∼25%–50% in humans [4 –6]. In rodents, hepatocyte polyploidization is induced at the time of weaning by the commencement of feeding [7 –9], and the frequency of polyploid hepatocytes increases with age [10]. Moreover, polyploid pancreatic β cells have been reported in mice [11] and humans [12], which are also thought to be induced at weaning followed by increases due to glucose challenge and age [13].
Somatic Cells with Polyploidy
perhaps some rare 16N.
not yet proven with lineage tracing.
perhaps up to 128N.
perhaps up to >800N.
mononuclear polyploidy never assessed.
RPE, retinal pigment epithelial.
Somatic polyploidy can arise two ways (see Orr-Weaver [14] for an in-depth review): from variant cell cycles wherein cells complete G1- and S-phases, thereby replicating their genomes, but fail to either enter or complete mitosis (termed endocycle and endomitosis, respectively), or by fusion of two or more diploid cells as is the case with skeletal myoblasts. Causation by variant cell cycles is the subject of this review.
Beyond the variability in frequency and age of induction described above, there is substantial heterogeneity in how polyploidy is displayed in cells. Polyploidy can come in the form of a single nucleus containing >2N DNA content (eg, 1 × 4N, 1 × 8N, etc.) or in the form of multinucleation (2 × 2N, 4 × 2N, etc.). In the case of a mononuclear polyploid cell, the variant cell cycle stalls before completion of karyokinesis (nuclear division), whereas a multinucleated cell successfully completes nuclear division but fails to complete cytokinesis. As one example, cardiomyocytes tend to be multinucleated in rodents (2–4 nuclei, Fig. 1) [1,15], pigs (8+ nuclei) [16], and rabbits, but are more commonly mononuclear and polyploid (1 × ≥4N) in humans [2]. It remains unclear if these different states are functionally relevant. For the sake of this review, “diploid” refers to 2N DNA content accounting for the entire cell, whereas “polyploid” refers to >2N DNA content due to increased numbers of nuclei and/or increased DNA content within a single nucleus.
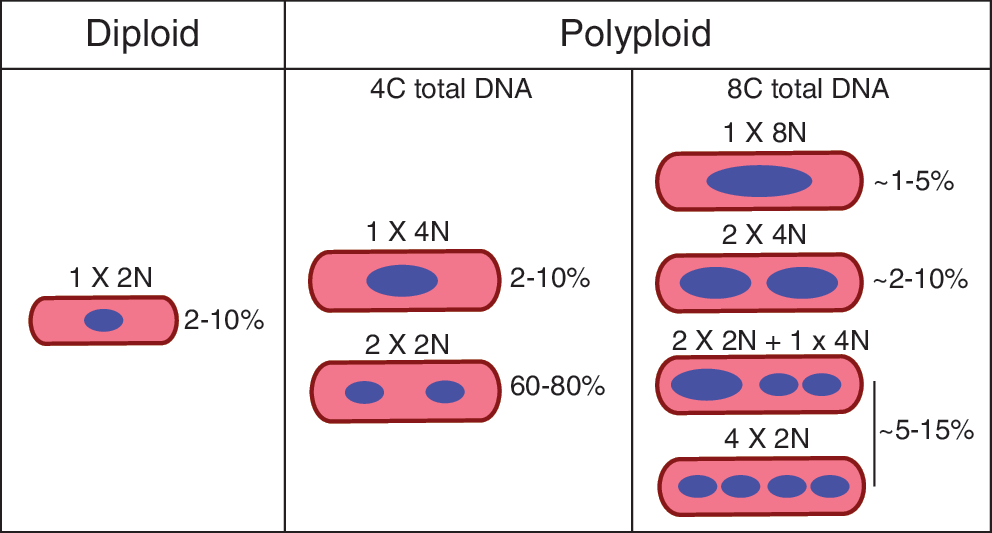
Frequency of various ploidy classes in normal murine heart. Schematic depicting the observed heterogeneity in ploidy content of adult mouse cardiomyocytes under basal conditions. Frequencies noted here are approximations collected from Patterson et al. [15] and personal observations.
In recent years, several studies spanning a variety of tissues have addressed the idea that the residual population of diploid cells in an otherwise polyploid tissue might possess a unique function within that organ. In other words, diploid cells are distinct from their polyploid counterparts. One such proposed distinction is that polyploidy is linked to restricted proliferative capacity; therefore, the residual subset of diploid somatic cells may be responsible for postnatal cell turnover within a tissue, including regeneration after injury and homeostasis. In this perspective review, we will discuss this model related to two specific tissues: heart and liver, where the two fields have attempted to address the concept directly. We also speculate on additional cell types that may participate in the regeneration of their respective tissues and discuss outstanding questions for each field to address.
Why become polyploid?
To understand the residual population of diploid cells within a tissue, it is important to first consider why a cell becomes polyploid in the first place. There are several speculations as to what physiological advantage polyploidy confers (reviewed in depth in Refs. [14,17]). Undoubtedly, this is tissue and species dependent. Research has frequently noted that polyploid cells tend to be larger in size than their diploid counterparts; thus, one argument is that there might be a functional advantage to being larger. This appears to be the case in polyploid megakaryocytes, from which platelets are produced through fragmentation of megakaryocyte cytoplasm [18]. In the heart, large cardiomyocytes might be better equipped to generate the force necessary to support circulation throughout the increased mass of the adult organism [19].
Another suggested advantage is that polyploid cells tend to have increased levels of transcription and therefore higher rates of biosynthesis to confer faster adaptive responses [17,19]. This hypothesis has certainly been proposed in the heart field where it is thought that polyploid cardiomyocytes might be able to generate a faster hypertrophic response (eg, biosynthesis of additional sarcomeres) after injury compared with their diploid counterparts. An extension of this biosynthesis hypothesis is that polyploid cells may also be capable of higher metabolic rates in situations where there is greater energy demand [20].
A second argument is that there might be a disadvantage to completing cell division. For example, because keratinocytes within the skin and trophoblast giant cells of the placenta provide a barrier function within their respective tissues, completing cell division might temporarily compromise these barriers [21]. In the case of cardiomyocytes, cell division could temporarily compromise the gap junctions required for electrical coupling of neighboring cells, thus potentially disrupting contractile function of the heart. In addition, because many polyploid cells arise during periods of rapid postnatal growth when limited resources are available, it is possible that cells conserve energy by not completing mitosis [17].
