
Abstract
Bone marrow-derived mesenchymal stromal cells (BM-MSCs) are being investigated for their potential in the treatment of musculoskeletal injuries, including tendon and ligament lesions, and cartilage lesions. Culture expansion of cells has traditionally been performed in medium supplemented with fetal bovine serum (FBS), however, concerns regarding the antigenicity and potential viral or prion contamination of FBS have prompted interest in alternative medium supplements. Platelet lysate (PL) contains elevated concentrations of growth factors, including transforming growth factor-β (TGF-β), platelet-derived growth factors, and fibroblast growth factor, released from the α-granules of platelets; therefore, PL could be an ideal medium supplement. The effect of PL on mesenchymal stromal cell (MSC) growth and differentiation has not been fully elucidated. We hypothesized that PL medium would contain significantly higher amounts of TGF-β1 than FBS medium and would be associated with enhanced osteogenic and chondrogenic differentiation. MSCs were isolated from bone marrow collected from five adult horses. Cells were cultured in traditional medium supplemented with FBS or in medium supplemented with fibrinogen depleted-PL (FD-PL). Immunophenotyping was performed using flow cytometry. Trilineage differentiation was assessed through histology and gene expression analysis using quantitative reverse transcription–polymerase chain reaction. TGF-β1 was quantified in both medium types. The immunophenotypes of BM-MSCs cultured in FBS and FD-PL medium were similar with both culture types containing cells positive for stromal cell markers [cluster of differentiation 29 (CD29), CD44, CD90, CD105, and major histocompatibility complex I (MHCI)] and negative for exclusion markers (CD45, CD79α, and MHCII). Despite significantly higher TGF-β1 concentration in FD-PL medium, chondrogenic and osteogenic differentiation were not significantly different between FBS and FD-PL supplemented cultures. PL is an appropriate alternative medium supplement for the culture of equine BM-MSCs up to passage 3. However, despite increased TGF-β1 concentration in FD-PL medium, significant changes in chondrogenic differentiation compared with FBS medium should not be expected.
Introduction
Bone marrow-derived mesenchymal stromal cells (BM-MSCs) are self-renewing progenitor cells that are found throughout different niches in the body [1]. Their use in the treatment of a variety of musculoskeletal injuries has been intensely investigated as mesenchymal stromal cells (MSCs) have the potential to differentiate into osteoblasts, chondrocytes, tenocytes and other stromal cells [1,2]. Additionally, MSCs have potent immunomodulatory properties which can coordinate and optimize healing in injured tissues [3]. Bone marrow (BM) is a common source of MSCs with culture expansion required after aspiration to provide sufficient cell numbers for clinical use.
Fetal bovine serum (FBS) has traditionally been used to supplement stromal cell culture medium due to a high concentration of nutrients and growth factors [4,5]. However, supplementation with FBS likely increases the immunogenic potential of cultured cells due to its xenogenicity [6]. Spees et al. reported the presence of retained animal proteins within the cytoplasm of human MSCs following culture of stromal cells in FBS [7]. The presence of intracytoplasmic xenogeneic proteins have also been confirmed following routine culture of equine BM-MSCs [6]. The presence of these xenogeneic proteins has been implicated in several adverse reactions that have been reported following administration of MSCs [6 –10]. Although the xenogenic potential of FBS has not been fully elucidated in the horse, recently presented data suggest that FBS-cultured MSCs, compared with xenogen-free MSCs, are recognized and targeted for cell death by recipient horses [11]. Potential microbial, viral and prion contaminants of FBS or allogeneic blood products present a further concern [12,13]. An alternative, autologous source of growth factors would therefore be ideal for MSC culture.
Platelet-rich plasma (PRP) is a whole blood derivative, processed to achieve an increased concentration of platelets and a reduction in other blood constituents [14 –16]. Alpha granules in platelets contain high concentrations of growth factors, including transforming growth factor-β (TGF-β), platelet-derived growth factors (PDGF), and vascular endothelial growth factor (VEGF), all of which play an important role in cell differentiation and tissue healing [17 –20]. Platelet lysate (PL), generated from PRP, can be used to supplement media in cell culture and has been demonstrated to potentiate MSC proliferation and viability [21], with significantly increased levels of PDGF-BB found in PL-supplemented equine umbilical blood MSC cultures [22]. Although allogeneic pooled PL has been most commonly investigated in human cell culture, autologous PL eliminates the concern of viral or prion contamination of pooled products [23].
