
Abstract
Recently, cell therapy has been developed as a novel treatment for perinatal hypoxic-ischemic encephalopathy (HIE), which is an important cause of neurological disorder and death, and stem cells from human exfoliated deciduous teeth (SHED) express early markers for mesenchymal and neuroectodermal stem cells. We investigated the treatment effect of SHED for HIE in neonatal rats. Seven-day-old rats underwent ligation of the left carotid artery and were exposed to 8% hypoxic treatment. SHED (1 × 105 cells) were injected via the right external jugular vein 24 h after the insult. The effect of intravenous administration of SHED cells was evaluated neurologically and pathophysiologically. In the evaluation of engraftment using quantum dots 655, only a few SHED were detected in the injured cortex. In the immunohistological evaluation 24 h after injection, the numbers of positive cells of active caspase-3 and anti-4 hydroxynonenal antiserum were lower in the SHED group than in the vehicle group. The number of Iba-1+ cells in the cortex was higher in the SHED group. However, the proportion of M1 microglia (Iba-1+/ED-1+) was significantly decreased, whereas M2 microglia (Iba-1+/CD206+) tended to increase in the SHED group. In the behavioral tests performed 5 months after hypoxic treatment, compared to the vehicle group, the SHED group showed significant elongation of the endurance time in the rotarod treadmill test, significantly ameliorated proportion of using the impaired hand in the cylinder test, significantly lower ratio of right/left front paw area in gait analysis, and significantly higher avoidance rate in the active avoidance test. In the in vitro experiment with cultured neurons exposed to oxygen-glucose deprivation, we confirmed the neuroprotective effect of the condition medium of SHED. These results suggested that intravenous administration of SHED exerted a treatment effect both histologically and functionally, possibly via a paracrine effect.
Introduction
Perinatal hypoxic-ischemic encephalopathy (HIE) causes injury affecting multiple organs, including neurological disorders (eg, cerebral palsy) throughout the life time of the organism [1]. Hence, HIE is positioned as one of the most important diseases in the perinatal period [1]. HIE has been reported to occur at a rate of about 1.5 per 1,000 births [2 –4], and its mortality rate was reported about 15% and also reported that severe seizures, sensory impairment, and developmental abnormalities occurred at high rates [4,5]. HIE lowers quality of life in patients and place a heavy burden not only on patients themselves but also on their families and society throughout their lifetime.
Hypothermia therapy has been shown to be effective in animal experiment models [6 –8] and long-term infarct sites and functional deficits were reduced in the neonatal HIE rat brain [8]. Although randomized trials on hypothermia therapy in clinical practice had been conducted and the effectiveness of neuroprotection of hypothermic therapy after HIE had been demonstrated, its mortality rate was still around 25%, and moderate to severe of neurological disorders were recognized about 20% even in the hypothermia group [9,10]. Currently, hypothermia therapy is the only established treatment option for HIE, but it is not effective for severe HIE [11]. Therefore, the development of new therapies for HIE is an urgent priority for perinatal care.
In recent years, several reports have shown the effectiveness of cell therapies for HIE using various kinds of stem cells, including neural stem cells [12] and umbilical cord blood cells [13 –15]. Mesenchymal stem cells (MSC) have been reported as a potential source for therapies in various diseases [16 –18], as it can differentiate not only into various mesodermal tissue cells but also into neuronal cells [19]. In addition, MSC has low immunogenicity [20], which facilitates allogeneic transplantations. MSC can be isolated from various tissues, including bone marrow, adipose tissue, placental tissue, and umbilical cord [21]. Among these tissues, bone marrow [22]- or adipose tissue [23,24]-derived MSC have been evaluated with the rat/mouse model of HIE.
Stem cells from human exfoliated deciduous teeth (SHED) are another unique and highly potential stem cell source for the following reasons. First, SHED is derived from ectoderm and has been identified as a highly proliferating population of clonogenic cells that can differentiate into a variety of cell types, including neuronal cells, adipocytes, osteoblasts, and endothelial cells [25 –27]. Moreover, SHED locate within the perivascular niche of the dental pulp and thought to originate from the cranial neural crest and express early markers for both mesenchymal and neuroectodermal stem cells [25,28], and it has been reported that SHED could survive and express neural markers when transplanted into mouse brain [25]. Second, SHED could change the hypoxic ischemia (HI)-induced inflammatory state to the anti-inflammatory state and suppressed apoptosis and reduced tissue loss [29]. Third, SHED is collected from falling teeth as a medical waste and the proliferation rate of SHED was significantly higher than that of bone marrow MSC [30].
As mentioned above, there are many advantages for treatment of HIE in the SHED treatment. We have shown a treatment effect of intracerebral injection of SHED in a mouse model by shifting the HI-induced inflammatory state to the anti-inflammatory state and resulting suppressed apoptosis and reduced tissue loss [29].
However, when considering the route of administration, the patients in the acute phase of HIE are likely to be very unstable [31]. For intracerebral administration, CT-guided injection should be the most appropriate approach; however, transportation to the X-ray room may lead to fluctuations in vital signs and interruption of hypothermia therapy; moreover, the risk of intracerebral hemorrhage is high in HIE [32]. In addition, our previous study with a mouse model indicated that injected cells themselves might cause undesirable gliosis in the host brain [33]. Therefore, considering the clinical applications, intravenous administration would be easier and safer if it can provide similar or more effective treatment compared with that provided by intracerebral administration.
The main objective of the present study is to determine if intravenous administration of SHED improves functional outcome, reduces lesion volume, and to elucidate mechanisms mediating these effects.
Materials and Methods
An expanded version of the Materials and Methods is available in the online Supplementary Data.
Animals
The Wister/ST dam and male pups were obtained from Japan SLC, Inc. (Shizuoka, Japan). The Institutional Review Board of Nagoya University approved the animal experiment protocols adopted in this study (Nagoya, Aichi Prefecture, Japan; permit numbers: 27191-2015, 28002-2016, and 29015-2017), and all animal studies were carried out in accordance with the Guidelines for Proper Conduct of Animal Experiments (Science Council of Japan, 2006).
We used 11 7-day-old rats for immunohistological evaluation (5 for vehicle group, 6 for SHED administration group) and 16 rats (8 for vehicle group and 8 for SHED administration group) were used for cytokine measurements in the acute phase (24 h after administration of SHED), and 24 rats were used for behavioral evaluations [10 for shams, 6 for vehicle group (2 died after injection), 8 for SHED administration groups] in the chronic phase. In total, 51 rats were used. Supplementary Fig. S1 shows the experimental design.
SHED cell culture
Naturally detached human deciduous anterior teeth were collected from children aged 6 to 12 at Nagoya University School of Medicine. All methods were performed in accordance with approved guidelines set by Nagoya University (H-73, 2003). Ethical approval was obtained from the Ethics Committee of Nagoya University (permission number 8–2). All the parents or legal guardians for study participations provided written informed consents.
The pulp was separated from the remaining crown and digested at 37°C for 1 h in a solution of 3 mg/mL collagenase type I (Worthington Biochem, Freehold, NJ) and 4 mg/mL dispase (Roche Molecular Biochemicals). Single-cell suspensions were obtained by passing the cells through a 70-μm strainer (Falcon). Dulbecco's modified Eagle's medium (D6429; Sigma-Aldrich, St. Louis, MO) and supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA) were used for subculture. We used the cells after ninth cell passage for this study.