A final proposed explanation is that polyploidy confers resistance to damage at the cellular level. There is evidence in various cell types that polyploidy induces a state of proliferative senescence [22]. In the absence of proliferation, polyploid cells maintain a relatively stable differentiated state. In contrast, diploid cells can acquire new attributes as they divide (eg, somatic mutations, passive dilution of proteins, and activation of alternative cell states), thus allowing them to be more plastic [14]. In addition to proliferative senescence, polyploidy can inactivate apoptotic pathways, thereby lengthening the life span of the cell while providing a steady-state environment for the tissue [23]. Finally, increased DNA content can serve as a genomic buffer against DNA damage or somatic mutations, which could lead to cancer. This has been most closely studied in the liver field where it has been reported that polyploid hepatocytes protect against loss of heterozygosity of tumor suppressors [9,24].
In some tissues, polyploidy is the predominant cellular phenotype. While the reason polyploidy arises remains primarily speculative at this point, it does suggest that polyploidy represents a functional state that is distinct from the residual diploid population. The tissues discussed below have several attributes in common (highlighted in Table 1). First, best evidence from the literature suggests that newly derived cells in these tissues arise from pre-existing differentiated cells (ie, the parenchymal cell of the tissue), rather than from multipotent stem or progenitor cells as occurs in the blood, skin, skeletal muscle, and intestinal lineages. Also, polyploidy in each of these tissues arises as part of a natural developmental process by way of an alternative cell cycle such as endocycling or endomitosis, rather than from cell fusion. Finally, polyploidy may increase with age or disease, perhaps coinciding with age-related decline in tissue homeostasis.
Diploid Cardiomyocytes and Myocardial Regeneration
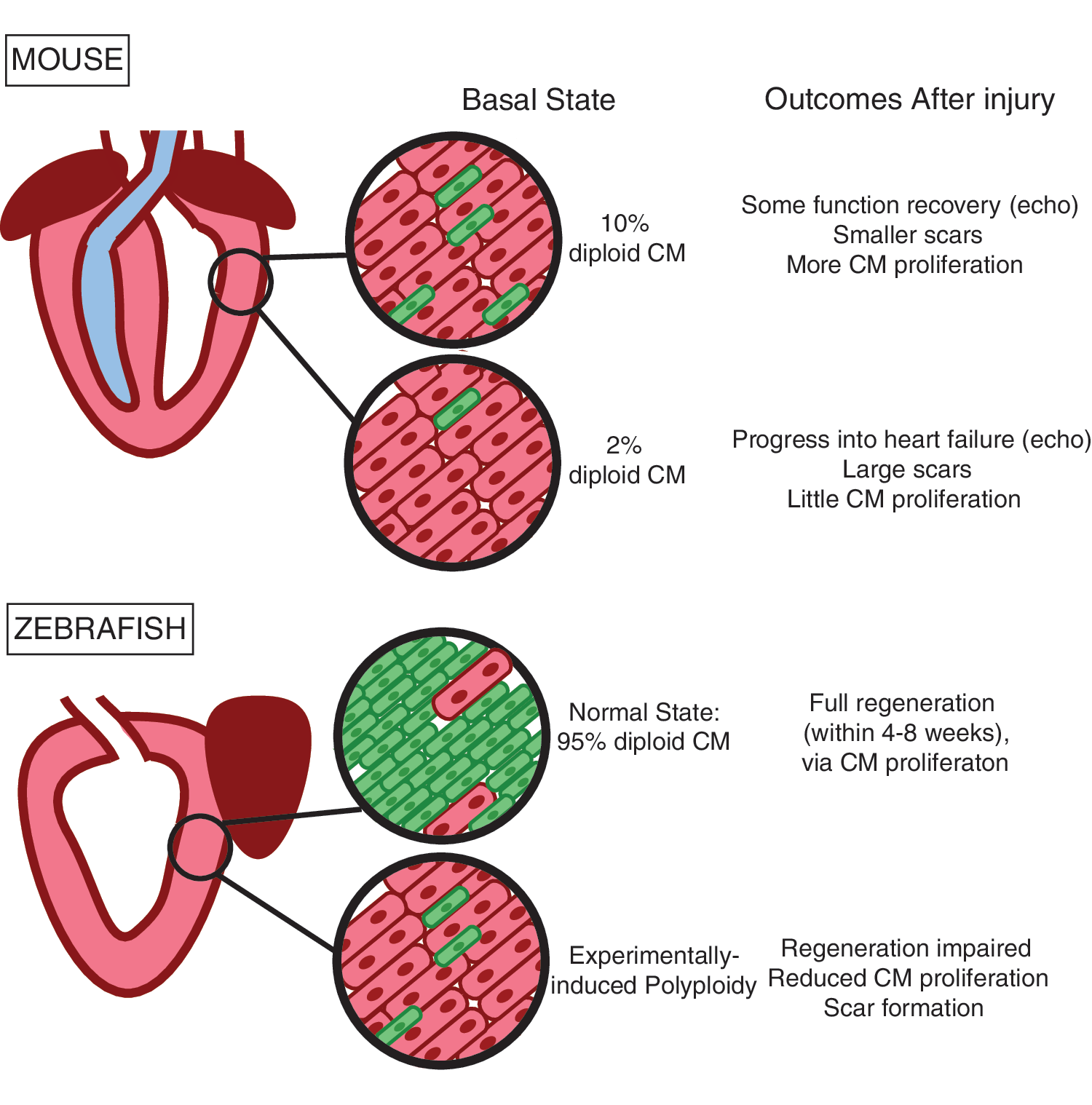
Historically, the cardiac field held that the adult mammalian heart is incapable of regeneration after insult. However, during the last decade, several studies demonstrated a small but significant degree of endogenous turnover of cardiomyocytes, measured at ∼1% per year in both adult mice [25,26] and humans [27], with a slight increase after ischemic injury [25]. More recently, an exhaustive survey comparing genetically diverse inbred mouse strains established that regeneration of the mammalian adult heart is a variable trait [15], suggesting reconsideration of the dogma that the heart cannot regenerate is in order. Regardless, adult mammals contrast with zebrafish [28], newts [29], and neonatal mice [30], all of which display a very high capacity for heart regeneration.