Although PL could provide an autologous alternative to FBS, the effects of fibrinogen-depleted PL (FD-PL) on equine BM-MSC differentiation have not been presented. PL contains increased concentrations of TGF-β1, a growth factor that has been associated with both osteogenesis and chondrogenesis. The TGF-β superfamily, which includes the bone morphogenetic proteins (BMPs), are known to play an important role in osteoblast differentiation and bone formation with TGF-β1 and TGF-β2 increasing chemotaxis and mitogenesis of osteoblast precursors [24]. Additionally, studies have demonstrated increased osteogenesis of MSCs in response to TGF-β in culture [25] and increased bone healing following application of PRP to cancellous bone grafts used in human mandibular defects [26]. TGF-β1 is also involved in growth and maintenance of articular cartilage [27] and several studies have found that TGF-β1 enhances chondrogenic differentiation of stromal cells [28,29]. The effect of TGF-β1 on MSC differentiation may be variable depending on species, cell source, and other culture conditions, and requires further investigation as the need to improve in vitro chondrogenesis for cartilage repair remains vital.
The effects of PL on BM-MSC differentiation is an important consideration when culturing cells for in vivo application. An additional consideration surrounding the effects of PL on BM-MSC differentiation includes combined intralesional injection of these products for soft tissue and chondral injuries. Interestingly, there have been some anecdotal reports of tendon and ligament calcification following intralesional injection of platelet-based therapeutics in equine patients. Furthermore, repair of full-thickness chondral defects with platelet-enriched fibrin + BM-MSCs appeared to stimulate bony proliferation in an experimental equine model [30].
Our objectives were to compare the viability and proliferative capacity of equine BM-MSCs cultured in 10% autologous fibrinogen-depleted medium (FD-PL) and standard complete BM medium containing 10% FBS (FBS). Additionally, we aimed to compare the trilineage differentiation potential of BM-MSCs expanded in FD-PL medium versus FBS medium. We hypothesized that there would be no significant difference between the proliferative or viability characteristics of equine BM-MSC cultured in FD-PL medium versus those cultured in FBS medium but that osteogenic and chondrogenic differentiation would be enhanced by FD-PL medium. Finally, we hypothesize that FD-PL medium would contain significantly higher amounts of TGF-β1.
Materials and Methods
Animals
Blood and BM samples were obtained from five, systemically healthy horses between the ages of 2–6 years. This study was approved and performed according to guidelines of the Institutional Animal Care and Use Committee of The University of Pennsylvania.
Preparation of PL
Blood (900 mL) was collected into citrate phosphate dextrose adenine collection bags from the jugular vein of each horses following sterile preparation. Within 2 h of collection, the blood was transferred to 50-mL conical tubes and PL was obtained as previously described [31]. Briefly, blood was centrifuged at 200 g for 15 min before plasma was collected and transferred to new tubes (Sorvall ST16 Benchtop Centrifuge; Thermo Fisher Scientific, Waltham, MA). Plasma was then centrifuged at 400 g for 15 min following which platelet-poor plasma (PPP) was removed leaving ∼1 mL of PRP. The PPP was then used to resuspend PRP to a concentration of 1 × 106 platelets/μL. Centrifuge acceleration and deceleration were set to 0 for all centrifugation steps. PL was generated using a single freeze/thaw cycle at −80°C overnight followed by thawing at 37°C. PL was then centrifuged at 4,000 g for 15 min and the supernatant collected and stored at −20°C.
BM collection and MSC culture
BM was collected aseptically from the sternebrae of horses being euthanized for unrelated reasons immediately following euthanasia. Using an 11-gauge Jamshidi BM biopsy needle (VWR Scientific, Bridgeport, NJ) and 60-mL syringe containing 10,000 U of heparin, 30 mL of BM was aspirated. BM samples were seeded into flasks containing medium consisting of Dulbecco's modified Eagle's medium with 1 g/L of
Fibrinogen depletion of PL medium
Mechanical fibrinogen depletion was performed by intentional hydrogel formation following addition of PL to medium as previously described [32]. Briefly, once the medium and PL were combined, they were incubated at room temperature for 4 h followed by overnight incubation at 4°C. The medium was then incubated for 1 h at 37°C to allow complete fibrin clotting and then the tube was shaken vigorously to disrupt the clot that had formed, and the tube centrifuged at 3,000 g for 10 min. The supernatant was then filtered through a 0.22 μm filter.
Proliferation and viability assay
P2 cells were seeded into six-well tissue culture plates at a density of 3,000 cells/cm2. For the first 48-h all wells contained FBS medium to ensure plastic adherence. Thereafter, two types of culture medium were used: (1) FBS medium consisting of MSC basal medium as described above with 10% FBS or (2) FD-PL medium consisting of MSC basal medium with 10% PL. At 24, 48, 72, and 96 h thereafter, cells were detached using 0.25% trypsin-EDTA dissociation reagent. Cell number and viability was determined using the Cellometer™ Auto 2000 Cell Viability Counter and ViaStain AOPI staining solution. All assays were performed in triplicate.