Hypoxic-ischemic insult
Rats were anesthetized and the left carotid artery was exposed, separated from the vagus nerve, double ligated, and cut between the ligatures. In the sham operation, the artery was exposed, and the wound was closed without ligation or cutting. After a 1-h break, they were placed in an incubator under 8% oxygen, 92% nitrogen environment for 1 h at about 37°C. A hypoxic load was applied to the rats in the vehicle and the SHED groups, but not the sham group.
Magnetic resonance imaging
Each rat underwent Diffusion-weighted magnetic resonance imaging (MRI) and severity was evaluated at 4 h after HI insult to assign rats to equivalent groups. For severity discrimination, we graded each image according to signal area: Grade 0 was almost unrecognized high signal area, grade 1 was limited only to the cortex and grade 2 recognized the high signal in the cortex and basal ganglia (Fig. 1).
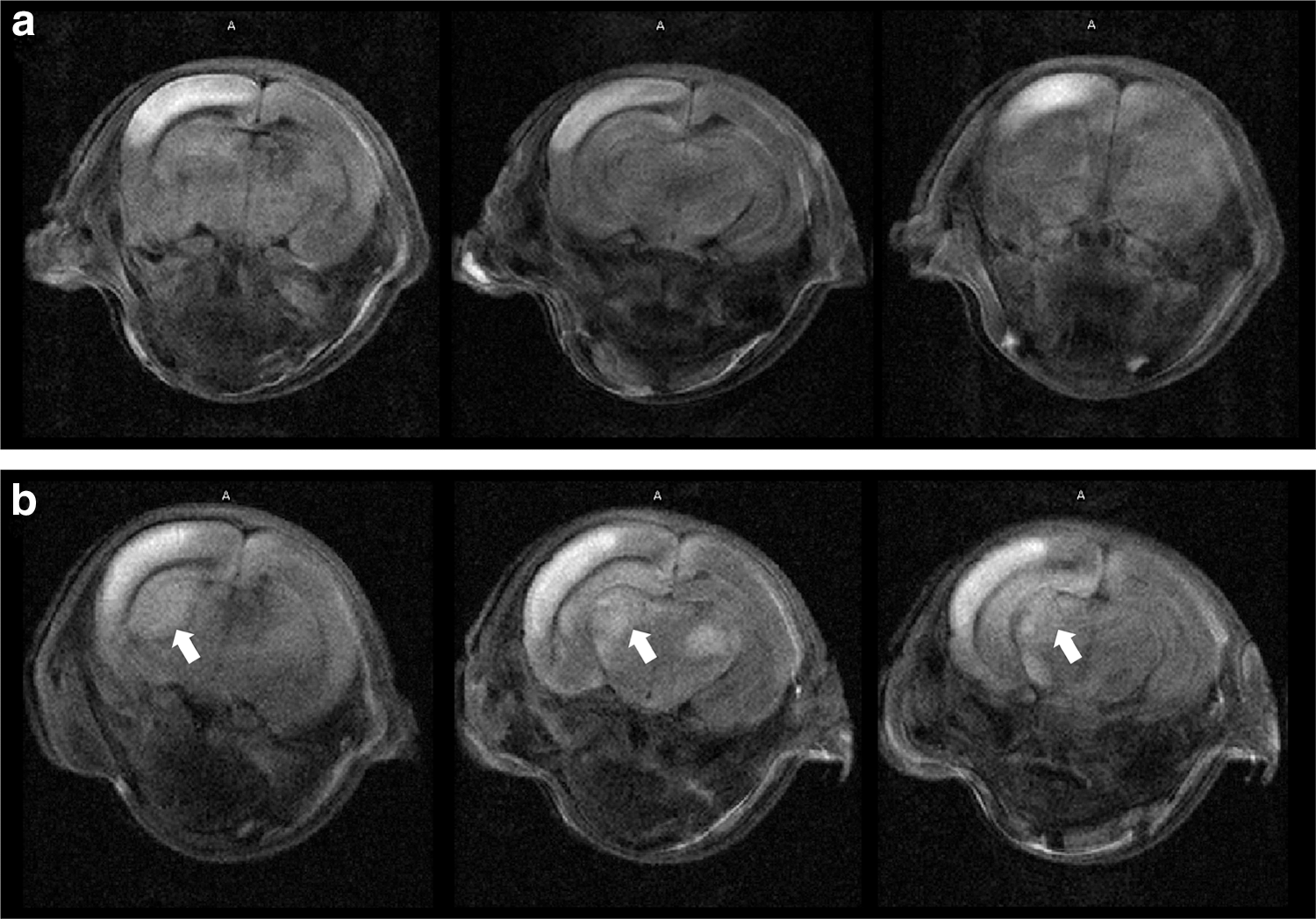
Examples of brain scanning by diffusion-weighted MRI after 4 h of HI.
These rats were divided into control (vehicle) and treatment (SHED) groups such that the numbers and severity were almost equal: in the immunohistological evaluation, two grade 1 pups (40.0%) and three grade 2 pups (60.0%) in the vehicle group and two grade 1 pups (33.3%) and four grade 2 pups (66.7%) in the SEHD group. In the behavioral and head MRI evaluations, both vehicle and SHED group included three grade 1 pups (37.5%) and five grade 2 pups (62.5%). In addition, the T2-weighted image was used to evaluate the brain volume in the chronic phase to see the treatment effect.
Administration of treatment
Twenty-four hours after HI, the treatment group had 1 × 105 SHED suspended in 0.1 mL phosphate-buffered saline (PBS) administered, and the vehicle group had 0.1 mL of PBS administered. The right external jugular vein was exposed under general anesthesia using isoflurane and proper body temperature, and injection was carried out using a 35-gauge needle.
Engraftment of SHED in the brain
In this study, we investigated the engraftment of SHED after intravenous administration of SHED using quantum dots (ODs, Qdot ITKCarboxyl Quantum Dots, Thermo Fisher Scientific, Tokyo, Japan) [34,35]. Based on the results of the pilot experiment (Supplementary Fig. S2), we determined that 4 nM QDs655 labeling for 24 h provided the optimal conditions as the introduction rate of QDs655 in SHED reached over 90% with these parameters. In previous studies, we confirmed that labeling stem cells using QDs655 at a concentration of 8 nM had no toxic effect on the cells [34].
In this investigation of the engraftment of SHED in vivo, 4 nM/8 mL of QDs655 was added to cultured SHED in a 25-mL flask at ∼80% confluency, and the cells were washed to remove excess labels and were collected 24 h later. Then, SHED containing QDs655 was administered 1 × 105 cells from the external jugular vein to HIE model rats at 24 h after HI treatment.
Tissue preparation and immunohistochemistry
Histological and immunohistochemical procedures were performed on day 9 after birth. All the rats were intraperitoneally administered pentobarbital sodium to anesthetize deeply and sacrificed. Serial (5 μm) coronal sections were cut throughout the brain embedded in paraffin at every 250 μm, and deparaffinized and rehydrated; then antigen retrieval was performed by heating for 10 min in 10 mM citrate buffer (pH 5.8).
These sections were then incubated overnight at 4°C with a primary antibody [rabbit anti-active caspase-3, 1:200; BD Pharmingen, Franklin Lakes, NJ, rabbit anti-4 hydroxynonenal antiserum (4HNE), 1:400; Alpha Diagnostic Intel, San Antonio, TX or goat anti-Iba-1, 1:1,000, Abcam, Cambridge, United Kingdom] in PBS containing 4% donkey serum (Jackson Immuno Research Laboratories, Baltimore Pike, PA) and 0.1% Triton X-100. Then, sections were incubated with the appropriate biotinylated secondary antibody for 60 min, followed by blocking of endogenous peroxidase activity with 3% H2O2 for 10 min. Binding was visualized using a VECTASTAIN ABC Kit (Vector Laboratories, Burlingame, CA) with peroxidase detection for 10 min (0.12 mg/mL 3,3′-diaminobenzidine, 0.01% H2O2, and 0.04% NiCl2).