Extensive analyses based on lineage tracing strategies in zebrafish [31,32], neonatal mice [30], and adult mammals [25,26] have conclusively established that new cardiomyocytes are derived from pre-existing cardiomyocytes. Although other work has indicated that adult cardiac stem cells (which appear to have their own unique interspecies ploidy dynamics [33]) may contribute to myocardial regeneration, this hypothesis has fallen out of favor in recent years [34 –36] and even been refuted by several studies [37 –40]. The observation that pre-existing cardiomyocytes give rise to new cardiomyocytes in both examples of efficient (ie, zebrafish and neonatal mice) and inefficient (adult mammals) regeneration suggests that there are differences between the cardiomyocytes of efficient and nonefficient regenerators, which in turn explain their differential regenerative capacities. These differences could be intrinsic to the cardiomyocytes themselves or could reflect differences within the anatomic niches that then affect the endogenous cardiomyocytes ability to proliferate.
One proposed characteristic distinguishing efficiently from inefficiently regenerative cardiomyocytes is cellular DNA content. Considering the murine developmental time course, one can observe that cardiomyocytes in the embryonic heart are entirely diploid and proliferative, maintaining diploidy until the time of birth, after which most cardiomyocytes undergo endoreplication resulting in a primarily binucleated polyploid population by postnatal day 7 (P7) [1]. This switch in ploidy state precisely coincides with loss of regenerative capacity, where murine neonatal hearts injured on postnatal day 1 (P1) can fully regenerate, while neonates injured on P7 cannot [30]. By contrast, zebrafish hearts, which maintain the ability to regenerate throughout the life span of the organism [28], are comprised primarily of diploid cardiomyocytes [15,41].
Our own work across 120 inbred mouse strains demonstrates that strains with high frequency of diploid cardiomyocytes in the naïve adult myocardium correlate with improved function, reduced scar size, and increased myocyte proliferation after myocardial infarction (Fig. 2) [15]. Thus, a thorough survey of cardiomyocyte ploidy spanning species, developmental ages, and genetic backgrounds links diploid cardiomyocyte frequency with regenerative capacity. Of course, many other cellular changes take place in the same neonatal window, including, a metabolic switch from glycolysis to fatty acid oxidation; formation of T-tubules, minor invaginations in the plasma membrane responsible for calcium dynamics; organization and lengthening of sarcomeres; and alterations in RNA splicing, resulting in postnatal isoforms of many cardiomyocyte-specific genes. It is not yet known how these events, referred to as maturation, collectively contribute to cell cycle arrest. Nevertheless, the correlation-based data presented above form the foundation for many in the field to hypothesize that diploid cardiomyocytes comprise a subpopulation of cardiomyocytes that maintain the capacity to proliferate in response to insult and are the source of the 1% per year turnover observed in the adult mammalian heart.
Importantly, however, these observational correlations are now supported by several studies that challenge the hypothesis through direct experimental methods. In our study, using a forward genetics approach across 120 inbred mouse strains, we identified one gene, Tnni3k, as being linked to the frequency of the diploid population. Using loss-of-function experiments in the mouse model, we definitively showed that Tnni3k is one of likely multiple genes that regulates the size of the diploid cardiomyocyte population; specifically, depletion of Tnni3k resulted in an increase in diploid cardiomyocytes, suggesting that Tnni3k drives polyploidization through unknown mechanisms [15]. In another study, the authors obtained a similar result by experimentally blocking thyroid hormone signaling with a dominant negative receptor, again observing an increase in diploid cardiomyocytes and improved regenerative capacity [42].
Perhaps most convincingly, we also employed gain-of-function experiments to demonstrate that the mouse homolog of Tnni3k drives cardiomyocyte polyploidization in the zebrafish heart, and that this impairs cardiac regeneration [15]. Recent studies reported by Gonzalez-Rosa et al. elegantly addressed this concept as well by artificially inducing polyploidy in the zebrafish myocardium with cardiomyocyte-specific transient expression of an Ect2 dominant negative transgene. Their results not only demonstrated that diploid cardiomyocytes outcompete their polyploid counterparts during proliferative challenges like regeneration, but also that possession of too many polyploid cardiomyocytes impairs regeneration (Fig. 2) [41]. Taken together, these studies strongly support the notion that cardiomyocyte polyploidy impairs myocardial regeneration and that diploid cardiomyocytes constitute one cardiomyocyte subpopulation that is capable of proliferation.
Additional subtle evidence in the literature provides further support for the ploidy model presented here. For example, Bersell et al., using video microscopy on P7 neonatal cardiomyocytes, observed that only mononuclear cardiomyocytes could complete cytokinesis after Neuregulin 1 stimulation, while binucleated cardiomyocytes were never observed to complete this process [43]. Another study induced cardiomyocytes to divide by viral introduction of G1/S and G2/M cell cycle regulators and followed their ability to complete cytokinesis using Mosaic Analysis with Double Markers reporter mice. Their results showed that ∼90% of cells that definitively divided after viral induction of cell cycle regulators were mononuclear after division [44], suggesting that cells that enter the cell cycle (ie, DNA synthesis) and proceed to complete cell division (ie, cytokinesis) while remaining diploid must have originated from a diploid state. Admittedly, this logic does not account for a situation where a binucleated cardiomyocyte, stimulated to proliferate through viral induction, undergoes one round of DNA synthesis thereby becoming labeled (this cell would transiently be 8N). If that now 8N cell either forms multiple cleavage furrows to generate four mononuclear diploid daughter cells, or divides resulting in two mononuclear tetraploid daughter cells, investigators would also observe that labeled cells are primarily mononuclear. To the best of our knowledge, such an occurrence has yet to be noted in the cardiomyocyte field; however, it has been proposed for the hepatocyte field (see Fig. 3 “ploidy conveyor” inset) [45,46]. In a similar vein, Senyo et al. also identify that 17% of isotope-labeled cardiomyocytes (indicating DNA synthesis) were both mononuclear and diploid, again possibly suggesting a diploid origin [25]. Finally, a fourth study has argued that a subpopulation of hypoxic cardiomyocytes is responsible for regeneration. In this study, the authors pointed out that the hypoxic fate-mapped cardiomyocytes “were more likely to be mononucleated” [47], suggesting possible overlap between the two populations.