Immunophenotyping
Flow cytometric analysis of P2 cells cultured in FBS or FD-PL medium was performed to evaluate the immunophenotype of cells. Before flow cytometry, cells were collected using Accutase® Cell Detachment Solution (Innovative Cell Technologies, Inc., San Diego, CA) to preserve cell surface markers [33]. Cells (1 × 105) were placed in 96-well round bottom plates and washed twice with PBS and then resuspended in 100 μL of PBS with 0.5% bovine serum albumin (Sigma Aldrich, St. Louis, MO) and 0.02% sodium azide (Thermo Fisher Scientific, Waltham, MA) and incubated at 4°C for 20 min. Cells were then incubated with 50 μL of the appropriate primary antibody at 4°C for 45 min, rinsed twice with PBS, then resuspended in the secondary antibody (50 μL) when appropriate, and incubated at 4°C for 45 min. After the final PBS rinse, the pellets were resuspended in 200 μL of PBS containing 7-Aminoactinomycin D (7-AAD; Thermo Fisher scientific, Waltham, MA). Cells were stained with anti-CD29, CD44, CD90, CD105, CD45, CD-79α, MHCI, and MHCII antibodies and isotype controls were used to establish fluorescent gates (Table 1). Flow cytometry and subsequent analysis was performed using the Cytoflex S Benchtop Flow Cytometer and CytExpert Software, version 1.0 (Beckman Coulter, Inc., Brea, CA).
Antibodies Used for Flow Cytometry Analysis of Equine Cell Surface Markers
Gifts from Dr. Doug Antczak, Cornell University, Ithaca, NY.
CD, cluster of differentiation; MHC, major histocompatibility complex.
Trilineage differentiation
Trilineage differentiation assays were performed using adipogenic, osteogenic or chondrogenic medium containing either FBS or PL. PL medium underwent the fibrinogen depletion process for all media types. For adipogenic differentiation, cells were resuspended in FBS or FD-PL growth media and seeded in to flat-bottomed 12 well plates at a cell-density of 5,100 cells/cm2. Growth medium was substituted by adipogenic induction medium 48 h later, this contained basal medium supplemented with biotin (8 μg/mL; Sigma-Aldrich), calcium pantothenate (4 μg/mL; Sigma-Aldrich), insulin (5.8 μg/mL; Sigma-Aldrich), dexamethasone (4 μg/mL), isobutylmethylxanthine (0.1 mg/mL; Sigma-Aldrich), rosiglitazone (0.0178 mg/mL; Sigma-Aldrich), and 5% rabbit serum (Thermo Fisher Scientific, Waltham, MA). Either 3% PL or 3% FBS was added to the medium depending on the treatment group. Medium was changed every 48 h. After 6 days in induction medium, the medium was changed to adipogenic maintenance medium using the same reagents as above without rosiglitazone or isobutylmethylxanthine. For each horse, an undifferentiated control was maintained in standard basal medium containing 10% FBS. Following 14 days of culture, cells were rinsed with PBS and fixed with 10% formalin before staining with Oil Red O (Sigma-Aldrich) for confirmation of lipid droplet accumulation in the cytoplasm of cells.
Osteogenic differentiation was performed with cells seeded into 12-well culture plates at a seeding density of 2,900 cells/cm2. Following 48 h of culture in FBS or FD-PL basal medium, osteogenic differentiation medium consisting of FBS or PL basal differentiation medium supplemented with β-glycerophosphate (2.2 μg/mL; Sigma Aldrich), dexamethasone (8 μg/mL; Sigma-Aldrich), and 2-phospho-
Chondrogenic differentiation was carried out in pellet cultures. MSC pellets were formed through centrifugation of 500,000 cells in 15-mL conical tubes at 400 g for 10 min. Pellets were maintained in FBS or FD-PL basal medium for 48 h and then chondrogenesis was induced with chondrogenic medium containing basal medium supplemented with TGF-β3 (0.01 μg/mL; Thermo Fisher Scientific, Waltham, MA), dexamethasone (0.4 μg/mL), 2-phospho-
Gene expression analysis
Following 14 days of osteogenic and adipogenic differentiation, cells were lysed and RNA was isolated using the Qiagen RNeasy Mini Kit (Qiagen, Germantown, MD). RNA was isolated from chondrogenically differentiated pellets after 28 days first through biopulverization in liquid nitrogen using a multisample stainless steel biopulverizer (BioSpec Products, Inc., Bartlesville, OK). RNA isolation was then completed using the Qiagen Fibrous Tissue Mini Kit (Qiagen). RNA concentration and purity were quantified using a UV microspectrophotometer (NanoDrop™ One; Thermo Fisher Scientific, Waltham, MA). Complementary DNA was prepared using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA) and an Eppendorf master cycler (Hamburg, Germany). Real-time quantitative polymerase chain reaction (PCR) was performed using TaqMan™ Master mix and the Applied Biosystromals™ QuantStudio™ 6 Flex Real-Time PCR Systromal (Applied Biosystromal, Foster City, CA). Primers and probes were designed using NCBI Primer-BLAST and Integrated DNA Technologies (IDT) PrimerQuest Tool software and synthesized by IDT (Coralville, IA) (Table 2). The following genes were analyzed: peroxisome proliferator-activated receptor (PPARγ) for adipogenesis; alkaline phosphatase (ALP) for osteogenesis; and aggrecan (ACAN), collagen type II (COL2b) and sex-determining region Y-box 9 (SOX9) for chondrogenesis. All samples were run in triplicate using 18S as a reference gene. Data were quantified using ΔΔCt comparisons.