To evaluate microglial polarization, one to two sections per pup at the hippocampal and basal ganglia levels were used such that the total number of cells was 50 or more. After antigen retrieval and blocking of nonspecific binding, sections were incubated with anti-Iba-1 (1:1000; Abcam), ED-1(1:300; EMD Millipore, Burlington, MA), and CD206 (1:100; Abcam) for primary antibodies at 4°C overnight. Sections were subsequently incubated with Alexa Fluor goat 546, Alexa Fluor mouse 488 and Alexa Fluor rabbit 647 (1:500) for 1 h at room temperature and mounted with ProLong Gold Antifade reagent containing DAPI (Thermo Fisher Scientific, Inc.).
Stereological quantification of cells
Sections were prepared at 5 μm and stained at intervals of 50 sections to count positive cells in each immunostaining. We counted the positive cells throughout the hippocampus and cortex using the Stereo Investigator version 10 stereology software (MicroBrightField Europe EK, Magdeburg, Germany); all positive cells were counted for the hippocampus and the Optical Fractionator, which is an unbiased stereological counting technique was used for the cortex.
For evaluation of microglial polarization, the percentage of M1 microglia (Iba-1+/ED1+) and M2 microglia (Iba-1+/CD206+) were evaluated in at least 50 Iba-1+ cells per animal using a fluorescence microscope (Olympus IX83).
Cytokine measurements
Sacrifice was performed by decapitation 24 h after administration, the brain cortex was quickly collected and frozen with liquid nitrogen. After the crushing using the Multi-beads shocker MB 2000 (Yasui Kikai Co., Osaka, Japan) and filtering the samples through a centrifugal filter (Millipore Sigma UFC 30 DV 00 Ultrafree MC DV 0.5 mL Centrifugal Filter Unit with Durapore PVDF Membrane, 0.65 μm; Merck, Darmstadt, Germany), we measured protein concentration by Pierce 660nm Protein Assay Reagent (Thermo Fisher Scientific) and adjusted with the homogenization buffer. Finally, various cytokines were measured using Multiplex analysis based on the xMAP Luminex technique with Rat Cytokine/Chemokine Kit (RECYMAG 65 K 27 PMX; Merck Millipore, Billerica, MA). Data were analyzed by xPONENT software (Luminex, Austin, TX).
Behavioral tests
Five months after HI, we performed the rotarod treadmill test at P162–163, cylinder rearing test at P169, gait analysis at P197–200, and the active avoidance at P202–205 test for behavioral evaluation.
Rotarod treadmill
The rats were placed on rotating rods, which accelerated at 4–40 rpm for 5 min. We measured twice a day with a break time of 4 h, in total, four times for 2 days. The time that the animal remained on the rod was the measured parameter.
Cylinder rearing test
Cylindrical transparent glass with diameter 20 cm and height 45 cm was used for this examination. The rats were individually placed in a cylinder and observed for 5 min once a day for two consecutive days. The forefoot placement of the walls of each contact bearing weight during the complete rear was recorded as right (impaired), left (nonimpaired), or both. The preference of forelimb use in each trial was calculated as follows: (nonimpaired − impaired)/(nonimpaired + impaired + both) × 100. The score added from both trials was used to represent the overall foot preference of each rat.
Gait analysis
We measured and evaluated the natural walking of rats using CatWalk, quantitative gait analysis system (Noldus company, Netherlands). The natural walking was recorded three times in each rat and the average was calculated. The gait-related parameters measured using the CatWalk system were the following: print area, area of paw print; mean intensity, the mean intensity of each paw in the run (the intensity is proportional to the load on the paw); duty cycle, percentage of time the paw accounted for the total step cycle of that paw; stride length, distance the paw traveled from one step to the next.
Active avoidance
We conducted this test 20 times for each trial using an automated shuttle box (W43.8 cm × D17.1 cm × H21.3 cm Product Number: MED-APA-D1R Med Associates, Inc., St. Albans, VT), in which the interval time between each test varied from 10 to 90 s (30 s on average), and the evasion rate was, thus, calculated. This was performed once a day for four consecutive days.
Oxygen-glucose deprivation and assessment of neuroprotective activity
For assessment of neuroprotective activity in vitro, we prepared the primary cortical neuron cultures [36] and SHED-conditioned medium (SHED-CM) as described before [23]. To examine the neuroprotective effect of SHED, we added SHED-CM to the neuronal cultures 24 or 48 h before oxygen-glucose deprivation (OGD) and quantitatively assessed neuronal damage 24 h after OGD using lactate dehydrogenase (LDH). All the evaluations were blinded regarding group allocation.
Statistical analysis
All data are expressed as mean (standard deviation). One-way analysis of variance was performed when comparing three groups. Post hoc comparisons were made using Tukey's test when main effects were found. Independent sample t-tests were used when comparing two groups. A P value of <0.05 was considered statistically significant. All statistical analyses were performed using JMP 13 (SAS Institute, Inc., Cary, NC)
Results
Evaluation of SHED engraftment using ODs
After SHED intravenous administration, we investigated its engraftment using ODs. Before the investigation, we examined the in vitro labeling of SHED using QDs655. Before flow cytometry, introduction rates of 60.0% and 91.6% were confirmed for 8 and 4 nM administration at 4 and 24 h, respectively (Supplementary Fig. S2). Based on the pilot experiment results, we determined that 4 nM QDs655 labeling for 24 h provided optimal conditions since it allowed the introduction rate of QDs655 in SHED to reach over 90%.
One hour after administration, the SHED-treated group exhibited a strong fluorescence reaction in the cerebrum. In both the sham and vehicle groups, such reaction could hardly be detected. At 24 h postadministration, the SHED-treated group continued to show a stronger fluorescence reaction in the affected cerebral side. At that time, the fluorescence reaction could also be observed, to some extent, in vehicle group, presumably due to HI-induced inflammation (Fig. 2a).