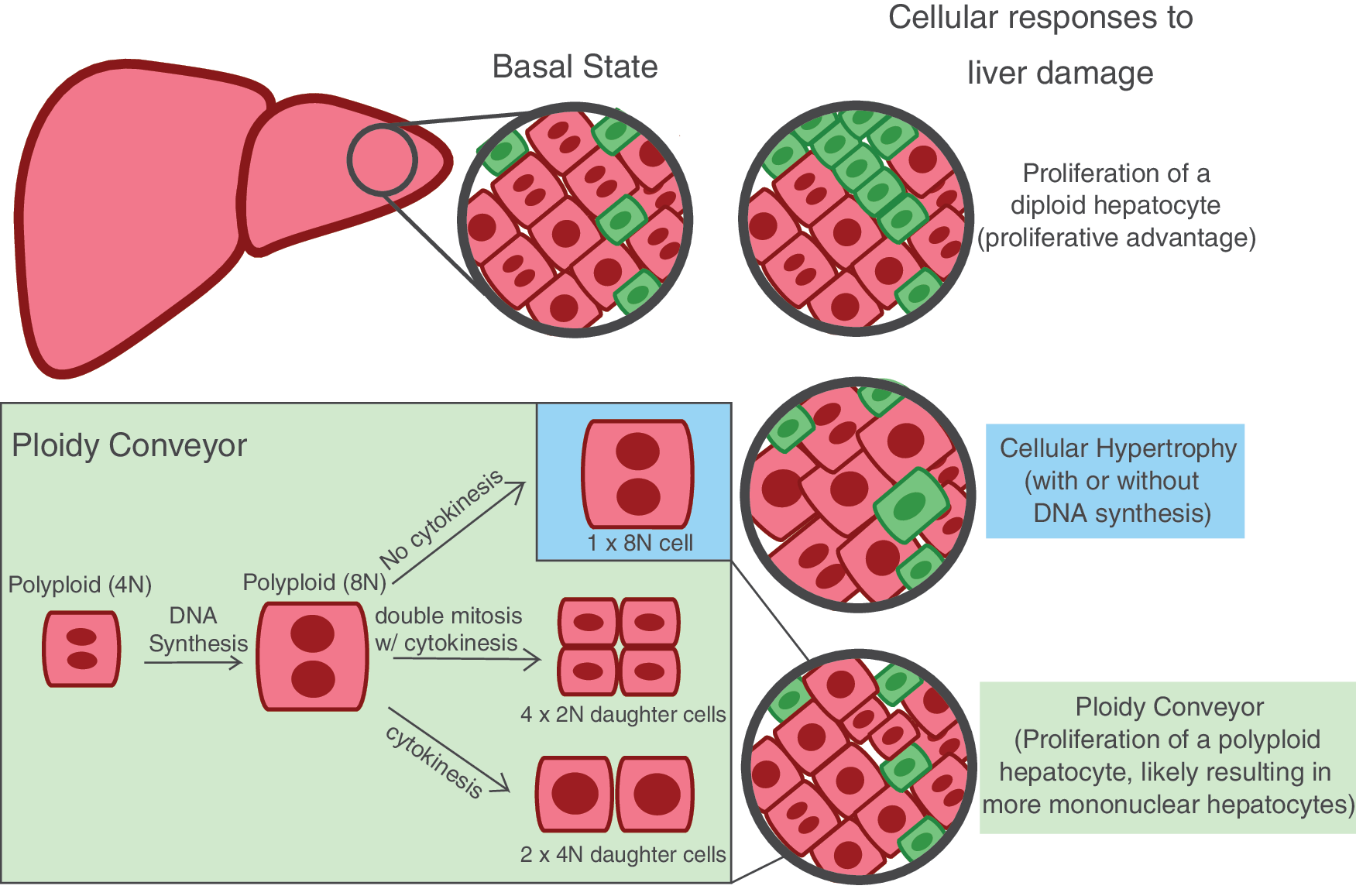
Diploid hepatocytes and the cellular processes accounting for liver regeneration. Summary schematic of the three cellular responses to liver damage presented by the literature, including proliferation of a diploid hepatocyte, cellular hypertrophy, and the “ploidy conveyor” model. Note: the inset depicting of the “ploidy conveyor” is a modest oversimplification. We summarize just three possible scenarios that could originate from a tetraploid hepatocyte reentering the cell cycle, where only the bottom two outcomes would represent proliferation of a polyploid hepatocyte. The top (blue) inset instead would be more aptly aligned with “hypertrophy.” These possible outcomes were chosen as they appear to be the best explanation for the observations made by Miyaoka et al. [45]. We refer readers to Duncan et al. [3] for more details.
Skeptics of the diploid subpopulation model argue that diploid cardiomyocytes are not a privileged subpopulation, and that binucleated cardiomyocytes are also capable of re-entering the cell cycle and dividing. This point of view was most recently propounded by Leone and Engel in a very thorough review [48]. The strongest data in favor of their argument come from several studies on P7 cardiomyocytes placed in culture and monitored for completion of cytokinesis using video microscopy [49,50]. In these studies, authors note that both mononuclear and binuclear neonatal cardiomyocytes are capable of proliferation. D'Uva et al. specifically quantified the frequency of mono- and binucleated cardiomyocytes isolated from constitutively active Erbb2 transgenic mice, noting that ∼8% of mononucleated and ∼4% of binucleated cardiomyocytes completed cell division through cytokinesis, perhaps challenging the Bersell et al. findings on multiple levels [43].
Of note, the cardiomyocytes in each of these studies were placed in vitro for video microscopy, which as Leone and Engel aptly argue is one of the only definitive ways to confirm true cell division through cytokinesis [48] (discussed further below); however, doing so could affect the senescent state of the myocytes allowing the cells to behave in a manner they would not normally in vivo. Furthermore, because P7 binucleated cardiomyocytes are just entering the state of proliferative senescence, it is possible that they retain some of the plasticity characteristics of early postnatal cardiomyocytes. Thus, while it is clear that neonatal binucleated cardiomyocytes can be forced to re-enter the cell cycle in vitro, it remains unknown if adult binucleated cardiomyocytes are capable in vivo. Nevertheless, even the results from D'Uva et al. [49] suggest that, at a minimum, mononuclear cardiomyocytes possess a twofold proliferative advantage over their binucleated counterparts. A final noteworthy study using adult newt cardiomyocytes in vitro showed that a small proportion (6%) underwent multiple rounds of endomitosis resulting in transiently multinucleated cells before advancing to cytokinesis [51]. Authors argue that this indicates that multinucleation is not a barrier to further proliferation; however, a majority of the cardiomyocytes that did become multinucleated in culture failed to progress to cytokinesis (80%). Furthermore, the rare cardiomyocyte that is capable of completing reductive division could represent a unique cell cycle distinct to newts, which display specialized strategies to regenerate other tissues as well.