Equine Primer and Probe Sequences Used for Gene Expression Analyses
Pellet glycosaminoglycan content
After 28 days of culture, pellets were collected and stored at −20°C in medium before analysis. The dimethylmethylene blue (DMMB) spectrophotometric assay (Sigma-Aldrich) was used to quantify glycosaminoglycan (GAG) content in the pellets following digestion in 0.5 mg/mL papain (Sigma-Aldrich) for 4 h at 65°C. Chondroitin-4 sulfate (Sigma-Aldrich) was used to establish a standard curve and the optical density determined at 525 nm [34].
Media TGF-β1 analysis
The concentration of TGF-β1 in FBS and FD-PL was quantified using a fluorescent bead-based multiplex assay (Luminex, Austin, TX) using an anti-human TGF-β1 antibody (EMD Millipore, Burlington, MA). Antibody specificity and crossreactivity with the horse was validated by EMD Millipore through western blot using single recombinant proteins. No or negligible crossreactivity was present with TGF-β2 or TGF-β3.
Statistical analysis
All data were analyzed using JMP14 (SAS, Cary, NC). A mixed effects model was used to analyze all continuous data, including cell proliferation and doubling time, cell surface marker expression, fold change gene expression, and GAG content. Cell proliferation, doubling time, cell surface marker expression, and GAG content are expressed as the mean ± standard error of the mean. Relative expression is expressed using a box and whisker plot with median (line), upper and lower quartiles (box), and 5% and 95% percentiles (whiskers). Horse was considered as a random effect. Significance was set at P < 0.05.
Results
Proliferation and viability