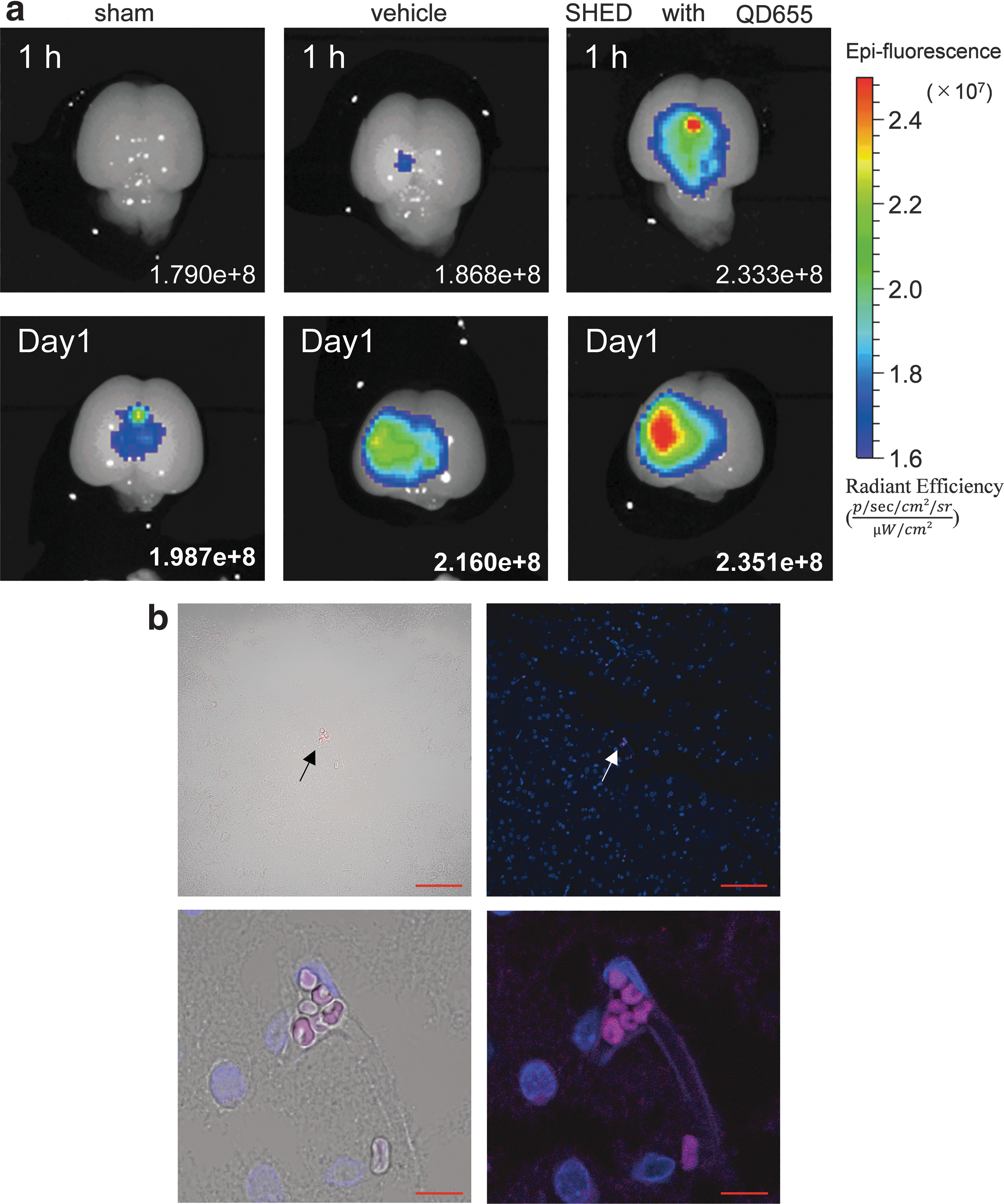
Evaluation of SHED engraftment using quantum dots.
To further examine the SHED engraftment in greater detail, paraffin sections were made and evaluated under a fluorescence microscope. At 24-h postadministration, almost none, less than 10 cells with QD655 in the cerebrum of SHED-treated animals, could be detected (Fig. 2b).
Effects of SHED treatment on acute injury markers after HI injury
Following HI injury, we immunohistologically evaluated the effects of SHED treatment on acute injury markers. SHED-treated animals exhibited a lower total number of hippocampal cells positive for active caspase-3, an apoptotic marker, than the vehicle group [2.958 (1,155) cells vs. 19,640 (3,002) cells, P < 0.001]. The number of positive cortical cells was also significantly decreased in this group [944,708 (410,432) cells vs. 2,172,610 (444,080) cells, P < 0.001].
The SHED-treated group also exhibited significant suppression of 4HNE, an oxidative stress marker, in the cortex [2,825,608 (498,923) cells vs. 3,784,420 (888,953) cells, P < 0.05], whereas significant difference was not observed in the hippocampus, although the number of positive cells was reduced by 23% [47,833 (14,838) cells vs. 62,240 (11,693) cells, n.s. not significant].
The number of hippocampal cells positive for Iba-1, a marker for whole microglia and macrophages, was 20% higher in the vehicle group than in the SHED-treated group [21,790 (3,877) cells vs. 17,442 (3,308) cells, n.s.]. Oppositely, in the cortex, the number of positive cells was 44% higher in the SHED-treated group than in the vehicle group [1,718,742 (359,323) cells vs. 1,191,510 (335,723) cells, P < 0.05] (Fig. 3a–c).
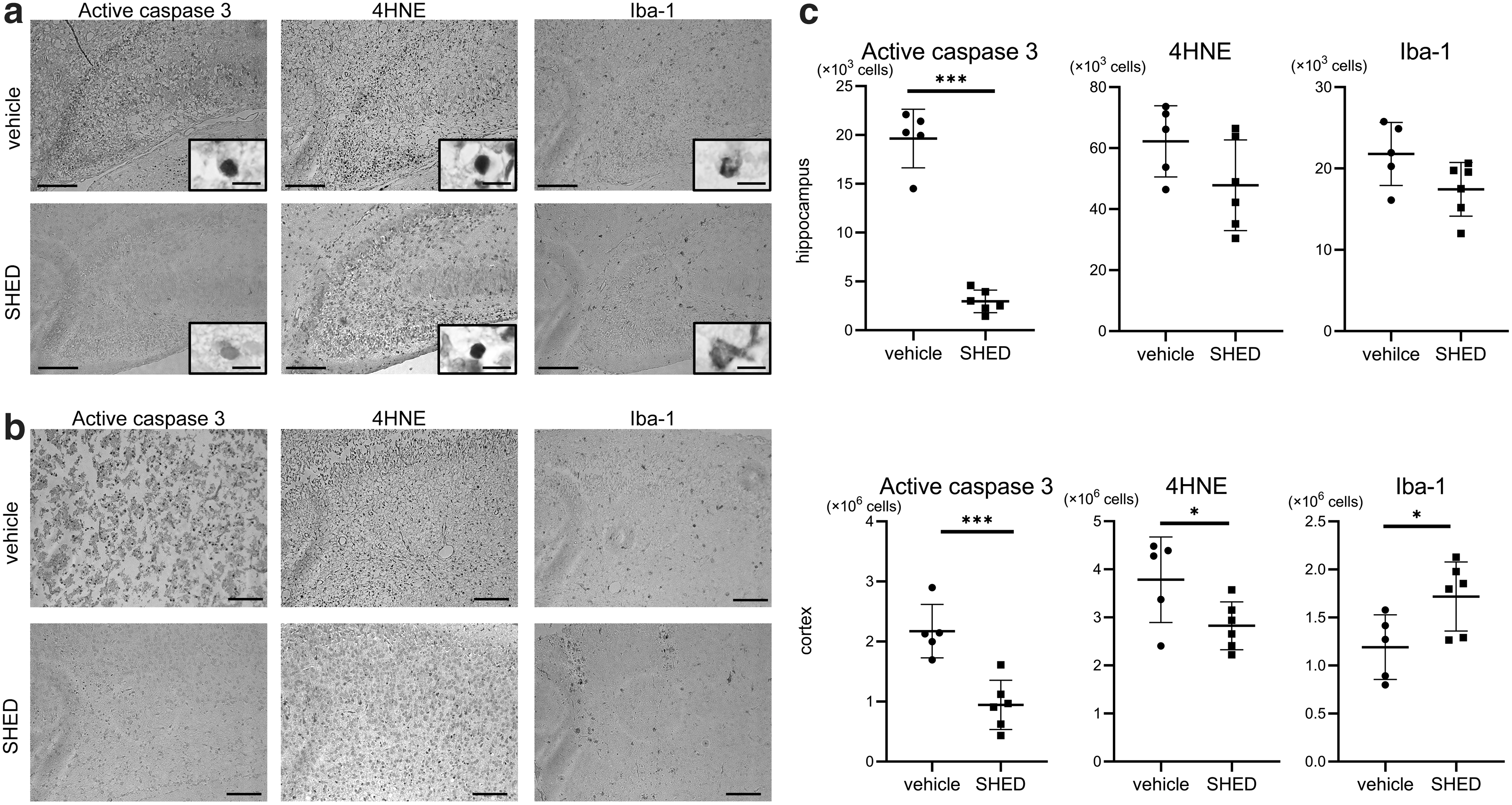
Effects of SHED treatment on acute injury markers after HI injury.