Diploid Hepatocytes and Liver Regeneration
Unlike the heart, the liver has an extraordinary capacity to regenerate. Even after substantial loss by two-thirds hepatectomy (PHx), the remaining tissue can expand to replace the lost mass and restore function to 100% efficacy within 5–7 days [52]. Similar to the heart field, substantial evidence supports the proliferation of pre-existing mature hepatocytes as the major driver of new postnatal hepatocytes [53,54]. Furthermore, hepatocyte polyploidy is a common phenotype that varies according to age and species. In mice and rats, hepatocytes undergo endoreplication resulting in various polyploid classes around the time of weaning, such that ∼70%–80% of adult murine hepatocytes become polyploid [55]. By contrast, only ∼25%–50% of human hepatocytes are polyploid [4 –6], although this increases with age [55] and disease [4].
In rodent models, several physiological and molecular mechanisms have been linked to the onset of polyploidization, including dietary changes [7], insulin/Akt signaling [10], E2f7 and E2f8 expression [9,56], and miR122 [57]. Furthermore, substantial work has been performed to ascertain the function of polyploidy in the liver, and to determine whether ploidy is linked to regeneration. Some of the original assumptions in the field were that polyploidy represented a terminally differentiated state with decreased proliferative capacity [58,59]; however, it is now clear that such a conclusion is not so straightforward.
The hepatic field offers many correlative observations, supporting the hypothesis that polyploidy in hepatocytes restricts their regenerative capacity. For example, in normal physiological circumstances hepatocytes are highly proliferative before weaning and this gradually declines with age. Thus, in both mouse and human, hepatocyte ploidy and senescence increase in parallel with age [60,61]. Further, distribution of hepatocytes across the zonation of the liver lobule in adult mice places most of the diploid hepatocytes near the portal and central veins, while the mid-lobular zone holds hepatocytes having the highest ploidy [62]. Two studies performing classic lineage tracing experiments identified subpopulations of hepatocytes that are capable of homeostatic liver renewal, which were located proximal to the central [63] or portal [64] veins. Notably, the Wnt-responsive hepatocytes in the pericentral zone (central vein) were found to be primarily diploid [63].
In addition, after two-thirds PHx, ∼80% of hepatocytes re-enter the cell cycle and synthesize DNA [45,65], resulting in increased ploidy of a single nucleus within 24 h after injury [22,66] and activation of signaling pathways associated with senescence [66]. Evidence suggests that polyploid hepatocytes isolated within 5 days of PHx are less proliferative than naïve hepatocytes by both cell culture and transplantation [22] measures, suggesting that PHx-induced polyploidy impairs proliferation. More recent research that measured total cell ploidy, accounting for both number of nuclei and DNA content within a nucleus, determined that by 28 days post-PHx the total cellular ploidy was comparable with the naïve state [67]. This finding suggests that either increased ploidy is only transient in nature, eventually correcting itself by 1 month, or perhaps there is a shift of cells displaying a binucleated phenotype (2 × 2N) in the naïve state to more mononuclear tetraploid cells (1 × 4N) after injury. The latter scenario would explain why nuclear ploidy appears increased as measured in the earlier studies [22,66], but this is not observed when whole cell DNA content is considered as measured in the more recent work.
On the other side of the coin, however, is the finding that there is no depletion of regenerative potential after either serial transplantation [68] or serial PHx (reviewed in Michalopoulos [52]), an observation that perhaps contradicts previous reports that PHx leads to decreased proliferative potential. Furthermore, identification of a mouse model (E2f7/E2f8 double knockout) displaying an abnormally high frequency of diploid hepatocytes (70%–80% diploid) failed to reveal heightened liver regenerative capacity or 5-bromo-2'-deoxyuridine (BrdU) incorporation after PHx [69], as the hypothesis posited here would predict.
Important to note is that the liver field frequently uses the term “regeneration” to indicate a replacement of tissue mass and function, but not necessarily through true cellular proliferation. While not inappropriate, this could explain at least some of the discrepancies observed in the field. Indeed, one study clearly demonstrated that after 30% PHx, liver mass was entirely restored through hypertrophy without cell division, while after 70% PHx restoration of liver mass was initially propagated by hypertrophy after which proliferation took over [45]. Further, the same study determined that <50% of hepatocytes that undergo DNA synthesis (as measured by thymidine analog incorporation) actually complete the cell cycle [45], suggesting that a considerable amount of cell cycle activity is endomitosis or endocycling and not true proliferation.
Beyond discrepancies in the literature that confound the ploidy hypothesis posed here, many in the liver field have detected a unique phenomenon coined the “ploidy conveyor” [3,45,55]. The ploidy conveyor model, originally described by Duncan et al., posits that during normal development, diploid hepatocytes undergo one to two rounds of DNA synthesis without completing mitosis or cytokinesis (ie, endomitosis) to generate polyploid hepatocytes. These polyploid hepatocytes can be displayed as 1 × 4N, 2 × 2N, 1 × 8N, or 2 × 4N. However, polyploid hepatocytes can also undergo ploidy reversal to return to lower ploidy classes. In one experiment, a population of 99.9% pure, fluorescence-activated cell sorted (FACS), 4N hepatocytes was allowed to expand in culture for 5 days, during which time ∼15% of the cells underwent endomitosis to become 8N, while ∼1% regressed into a 2N state. Video microscopy confirmed that in some instances, multipolar spindles formed in proliferating tetraploid hepatocytes, while in other cases two rounds of cytokinesis occurred, both of which could account for the observed ploidy reversal [3]. Thus, the entire process defining the “ploidy conveyor” includes the following: (1) originating in a diploid state; (2) undergoing endomitosis to become polyploid, possibly exiting the cell cycle (G0) at least transiently; and (3) once polyploid re-entering the cell cycle to undergo a second round of DNA synthesis while ultimately dividing into four daughter cells each with diploid content (ploidy reversal).
With additional insights, it is now clear that ploidy reversal frequently results in chromosomal segregation defects and aneuploidy, rather than producing a true diploid cell (euploidy) [3,55]. Utilizing the same E2f7/8 double knockout mice described above, two papers have demonstrated that polyploidy is an important cellular state within the liver in that it confers cancer resistance through protection from loss of heterozygosity [9], generation of genetic variation through ploidy conveyor-induced aneuploidy [24], and reduced proliferative capacity [24].