Proliferation and viability of P2 BM-MSCs were measured over a 96-h period. The rate of cell proliferation was similar in both the FBS treatment group and the FD-PL treatment group (Fig. 1). Cell viability was high in both treatment groups at all times. In the FBS treatment group cell viability remained >91%, while cell viability remained >93% in the FD-PL treatment group (Fig. 1).
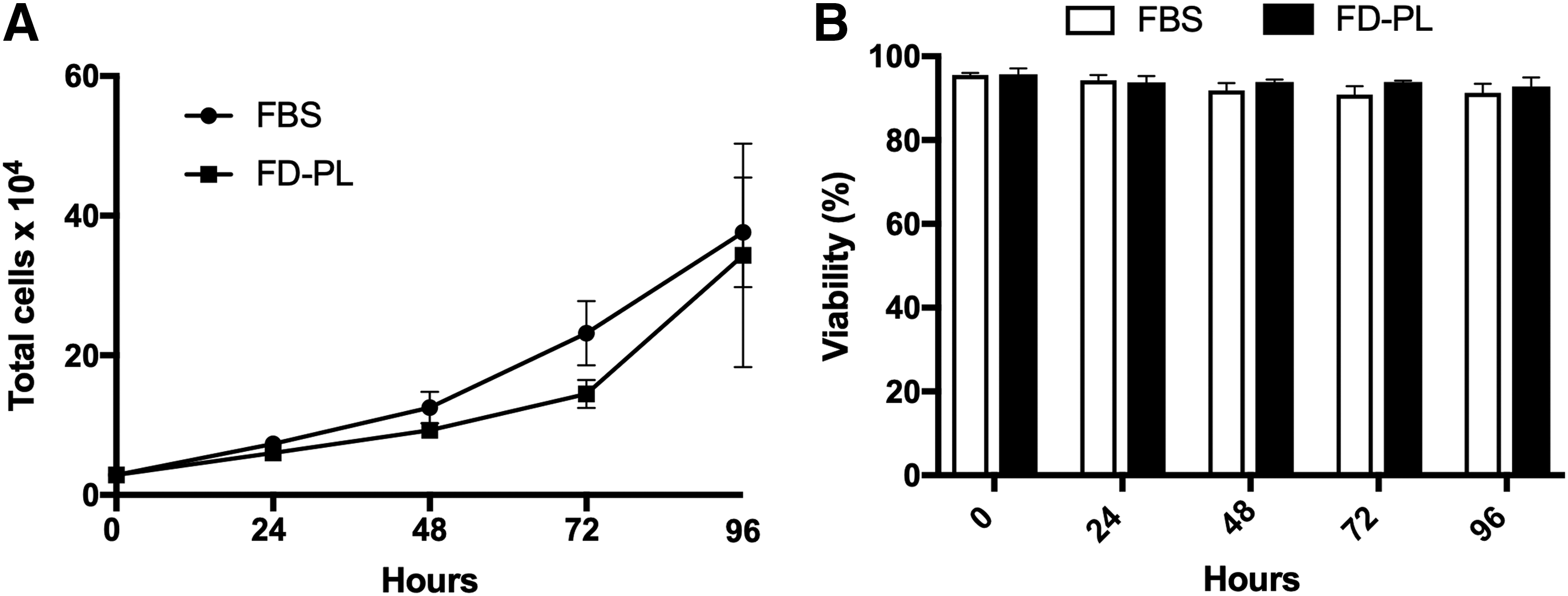
Immunophenotypic analysis
Flow cytometry was used to characterize the expression of cell surface markers on BM-MSCs cultured in FBS compared with BM-MSCs cultured in FD-PL medium. Cell surface markers included CD29, CD44, CD45RB, CD79α, CD90, CD105, MHCI and MHCII (Fig. 2). There were no statistically significant differences in expression of cell surface markers between BM-MSCs cultured in FBS or FD-PL. Both culture types contained cells that were strongly positive for the inclusion markers, CD29 (79.18% ± 0.05% FBS; 74.96% ± 0.09% FD-PL), CD44 (95.14% ± 0.02% FBS; 96.08% ± 0.01%), CD90 (87.14% ± 0.03% FBS; 89.56% ± 0.01% FD-PL), CD105 (82.52% ± 0.07% FBS; 88.58% ± 0.01% FD-PL), and MHCI (71.55% ± 0.05% FBS; 85.13% ± 0.02%). Minimal expression of exclusion markers was detected in both culture types, including CD45RB (0.60% ± 0.01% FBS; 0.97% ± 0.01% FD-PL), CD79α (0.62% ± 0.01% FBS; 1.23% ± 0.01% FD-PL), and MHCII (3.51% ± 0.02% FBS; 4.38% ± 0.02% FD-PL).
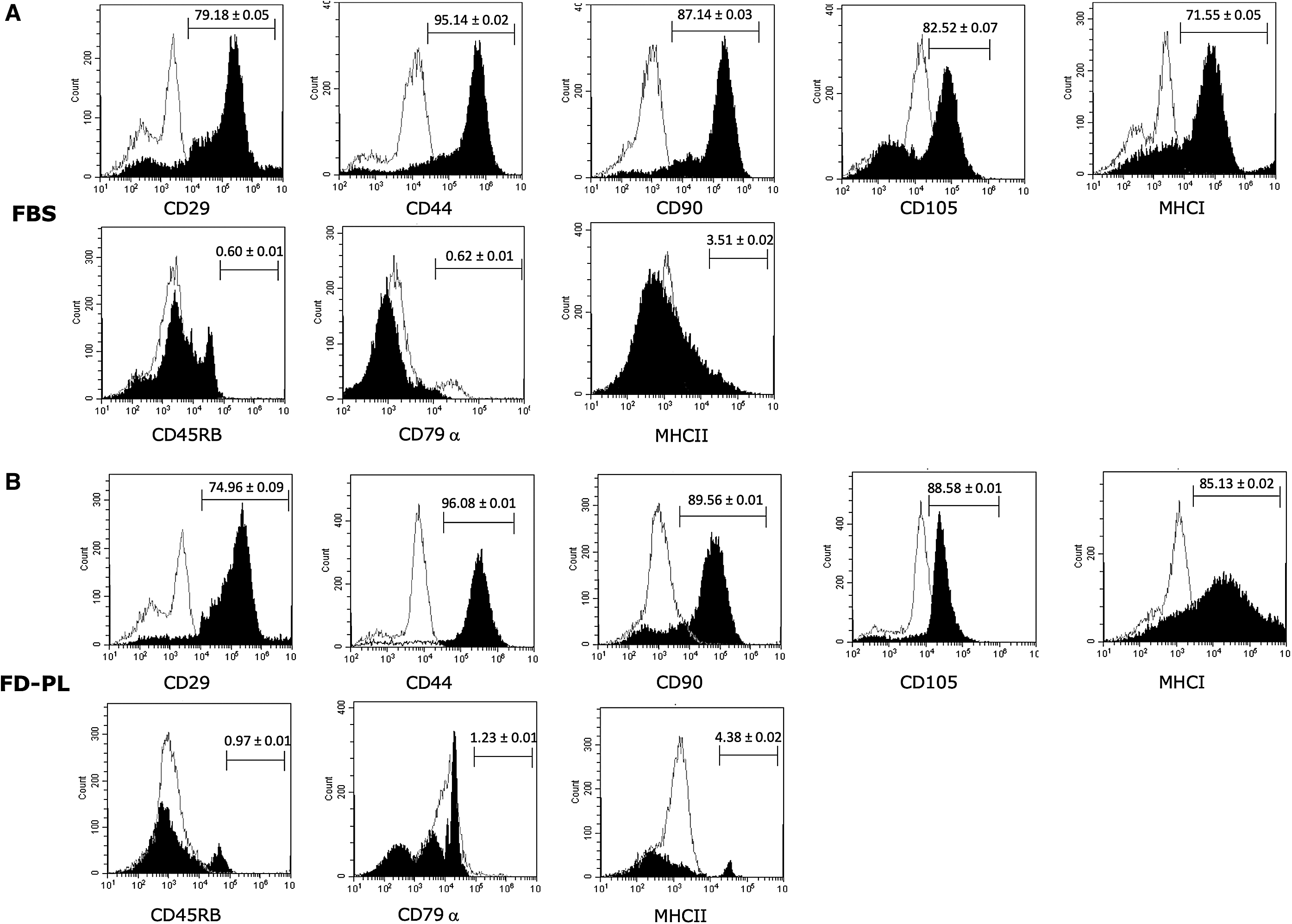
Flow cytometry histogram analyses of cell surface marker expression in P2 BM-MSCs cultured in
Trilineage differentiation
Trilineage differentiation assays were performed using noninductive complete BM-MSC medium (control), inductive medium with FBS (FBS), and inductive medium with FD-PL (FD-PL). Osteogenic and adipogenic differentiation assays were performed in monolayer cultures, while the chondrogenic differentiation assay was performed in three-dimensional pellet culture to allow for extracellular matrix (ECM) formation.