Effects of SHED treatment on microglial polarization
In addition, we used fluorescent immunostaining to calculate the percentage of Iba-1/ED-1 (M1 microglia) and Iba-1/CD206 (M2 microglia) double-positive cells among whole Iba-1+ cells. Representative pictures of the triple staining are presented in Fig. 4a.
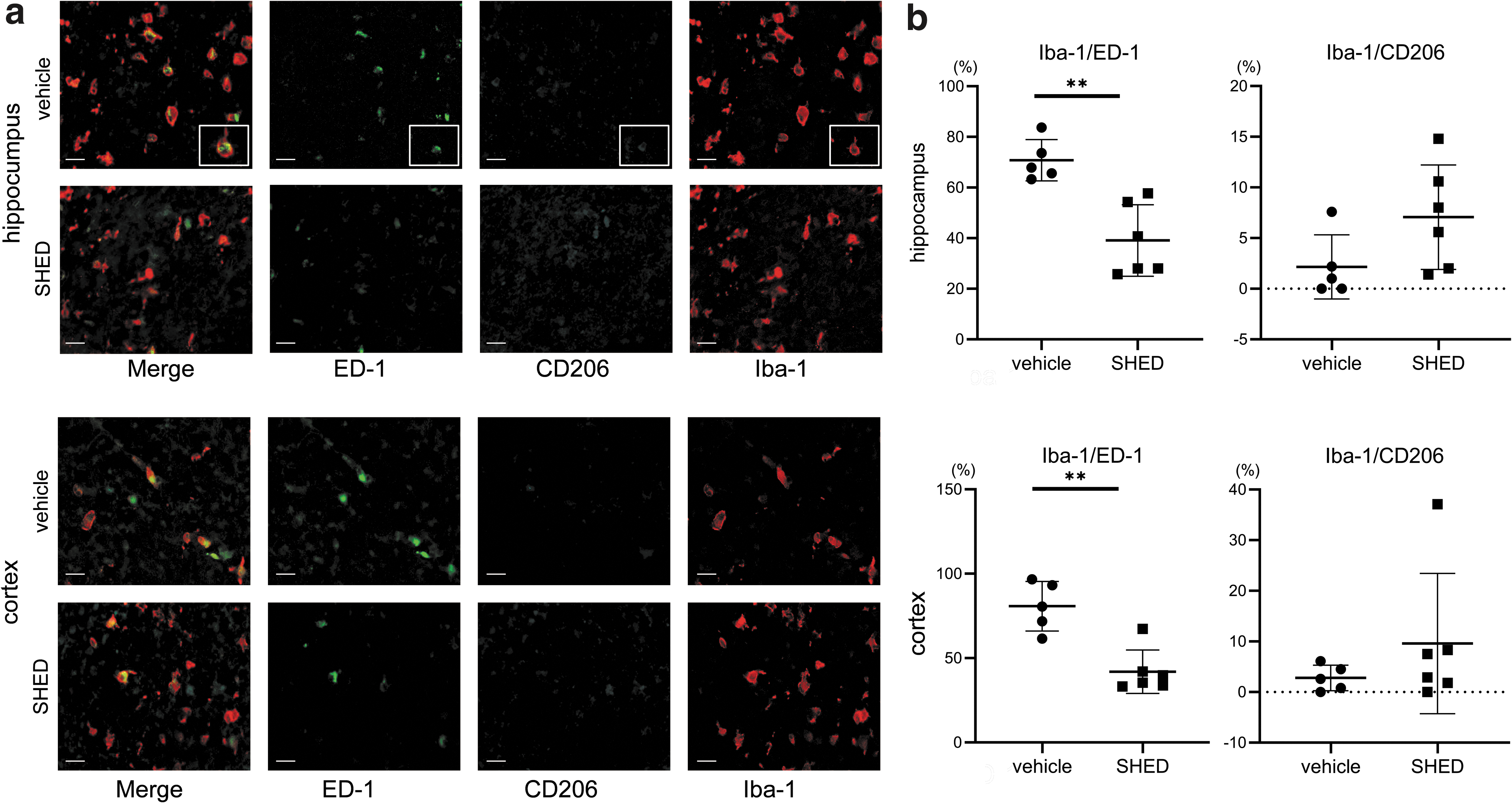
Effects of SHED treatment on microglial polarization.
The proportion of Iba-1+/ED-1+ in the hippocampus of the vehicle group was 70.8 (8.1) %, whereas in the SHED-treated group it was significantly decreased to 39.1 (14.2) % (P < 0.01). Similarly, in the cortex of the vehicle group, the proportion was 80.8 (14.7) %, whereas SHED treatment significantly decreased it to 41.9 (12.9) % (P < 0.01). On the contrary, the proportion of Iba-1+/CD206+ cells in the hippocampus in the SHED-treated group was more than double of that in the vehicle group, although it did not reach statistically significant difference [7.1 (5.2) % vs. 2.2 (3.2) %, n.s.], with a similar tendency observed in the cortex [9.6 (13.6) % vs. 2.8 (2.5) %, n.s.] (Fig. 4b).
Cytokine measurements
Next, we evaluated various cytokines in the brain 24 h after SHED administration using Multiplex analysis (Merck Millipore). The SHED-treated group (n = 8) had a significantly higher level of IL-1a [64.6 (18.7) pg/mL vs. 44.1 (12.9) pg/mL, P < 0.05], IL-18 [439.4 (127.3) pg/mL vs. 315.6 (38.2) pg/mL, P < 0.05], IP-10 [342.8 (170.2) pg/mL vs. 200.2 (74.0) pg/mL, P < 0.05], and MIP-1a [83.2 (24.1) pg/mL vs. 59.8 (7.4) pg/mL, P < 0.05] compared to the vehicle group (Fig. 5). On the contrary, no significant difference was observed between the two groups regarding the levels of IL-4, IL-1β, IL-2, IL-6, EGF, IL-13, IL-10, IL-12p70, IFN-γ, IL-5, IL-17A, IL-18, MCP-1, IP-10, GRO/KC, VEGF, Fractalkine, LIX, MIP-2, TNF-α, and RANTES.