Yet, another study utilizing a virally induced sparse labeling strategy to assess cell cycle and ploidy in liver regeneration in vivo observed a possible reduction in nucleation (ploidy of individual nuclei was not assessed) [45]. Before injury, most labeled hepatocytes were found as single cells, ∼70% of which were mononuclear. After two-thirds PHx, >95% of the labeled hepatocytes found in doublets, suggesting that cell division had occurred, were mononuclear [45]. Such a result could only be true if either (1) mononuclear hepatocytes preferentially divided; (2) binuclear hepatocytes had undergone reductive division without DNA synthesis (Note: this is not how Duncan et al. described the ploidy conveyor model, which required DNA synthesis [3]); or (3) binuclear hepatocytes underwent DNA synthesis and divided to generate two mononuclear tetraploid cells (as previously described in Guidotti et al. [46]). Because assessments of neither DNA synthesis nor DNA content within individual nuclei were incorporated into this analysis, it is impossible to distinguish between the three possibilities. However, authors did note a reduction in the ratio of binucleated hepatocytes in remaining singlets (those hepatocytes that had presumably not divided), suggesting that at least some of the observed divisions could have arisen from one of the latter two scenarios. Notably, doublets containing two binucleated hepatocytes were never observed [45], suggesting that binucleated hepatocytes, if capable of division, are not doing so through a conventional cell cycle.
To date, one study has addressed the model postulated here most directly. Using the E2f7/8 double knockout mice, Wilkinson et al. determined that double knockout (70%–80% diploid) hepatocytes could proliferate faster than wild type as measured both in vitro and in vivo by repopulation studies. To distinguish the genetic (ie, E2f7/8 knockout) versus ploidy contributions behind the observed differences in proliferative capacity, the authors sorted diploid and polyploid hepatocytes from wild-type animals, and again determined that diploid hepatocytes could re-enter the cell cycle and proliferate faster than polyploid counterparts. They note that all ploidy subsets were capable of proliferation (as measured by BrdU and phospho-HH3); however, at 72 h, 80% of mononuclear hepatocytes had cycled, while only 30% of binucleated hepatocytes had. Further, quiescent hepatocytes (those that did not stain positive for Ki67) were overwhelmingly polyploid [67].
Given these findings and the observations from the earlier literature, the judicious conclusion would be that diploid hepatocytes have a proliferative advantage over their polyploid counterparts; however, at least some polyploid hepatocytes can proliferate (Fig. 3). In vitro analysis of polyploid hepatocytes might suggest that only a rare hepatocyte can undergo reductive division (∼1% in the original Duncan et al. work [3]). However, this is possibly more extensive in vivo, where aneuploidy, requiring ploidy reversal (to the best of our knowledge), can be observed in ∼25% of mouse hepatocytes [3] and 25%–50% of human hepatocytes [5].
Are There Other Tissues with a Similar Subpopulation?
Multiple other tissues contain subpopulations that undergo endocycling or endoreplication resulting in polyploidy and/or multinucleation (Table 1), begging the question if polyploidy impairs regenerative capacity in these tissues as well. Here, we describe a couple additional parenchymal cell types where this question has not been tested directly, as was the case for the two tissues described above, however where there are several lines of evidence suggesting that the model could hold true.
Retinal pigmented epithelial cells
In rodents, retinal pigmented epithelial (RPE) cells, responsible for support, phagocytic maintenance, and providing a blood barrier for the neural retinal layer of the eye, begin to undergo multinucleation shortly after birth, a process that appears to plateau ∼2 weeks of age [70], similar to cardiomyocytes. Quintessential lineage tracing experiments have not been performed within the RPE; however, best evidence thus far suggests that newly born RPE cells are derived from pre-existing RPE [71,72].
Both regional and temporal evidence in laboratory animals suggest that there could be a link between DNA content and proliferative capacity. First, mononuclear cells are observed at the highest levels in the periphery of the retinal eye cup (∼75%), and this gradually decreases as you move to the central region (20%–30%) (Fig. 4) [73,74]. Meanwhile, cell cycle analysis of mammalian RPE, either in homeostatic conditions or after injury, suggests that there is some turnover of RPE cells [73,74] and the observed cell cycle activity is mostly restricted to the peripheral territory, where mononuclear RPE cells are most prevalent [73]. One study, following nucleation and cell cycle in aged retinas, determined that oxidative stress-induced multinucleation increases with age particularly in the central and equatorial (middle) regions, while total cell numbers decline [74], suggesting a switch to hypertrophic growth as multinucleation increases with age.