Cells cultured in both FBS and FD-PL osteogenic medium underwent osteogenic differentiation compared with control medium. Specifically, BM-MSCs cultured in either FBS or FD-PL osteogenic medium had increased calcium deposition noted following staining with Alizarin Red, whereas control cultures had no calcium present. Expression of ALP was significantly increased in both induced FBS and FD-PL cultures compared with control cultures (Fig. 3).

Comparison of osteogenic and adipogenic differentiation of BM-MSCs cultured in FBS medium or FD-PL medium.
Similar to the osteogenic differentiation assay, BM-MSCs cultured in both FBS and FD-PL adipogenic medium underwent adipogenic differentiation compared with control medium. BM-MSCs cultured in either FBS or FD-PL adipogenic medium demonstrated lipid droplet accumulation by positive staining with Oil Red O. Expression of PPARγ was increased in both FBS and FD-PL cultures compared with control cultures, although this did not reach statistical significance (Fig. 3).
Chondrogenesis, as demonstrated by Toluidine Blue staining of proteoglycan deposition in pellets, was noted in induced FBS and FD-PL cultures. No evidence of chondrogenesis was noted in control cultures. Proteoglycan deposition did not appear to differ between FBS and FD-PL cultures (Fig. 4). Expression of COL2b, ACAN, and SOX9 was upregulated in both the induced FBS and FD-PL cultures compared with the control cultures, however, no difference was detected between FBS and FD-PL cultures (Fig. 4). Finally, GAG content in pellet cultures as determined by the DMMB assay was increased in both FBS and FD-PL pellets compared with control, however, the increase was not statistically significant.

Chondrogenic differentiation of BM-MSCs cultured in control, FBS, and FD-PL medium.
Medium TGF-β1 concentration
The amount of TGF-β1 was significantly more in FD-PL (22.32 ± 0.23 ng/mL) compared with FBS (9.71 ± 0.23 ng/mL) (P < 0.0096) (Fig. 5).
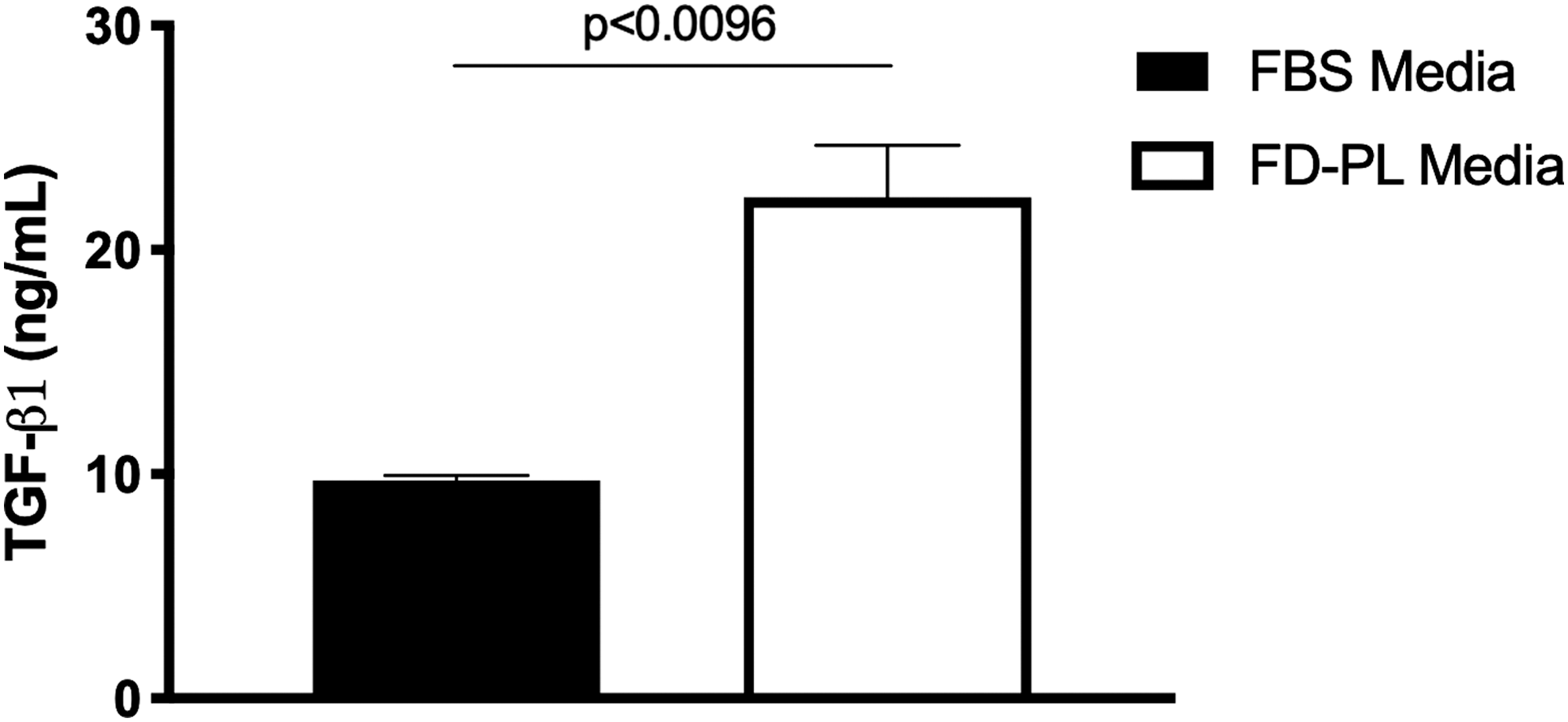
Mean ± SEM TGF-β1 concentration (ng/mL) in FBS and FD-PL (n = 5). TGF-β, transforming growth factor-β.
Discussion
In this study, we found that autologous fibrinogen-depleted PL can be successfully used to culture equine BM-MSCs up to passage 3 without significant changes in proliferation rates, viability, or immunophenotype. Despite significantly more TGF-β1 in PL-supplemented media, no significant differences were noted in chondrogenic or osteogenic differentiation. We specifically aimed to investigate the effects of FD-PL on BM-MSC differentiation due to increased TGF-β1 concentration in PL, which has been associated with both chondrogenesis and osteogenesis. Although the TGF-β superfamily of growth factors play a key role in bone development in vivo [24], conflicting reports have demonstrated that TGF-β1 can promote both osteogenesis and chondrogenesis of MSCs in vitro [25,28,29]. With the expansion of the clinical use of MSCs and the emergence of PL-supplemented media, we sought to further evaluate the effects on in vitro trilineage differentiation as this may significantly affect the behavior of cells used in vivo.