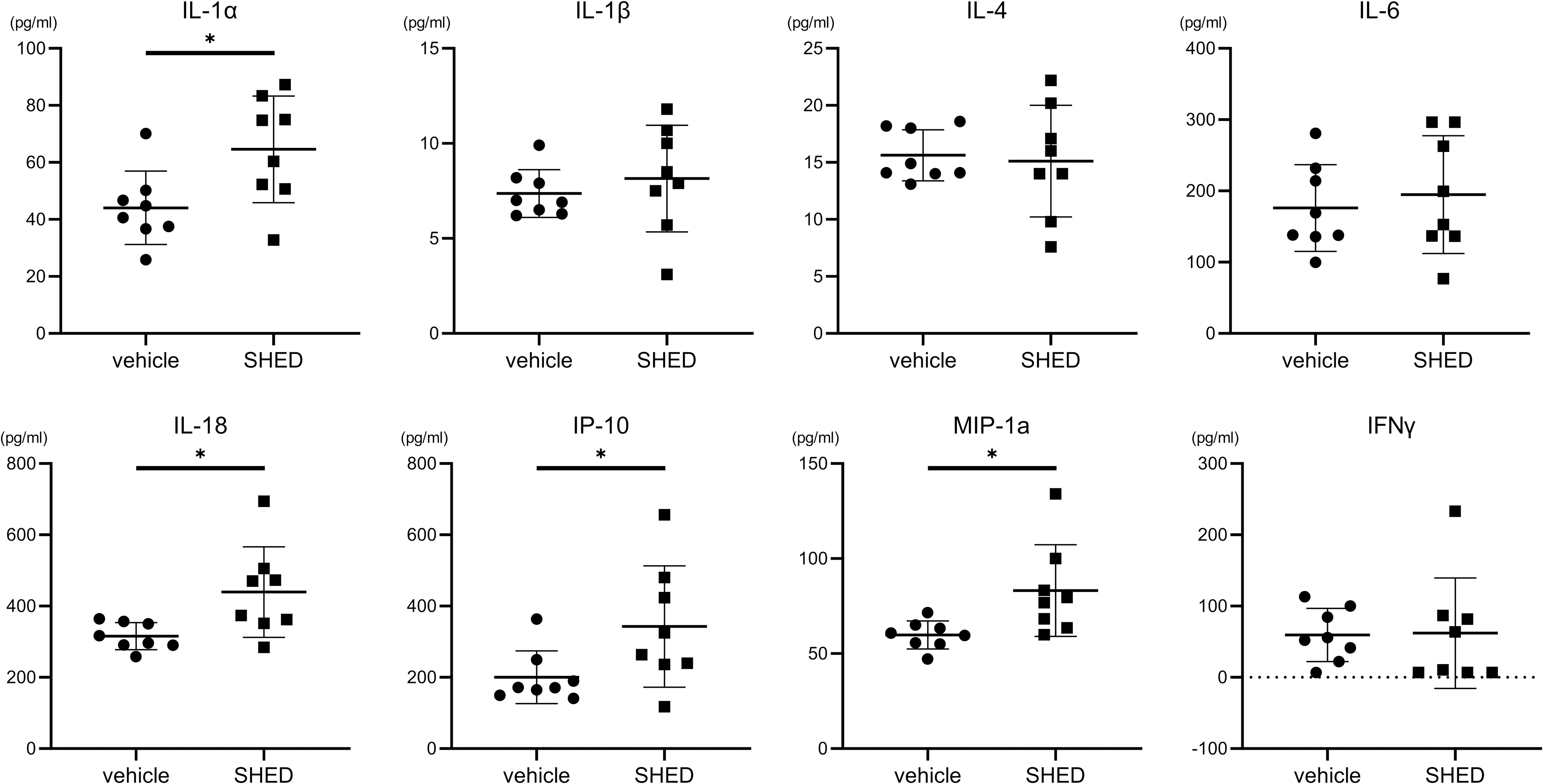
Cytokines in the brain. The SHED administration group had significantly higher IL-1a, IL-18, IP-10, and MIP-1a, compared to the vehicle administration group. Data are expressed as mean ± SD (vehicle, n = 8; SHED, n = 8). *P < 0.05.
Behavioral tests
The effect of SHED on the HI-induced behavior impairment was evaluated through four behavioral tests: the rotarod treadmill, the cylinder rearing, gait analysis, and active avoidance tests.
Rotarod treadmill
The rotarod was performed twice a day, for two consecutive days, between P162 and P163. The average daily scores were calculated. On the first day, both the sham- and SHED-treated groups tended to have a longer latency to fall than the vehicle group, although the difference did not reach statistical significance. However, on the second day, the vehicle group presented a significantly shorter latency to fall than the sham group, whereas the SHED-treated group presented a significant improvement of the shortened latency [vehicle vs. sham, 85.7 (29.3) s vs. 147.3 (25.1) s, P < 0.001; SHED vs. vehicle, 132.1 (19.4) s vs. 85.7 (29.3) s, P < 0.01] (Fig. 6a).
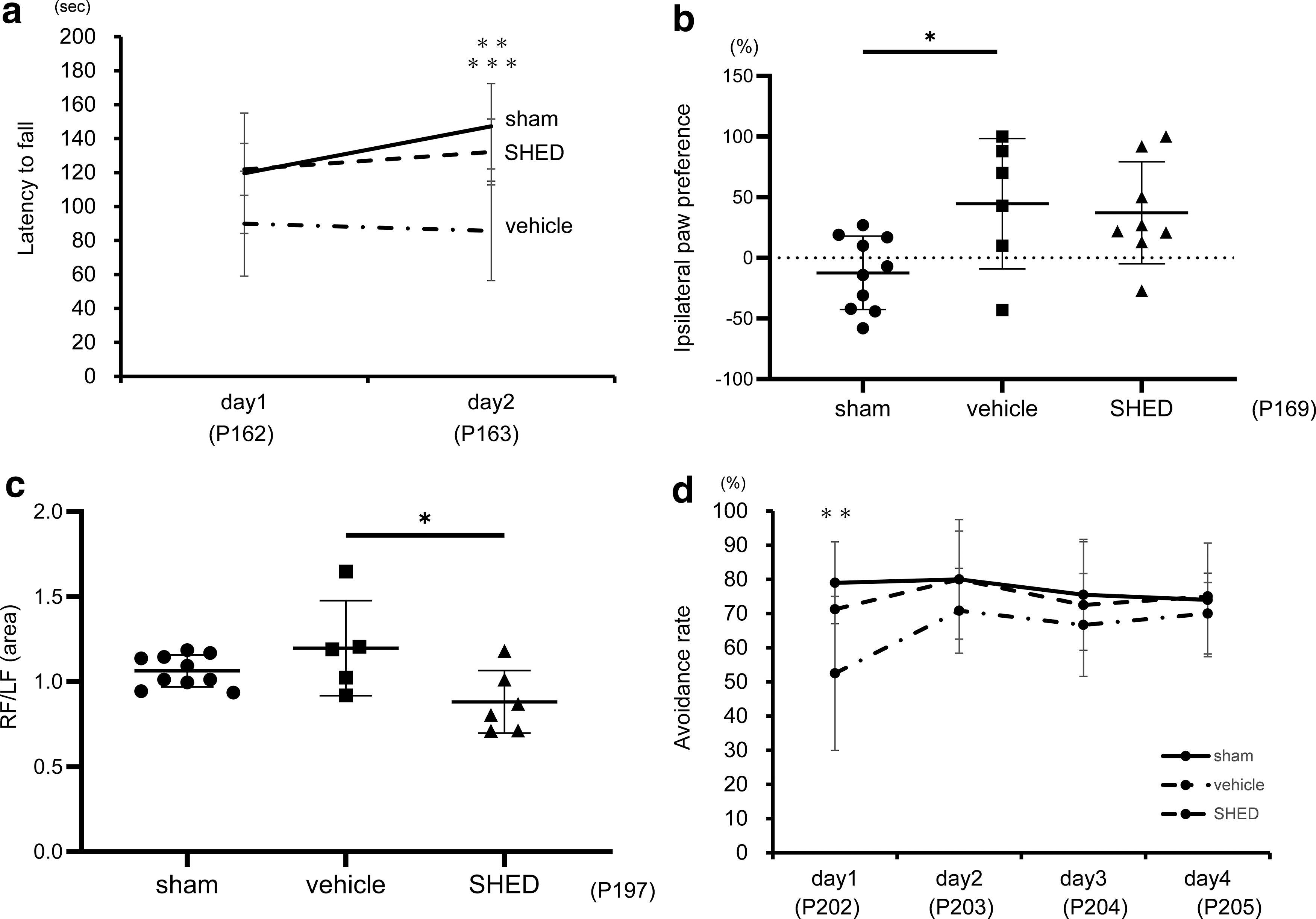
Behavioral tests. Behavioral tests: rotarod treadmill
Cylinder rearing test
The cylinder rearing test was conducted between P169 and P170. The average scores for 2 days were calculated and compared among the three groups. The vehicle group exhibited a significant preference for the noninjured forelimb (left) compared to the sham group [vehicle vs. sham, 44.7 (53.8) % vs. −12.4 (30.3) %; P < 0.05], whereas there was no significant difference between SHED group and sham group [SHED vs. sham, 37.2 (42.1) % vs. −12.4 (30.3) %; n.s.] (Fig. 6b).