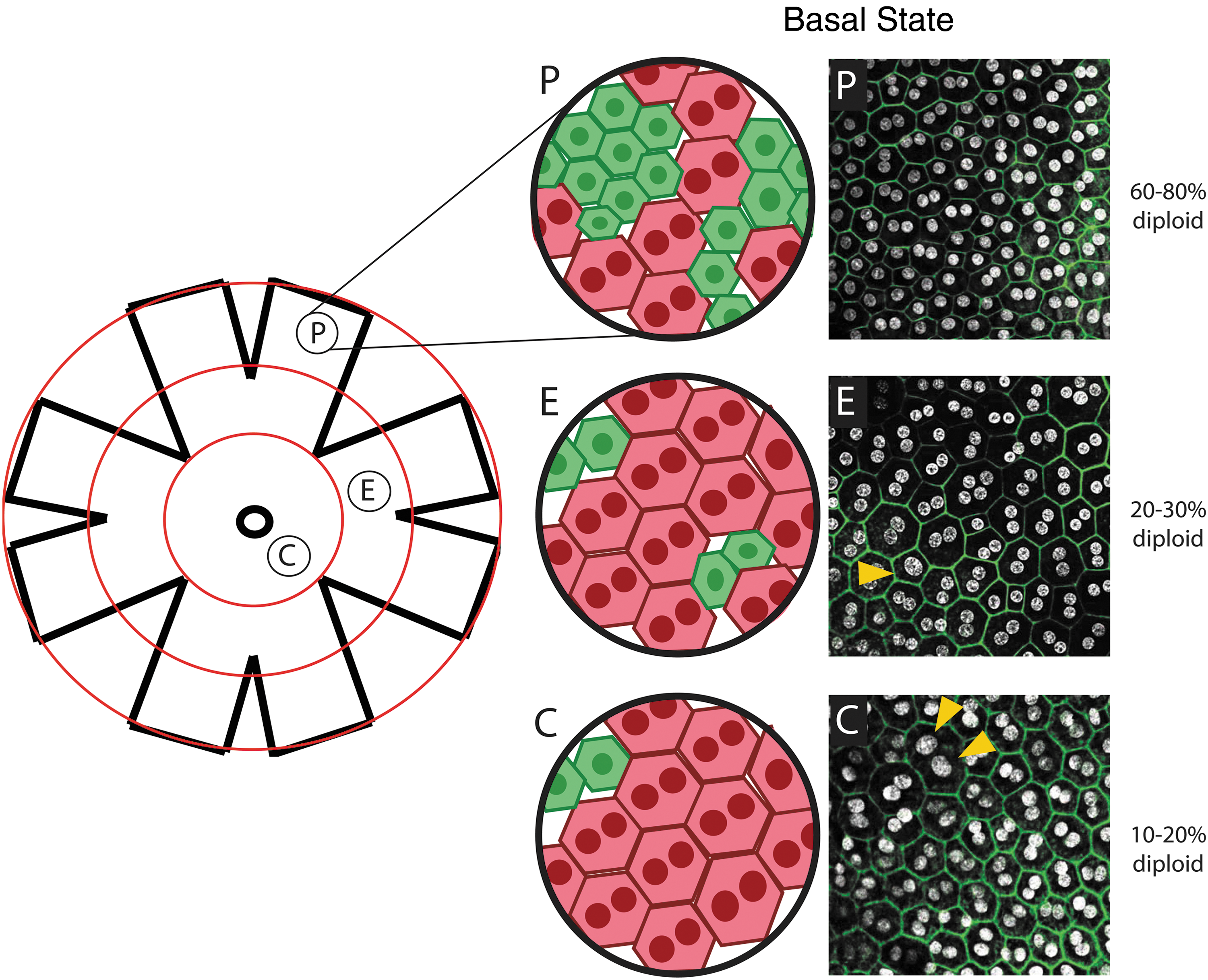
Polyploidy in murine retinal pigment epithelial cells. Schematic of a whole mount eye cup with concentric circles depicting the central (C), equatorial (E), and peripheral (P) domains. Example confocal images of a rat RPE within each domain stained for DAPI (white) and phalloidin (green). Yellow arrowheads point to possible examples of single nucleus tetraploidy. Frequencies of diploid RPE are approximations gathered from Refs. [73,74] and personal observations. DAPI, 4′,6-diamidino-2-phenylindole; RPE, retinal pigmented epithelial.
While the data on laboratory animals appear to support the hypothesis, observations in human RPE may appear to contradict the model. First, some have argued that there is a progenitor or stem cell within the human RPE that when isolated and put into culture can proliferate and generate both mesenchymal cell types and RPE cobble stone monolayers [75]; of course it is impossible to test whether this happens in vivo. In addition, <5% of human RPE are bi- or multinucleated [76], which at first glance might also challenge the hypothesis; however, to date there have been no analyses of DNA content within a single nucleus of RPE. This is worthy of note because in cardiomyocytes single nucleus polyploidy (ie, 1 × 4N or 1 × 8N) appears to be more the common cellular phenotype for humans [2]. In our own laboratory, we have observed occasional large nuclei (Fig. 4, arrowheads) within rat RPE, perhaps indicating potential nuclear polyploidy in rodents, at least. Certainly, more work in laboratory models and a better understanding of differences between humans and animal models are necessary to draw any real conclusions.
Pancreatic β cells
Polyploidy within the β cells of the islets of Langerhans was first reported more than four decades ago in both mice [11] and humans [12] alike. Best estimates suggest that these cells arise at weaning coinciding with the adoption of hypertrophic growth mechanisms [13]. However, even after adoption of hypertrophic growth, there is some low-level, homeostatic turnover of β cells [13,77], and this proliferation is absolutely required for pancreatic function [78]. As in other tissues described here, lineage tracing analysis links new pancreatic β cells to division by pre-existing β cells [77].
There is little direct evidence linking DNA content to regenerative capacity; however, the field has observed increased endoreplication and polyploidy with exposure to hyperglycemic states, and authors concluded that this resulted in a premature aging phenotype [79]. Further, endoreplication-based hypertrophy is insufficient to maintain β cell mass [78]. Taken together, this could suggest that as polyploidy increases in a disease state, β cells become less capable of proliferating and regenerating β cell mass, but of course much more work is necessary.
Polyploidy as an Injury Response
Thus far, we have discussed examples of somatic polyploidy, which occurs as part of a normal developmental process to certain parenchymal cells. Beyond this physiological polyploidy, there are examples in the literature where endoreplication can be induced after disease or injury, seemingly as a strategy for tissue repair. This would be most aptly defined as “hypertrophy”; however, we recognize that hypertrophy can also occur in the absence of DNA synthesis. We have already described such a scenario in the liver, where after 30% PHx liver mass and function are restored entirely through hypertrophic means and after 70% PHx restoration of liver mass is initially propagated by hypertrophy after which proliferation takes over [45]. In the latter example, it was determined that <50% of hepatocytes that undergo DNA synthesis actually complete the cell cycle, suggesting substantial polyploidization as an injury response [45]. And a similar response is presumed to hold true in the heart as well, where reports indicate that 40%–85% of cells that replicate their genomes are likely failing to divide [15,25].
At least two additional cell types appear to have a similar strategy for repair. Within the epicardium, the mesothelial layer of the heart, Cao et al. describe how two distinct cell cycle processes contribute to regeneration of zebrafish epicardium, where cells at the leading front undergo endoreplication resulting in dramatic hypertrophy and rapid restoration of surface coverage, while “followers” instead remain small and mononucleate choosing instead to complete cell division. Authors went on to describe how mechanical tension contributed to these differing cell cycle dynamics. Interestingly, by 14 days postinjury the multinucleated leader cells underwent apoptosis leaving behind the more permanent cells that arose by true proliferation [80]. Similarly, after acute kidney injury, Lazzeri et al. describe an initial recovery response through endocycle-induced hypertrophy of remnant tubular endothelial cells, which was then followed by proliferation of a progenitor cell [81].