We found that BM-MSCs cultured in FD-PL had similar proliferation rates and viability over a 96-h culture period. Culture expansion of MSCs is necessary to achieve an adequate number of cells for in vivo clinical application of cells in musculoskeletal injuries, therefore, it is important to determine the effects of different culture techniques on cell proliferation and viability. Several other studies have demonstrated similar or increased proliferation rates for MSCs cultured in PL compared with FBS [35 –37]. For example, Naskou et al. found that equine MSCs cultured in pooled PL produced through plateletpheresis had a similar proliferation rate and viability to MSCs cultured in FBS, although overall viability rates were lower in that study (FBS = 75% and PL = 84%) compared with the viability in our study [35]. The use of PL has also been described for the culture of equine umbilical cord-derived MSCs, however, this study found decreased proliferation rates in PL-supplemented cultures [22], suggesting that culture conditions may have variable effects on cells from different sources.
It has been suggested that the concentration of the growth factors found in PL, including TGF-β1, VEGF, fibroblast growth factor, and PDGF, support chondrogenesis of MSCs [12,32]. Enhanced chondrogenesis of MSCs would be of great benefit in cell-based cartilage repair since complete chondrogenic differentiation remains elusive and could be affected by alternative culture methods [38,39]. Few previous studies have evaluated the effect of equine PL on chondrogenic differentiation of BM-MSCs cultured in three-dimensional cultures. Naskou et al. showed enhanced chondrogenesis in micropellets (100,000 cells/pellet) cultured in pooled PL with increased proteoglycan production noted following Alcian Blue staining [35]. Additionally, few studies have investigated the effects of equine PL on gene expression associated with chondrogenesis. TGF-β1 has been associated with increased synthesis of ECM proteins, including ACAN and collagen type II b [28]. Although the FD-PL in the study reported here had significantly increased concentrations of TGF-β1, we did not find increased expression of chondrogenic genes, including SOX9, ACAN, or COL2b, when compared with FBS-supplemented cultures. Additionally, we did not find significantly increased GAG content in FD-PL pellets compared with FBS pellets. Despite the fibrinogen depletion step in the study reported here, the concentration of TGF-β1 was comparable to previously reported concentrations [22].