Gait analysis test
In the gait analysis, performed at P197, the SHED-treated group exhibited a lower right forepaw/left forepaw (RF/LF) area ratio than the vehicle group [vehicle vs. sham 1.20 (0.28) vs. 1.06 (0.09), n.s. SHED vs. vehicle 0.88 (0.18) vs. 1.20 (0.28), P < 0.05] (Fig. 6c). No significant difference was found among the three groups regarding the RF/LF mean intensity ration or the RF duty cycle. The vehicle group presented a longer RF stride length than the sham group, which remained unchanged in the SHED-treated group [vehicle vs. sham 4.1 (0.2) cm vs. 3.3 (0.4) cm, P < 0.01. SHED vs. vehicle 4.3 (0.3) cm vs. 4.1 (0.2) cm n.s.] (Supplementary Fig. S3).
Active avoidance
The active avoidance test was performed once daily, for four consecutive days, between P202 and P205. On the first day, the vehicle group presented a significantly lower avoidance rate than the sham group [52.5 (22.5) % vs. 79.0 (12.0) %, P < 0.01], whereas the SHED-treated group had no significant difference in avoidance rates compared to the sham group [71.3 (7.5) % vs. 79.0 (12.0) %, n.s.]. The same trend was observed over the three subsequent days, although no significant differences were found (Fig. 6d).
Evaluation of the cerebral volumes measured with MRI in chronic phase and their correlation with the injury grades in the acute phase MRI
Upon completion of behavioral tests, we performed head MRI at P217. Within each slice, the brain parenchymal area was measured using 24 slices per brain with the ImageJ software [version:1.52a; Wayne Rasband (NIH), Bethesda]. Brain volumes were calculated using the Cavalieri principle. The vehicle group exhibited a significantly smaller cerebral volume than the sham group. No significant difference was found between the vehicle group and the SHED-treated group [sham vs. vehicle 1,426.5 (63.3) mm3 vs. 1,167.8 (114.5) mm3, P < 0.001, sham vs. SHED 1,426.5 (63.3) mm3 vs. 1,153.3 (157.2) mm3, P < 0.001] (Fig. 7).

Evaluation of the cerebral volume. Cerebral volumes measured with MRI at 30 weeks after HI injury. SHED treatment did not affect brain tissue loss compared to vehicle-treated group. Data are expressed as mean ± SD (sham, n = 10; vehicle, n = 6; SHED, n = 8). ***P < 0.001.
We also evaluated the relationship between the injury grades in acute MRI and brain volumes in the chronic phase MRI. There was no significant difference in the volumes in the chronic phase: the average of the volumes in grade 1 was 1,211.4 (116.9) mm3 and that in grade 2 was 1,120.8 (130.8) mm3 (n.s.) (Supplementary Fig. S4).
Reduction of OGD-induced cell death by SHED-CM
The neuroprotective effect of SHED-CM was investigated by performing experiments on neurons cultured with OGD to replicate the HI pathophysiological conditions in vitro. Primary cultures of rat cortical neurons were treated with 5% SHED-CM, 10% SHED-CM, or control (CM-) at 24 or 48 h before OGD. Under each condition, LDH was measured after OGD. Adding SHED-CM to the neurons at both 24 and 48 h before OGD significantly reduced LDH release by 55.9% and 67.2% at 5% CM (P < 0.001) and 10% CM (P < 0.05), and by 49.5% and 58.9% at 5% CM (P < 0.001) and 10% CM (P < 0.001), respectively (Fig. 8).
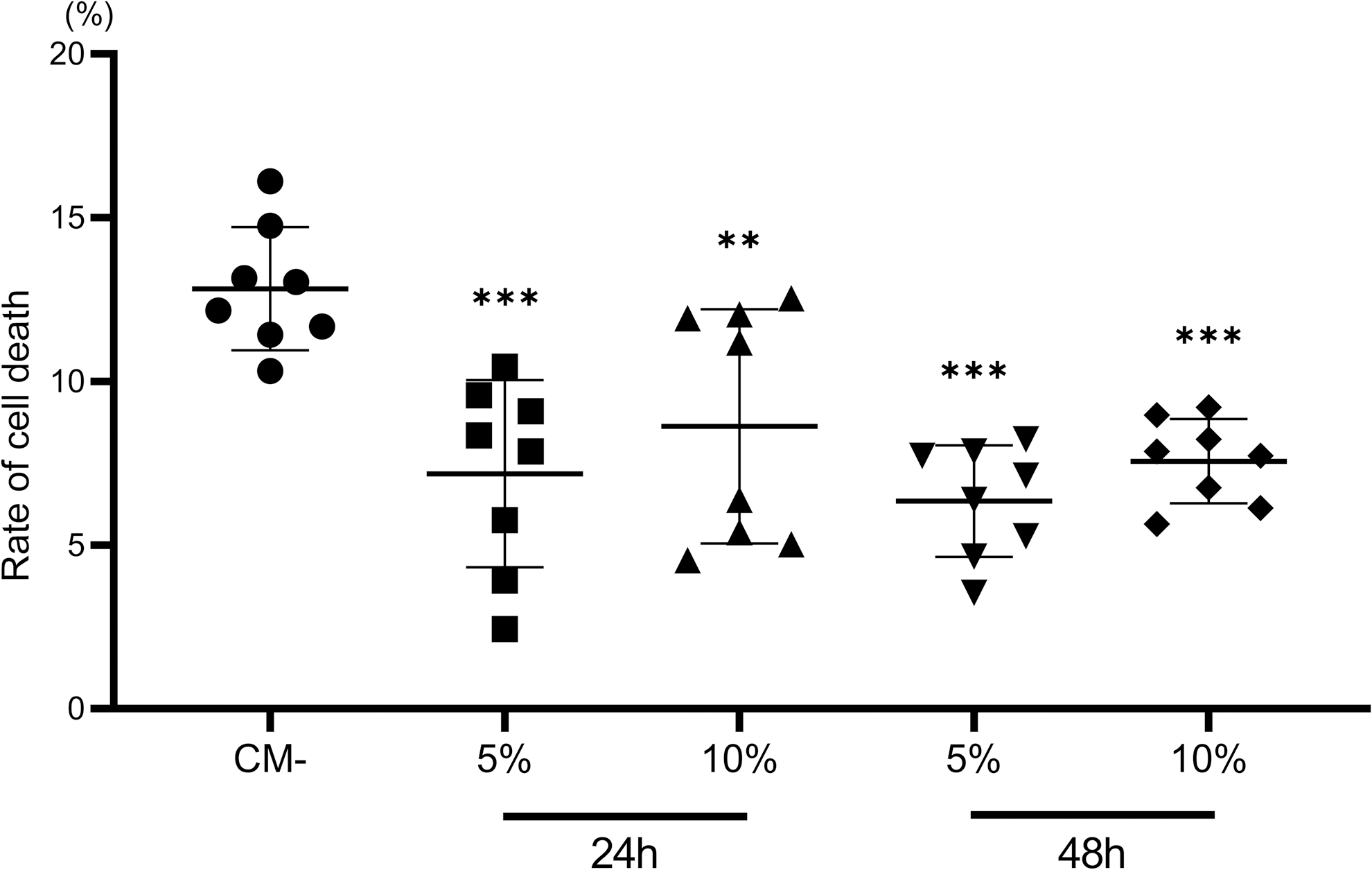
Reduction of OGD-induced cell death by SHED-CM. Effect of SHED-CM on OGD-induced cell death in primary cortical neurons. Neuronal cell cultures were treated with SHED-CM at concentrations of 5%, 10% for 24 or 48 h, then exposed to OGD for 3 h. Cell death was measured as LDH release in the medium at 24 h after OGD, and was significantly lower at both concentrations of SHED-CM. Data are expressed as mean ± SD (n = 8, each group). LDH, lactate dehydrogenase; OGD, oxygen-glucose deprivation; SHED-CM, SHED-conditioned medium. **P < 0.01, ***P < 0.001.