Thus, in these examples, injury response is supported by two synergistic mechanisms: an initial and rapid hypertrophic event, frequently involving endoreplication or endocycling, followed by a slower proliferative response meant to sustain more permanent repair [82]. This concept of an initial hypertrophic response aligns nicely with many of the reasons a cell might become polyploid as presented above in the introduction: conservation energy and resources by not completing cytokinesis, enlargement to fill spaces left by injury, and preservation of barrier function (tissue specific). It could also suggest that there are two distinct and targetable waves of cell cycle-based responses after injury.
Conclusions and Future Directions
In this review, we present a developing model of tissue regeneration whereby in some tissues a residual population of diploid parenchymal cells retain proliferation competence making them the privileged subpopulation capable of facilitating tissue regeneration, specifically through cell proliferation. We present an in-depth analysis of the existing literature for two tissues that support this model and speculate where the model could be relevant in at least two other tissues. Certainly, the evidence for this model is not without weaknesses and inconsistencies, which we have tried to judiciously describe. One avenue which would definitely prove the model posited here would be classic lineage tracing experiments. To date, no molecular marker of the diploid subpopulation of any tissue has been identified, thus rendering this strategy currently unfeasible. Further, attempts to distinguish diploid hepatocytes from polyploid counterparts by transcriptomic analysis have uncovered very few differences between the two populations [83]. This result could have multiple explanations. First, the comparison was carried out by microarray analysis. In the advent of in-depth sequencing strategies, it may now be possible to identify more subtle differences between the two cell states, which a microarray-based approach was unable to identify. Alternatively, even with more detailed analysis the result may still hold true for the reason that such an analysis would simply represent a comparison of a quiescent cell with a senescent cell. Advances in this regard would be vital for validating the model.
The data presented by the field thus far highlight shortcomings where more in-depth analyses are needed. First and foremost, it has become evident in the cardiomyocyte field that the traditional markers of proliferation (ie, Ki67, thymidine analogs, proliferating cell nuclear antigen (Pcna), and phosphohistone H3) are insufficient to claim proliferation because each of these markers is also present during alternative cell cycles, such as endocycling and endomitosis, neither of which results in a new daughter cell. Even Aurora B Kinase, a supposed definitive marker of cytokinesis, can form at the cleavage furrow in binucleating cardiomyocytes that fail to complete cytokinesis; thus, it is also inadequate to claim proliferation. This appears to be no different for the hepatocyte field, where livers can restore tissue mass by both proliferation and hypertrophic means and lack of rigor to discern the two could explain discrepancies in the field.
Multiple groups are now acknowledging this weakness in the data [48,84,85]. In our work, we developed a strategy that definitively quantifies completion of the entire cell cycle [15]. In brief, we paired thymidine analog incorporation to label cardiomyocytes that had undergone DNA synthesis in response to injury with single cell analysis of ploidy, and reasoned that if an EdU+ cardiomyocyte was both mononuclear and diploid it must have successfully completed cytokinesis. In contrast, if an EdU+ cardiomyocyte was binucleated or mononuclear tetraploid it could instead represent cell cycle activity with failed cytokinesis (ie, endoreplication). Admittedly, this logic does not account for scenarios like those perhaps observed in the hepatocyte field whereby a binucleated cell undergoes DNA synthesis and divides to generate two daughter cells each being 1 × 4N [45,46]. In other words, we can speak to the EdU+ diploid cells as having completed cytokinesis, but we cannot conclusively discern if the EdU+ polyploid cells are the result of endoreplication (ie, a 1 × 2N undergoes DNA synthesis to become 1 × 4N or 2 × 2N), or an alternative division strategy like those observed in the hepatocyte field (ie, a 2 × 2N cell undergoes DNA synthesis and divides to become two 1 × 4N cells; Fig. 3, “ploidy conveyor” inset). Several groups in both the hepatocyte and cardiomyocyte fields have utilized video microscopy of primary cells in vitro [3,43,48 –50,86], which we have to agree is one of the few ways to definitively demonstrate cytokinesis and has been instrumental to our understanding by what mechanisms cells become binucleated. However, it does require taking the cells out of their natural environment perhaps affecting their behavior. Certainly, new strategies addressing this concern are warranted.
Another fundamental question for the model is what are the molecular drivers of replicative senescence? Does each tissue have its own methodology for initiating the process or are the mechanisms universal? This is an ongoing field of investigation; however, one common theme arising from multiple tissues is a possible link to oxidative stress and DNA damage, although in distinct settings. In cardiomyocytes, for example, work by Puente et al. suggested that oxidative stress at birth resulted in physiological cell cycle arrest, which takes place over the first postnatal week of life [87]. Our own work identifying Tnni3k as a genetic mechanism of cardiomyocyte polyploidization [15] might also converge onto oxidative stress signaling [88,89]. Further work in both hepatocytes and RPE links oxidative stress to pathological polyploidization observed in aging and diseased states [74,90,91]. However, to the best of our knowledge, oxidative stress has not been tied to physiological polyploidy of hepatocytes, where research has instead indicated insulin/Akt signaling [10], E2f7 and E2f8 expression [9,56], and miR-122 [57]. Beyond oxidative stress and DNA damage, the relationship of transcription and epigenetics to somatic polyploidy remains an almost entirely unexplored avenue, with examinations of such just brushing the surface [33,92].
A final and equally intriguing idea is that perhaps the diverse ploidy classes, beyond diploid versus polyploid, are not all created equal. For example, are bi- and multinucleated cells (with diploid nuclei) comparable with single nucleus polyploidy, or do each of these states exist for a unique reason [4,48,93]? Furthermore, what is the extent of aneuploidy in these tissues and how is it displayed? The liver field has begun to address this, suggesting that aneuploidy displays as whole chromosomal gains or losses due to imperfect segregation at the time of mitosis [3,24]. It remains unknown if smaller abnormalities (ie, regional duplications/losses through mitotic recombination) exist simply due to the depth in which this subject has been addressed thus far. Finally, to the best of our knowledge, it remains unknown if aneuploidy affects proliferative capacity. Here, the possibilities are boundless, and strategies to approach such questions will only be improved in the current age of advancing single cell technology.
Footnotes
Acknowledgments
We thank Dr. Caitlin O'Meara, Dr. John Lough, and Dr. Allison Ebert for their time and constructive feedback on this article.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work is supported in part by the Research and Education Program Fund, a component of the Advancing a Healthier Wisconsin endowment at the Medical College of Wisconsin and by the Cardiovascular Center at the Medical College of Wisconsin. Further support comes from the American Heart Association (18CDA34110240).