In the study reported here, TGF-β3 was added to all chondrogenic differentiation media as per our standard laboratory protocol, whether the media were supplemented with FBS or PL. We routinely add TGF-β3 to our chondrogenic differentiation medium as we have achieved consistently better differentiation results when compared with TGF-β1 (unpublished data). TGF-β3 has been shown as an effective driver of MSC chondrogenesis in several other studies, with superior type II collagen expression and deposition [40 –42]. Of the three TGF-β isoforms, PRP contains large quantities of TGF-β1 and small quantities of TGF-β2, therefore, the majority of the focus on the impact of PL on MSC differentiation has been placed on TGF-β1 [43]. Further studies could aim to directly compare TGF-β1 and TGF-β3 PL-supplemented differentiation.
The effects of PL on the osteogenic differentiation of equine MSCs is also intriguing due to the common clinical practice of using both biologics in combination in equine medicine. Goodrich et al. reported bone formation in the repair tissue of full-thickness chondral defects created on the lateral trochlear ridges of the femur in 4/12 horses [30]. Interestingly, this effect has not been reported in other species with a recent study demonstrating improved cartilage repair in rabbit osteochondral defects filled with BM-MSCs in a PRP scaffold compared with BM-MSCs alone [44]. Clinically, there are several anecdotal reports of mineralization of equine soft tissue injuries following injection of PRP and PRP + MSCs, although these findings have not be reported in experimental equine models of tendonitis [45,46]. Moreover, mineralization or calcification has not been reported as a side effect of PRP injection for human soft tissue pathology [47]. In the study reported here, we did not find evidence of increased osteogenesis in MSCs cultured in FD-PL, although further in vivo assessment is indicated.
Mechanical fibrinogen depletion was initiated in this study due to issues with initial clot formation in cultures being supplemented with PL medium. This was particularly apparent in chondrogenic differentiation assays as the pellets would become encased in the clotted material. Following institution of fibrinogen depletion, cells attached normally to culture plates and pellets were not encased in clotted material. Although porcine-derived heparin is a conventional additive to PL medium to prevent clot formation, Laner-Plamberger et al. described a method of mechanical fibrinogen depletion of human allogeneic PL medium in an effort to eliminate xenogeneic substances from the culture media [32]. Additionally, fibrinogen depletion may be beneficial due to reported detrimental effects of fibrinogen on the immunomodulatory properties of MSCs [32]. Laner-Plamberger et al. found that their mechanical fibrinogen depletion protocol led to a 1,000-fold decrease in fibrinogen in PL [32]. In the study we report here, we did not quantify the reduction in fibrinogen following mechanical depletion, however, no issues with clot formation were noted when this protocol was initiated suggesting that fibrinogen was sufficiently depleted.
Pooled PL has been investigated as an alternative and, despite being allogeneic, does not appear to be associated with significant immunogenic reactions [35,48]. Although studies have demonstrated a potential additive effect of pooled PL when compared with single source PL [23,49] the risk of disease transmission through plasma-borne viruses continues to be a serious concern and requires viral inactivation and reduction steps, which can result in protein inactivation [50]. For this reason, we chose to investigate the effects of autologous PL in this study, however, the production of large volumes of autologous PL for large-scale MSC expansion will likely be a significant limitation.
One major limitation we encountered in this study was the need to culture expand BM-MSCs to P2 in traditional FBS medium because we were unable to produce enough PL to maintain all cultures in FD-PL medium throughout all expansion and differentiation assays. Another limitation in this study is that our undifferentiated control cultures were maintained in basal medium containing FBS only, as our main objective was to determine if the use of PL would increase differentiation compared with FBS in a standard differentiation assay. In future studies, inclusion of an undifferentiated control with PL supplementation in addition to FBS supplementation should be considered. Additionally, using allogeneic pooled PL, instead of autologous PL, would be a simple way of overcoming the lack of PL volume and would allow all cultures to be carried out in PL.
In conclusion, FD-PL should be considered as an appropriate alternative medium supplement for the culture of equine BM-MSCs. However, despite increased TGF-β1 concentration in FD-PL medium, significant changes in chondrogenic or osteogenic differentiation compared with FBS medium should not be expected with the current protocol. Further studies are required to assess the combinatorial in vivo application of PL + MSCs, although we did not find evidence to support PL-associated osteogenic differentiation of MSCs in this study.
Footnotes
Acknowledgments
The authors would like to thank Karie Durynski for her technical assistance with histology and Dr. Keith Russell for his assistance with the platelet lysate protocol.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Raymond Firestone Trust Research Grant at the University of Pennsylvania.