Discussion
In the present study, we demonstrated that an intravenous administration of SHED to HIE model rats strongly suppressed apoptosis and might relieve oxidative stress and could possibly reduce harmful M1 microglial activation. The cell dynamics observed using QDs revealed only a few cells in the brain 24 h after HI treatment, and the in vitro experiments with cultured neurons exposed to OGD demonstrated the therapeutic effect of SHED-CM. In addition, improvements in various behavioral experiments were observed.
First, we showed that intravenous administration of SHED significantly decreased several markers of tissue injury, including apoptosis, in acute phase injury after HIE. In previous reports, SHED strongly suppresses apoptosis of all neuronal lineages and minimizes the spread of tissue injury [37]. In addition, SHED shifted the HI-induced inflammatory to anti-inflammatory state and decreased tissue loss [29]. Some mechanisms which underlie the physiological mechanism of SHED have been demonstrated, such as intravenous injection of SHED expresses Fas ligand and induces apoptosis of T cells, thereby causing immune tolerance [38]. Moreover, SHED inhibits helper T17 inflammatory T cells, thus, raising the proportion of regulatory T cells and resulting in an anti-inflammatory action [39].
Next, we showed that oxidative stress was significantly suppressed by SHED administration. In our previous studies involving human umbilical cord blood cells [14] or dedifferentiated fat cells [23], we demonstrated that these cells significantly suppressed oxidative stress markers, including 4HNE.
Oxidative stress is strongly related to the onset and progression of HIE [40], and neonatal brains are particularly vulnerable to oxidative stress because the free radical elimination system has not matured yet and, thus, causes insufficient synthesis of antioxidant enzymes and scavengers following injury [41]. Therefore, a therapeutic effect observed against oxidative stress by SHED was considered immensely significant for neuroprotection against HIE. Since excessive oxidative stress causes impairment of neurogenesis, leading to cognitive deficit [42], significant suppression of oxidative stress by SHED has the potential to promote future neurodevelopment and/or a therapeutic effect on cognitive function.
Furthermore, we examined microglial polarity. In ischemia, activation of microglia causes phagocytosis, releases inflammatory and anti-inflammatory cytokines, and matrix metalloproteinases, which induce antigen presentation and fracture of the blood–brain barrier [43]. Ischemia also causes brain infiltration of peripheral leukocytes, resulting in further deterioration of inflammation and brain damage [44]. A recent report showed that classical activated microglia (M1) releases cytokines, chemokines causing subsequent inflammation, followed by microglia eliminating reactive oxygen species by M2 activity with anti-inflammatory action and, thus, causing wound healing [45].
Regarding this cascade of inflammation and changes in microglia, previous reports also showed that MSC could rescue injured neuronal cells by regulating the inflammatory/immune response [46]. Moreover, MSC transplantation suppressed activated macrophages (M1 phenotype) and increased alternately activated macrophages (M2 phenotype) [47]. In the present study, we also revealed that the ratios of ED-1+ cells in Iba-1 were significantly lower, and the ratio of CD206+ cells in Iba-1 tended to be higher in the SHED group than those in the vehicle group, which could lead to neuroprotection for HIE.
In particular, the pathway in which SHED induces M2 macrophages is unique. In a previous report, we showed that MCP-1/ED-Siglec-9 secreted from SHED was very much involved in the induction of M2 cells. The M2 bone marrow macrophages induced by MCP/ED-siglec-9 suppressed apoptosis of neurons [48], also increased various factors related with neuroprotection/repair, including vascular endothelial growth factor, insulin-like growth factor [49], brain-derived neurotrophic factor, and fibroblast growth factor 2 [50]. Consequently, the MCP-1/ED-Siglec-9 exerts multifaceted actions for neuronal protection/repair other than suppression of inflammation. From these evidences, SHED can bring multifaceted neuronal protection/repair by changing the polarity of microglia via trophic factors, and this property might be consistent with the results in the present study.
The optimal cell number was not verified in the present study. It is necessary to assess the dose–response to develop a novel therapy. Because we needed a very large number of rats to assess the dose–response, the number of cells to be administered was decided based on the results of our previous studies using other stem cells.
In a study with human umbilical cord-derived MSCs using neonatal stroke mice model, we confirmed that 1 × 104 was ineffective, but 1 × 105 was effective [51], and in the studies with bone marrow-derived MSCs and dedifferentiated fat cells, we confirmed the therapeutic effect on HIE-rat model with the same number of cells, 1 × 105 [23,24]. Furthermore, considering the clinical application, the higher number of cells administered, the higher was the risk and cost for the patient. Therefore, we used 1 × 105 cells in the present study. Nevertheless, an optimal dose needs to be determined before clinical application. We plan to assess the dose–response in subsequent studies.
In addition, we did not evaluate the therapeutic effect of SHED combined with hypothermia therapy, which is an established treatment for HIE. It has been reported that the outcome of combination therapy of stem cell therapy using human umbilical cord blood and hypothermia therapy was better than individual therapy and was effective even in severe cases, in which the therapeutic effect of hypothermia therapy was poor [52]. When SHED is used in combination with hypothermia therapy, the therapeutic effect could be enhanced and the scope of treatment may be expanded; however, further studies are required to verify this effect.
In conclusion, intravenous administration of SHED suppressed apoptosis, oxidative stress, and M1 microglial activation in HIE model rats. Furthermore, SHED ameliorated the impaired behaviors of these rats. The present study indicates that cell therapy using intravenous SHED is a good candidate for a clinical application in the treatment of HIE.
Footnotes
Acknowledgments
We are grateful to Ms. Kimi Watanabe, Ms. Eiko Aoki, Ms. Tokiko Nishino, Ms. Azusa Okamoto, Ms. Tomoko Yamaguchi, and Ms. Tomoko Arimoto for their skillful technical assistance. We also wish to acknowledge Division for Medical Research Engineering, Nagoya University Graduate School of Medicine, for usage of the pulverizer.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by Japan Society for the Promotion of Science KAKENHI (grant no.: 15K19650, grant no.: 17H02731).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.