
Abstract
Stem cells derived from dental apical papilla (SCAPs) can secrete various soluble factors, which may stimulate tissue repair and regeneration in vivo. The aim of this study was to elucidate the effect of the soluble factors released by SCAPs on the proliferation and differentiation of dental pulp cells (DPCs). We compared the osteo/odontogenic, angiogenic, and neurogenic effects of soluble factors released from SCAPs and bone marrow mesenchymal stem cells (BMSCs) in vitro. Conditioned media (CM) were collected from human SCAPs and BMSCs cultures, and their effects on human DPCs proliferation and differentiation were evaluated. Cellular proliferation was unaffected by SCAPs-CM and was inhibited by BMSCs-CM. Cells treated with osteo/odontogenic inducing medium (OM) plus SCAPs-CM showed higher alkaline phosphatase activity than did cells in the OM group. The expression level of osteo/odontogenic markers were higher in the SCAPs-CM plus OM group than in the BMSCs-CM plus OM and OM groups. SCAPs-CM and BMSCs-CM significantly promoted DPCs migration. DPCs angiogenic differentiation was not affected by SCAPs-CM but was significantly enhanced by BMSCs-CM. In DPCs cultured in media optimized for neural stem cell growth for 2 weeks, the expression levels of neurogenic markers were significantly enhanced by the addition of SCAPs-CM. Neuronal markers expression was significantly reduced, while neurotrophic marker expression significantly increased by the addition of BMSCs-CM. In conclusion, SCAPs-CM significantly enhanced osteo/odontogenic differentiation, migration, and neurogenic differentiation potential of DPCs, but have no effect on DPCs proliferation and angiogenic differentiation in vitro. CM released from SCAPs have a greater osteo/odontogenic and neurogenic inductive effect on DPCs than BMSCs-CM. It indicates that SCAPs-CM can serve as additive to improve pulp tissue repair and regeneration.
Introduction
Dental pulp contains vasculature, nerves, and connective tissues to ensure the viability of teeth [1]. Lack of nourishment provided by the pulp tissue can increase the risk of tooth fracture. In immature teeth, pulpal necrosis terminates root development and results in poor long-term survival of teeth. In the procedure of pulp therapy, maintaining the viability of the residual pulp tissue as far as possible is the main consideration.
For exposed vital pulp, direct pulp capping and pulpotomy are traditional therapeutic approaches. However, the differentiation and proliferation of dental pulp cells (DPCs) might be influenced by the activity of dental materials after direct pulp capping and pulpotomy [2,3]. For necrosis of dental pulp, stem cell-based tissue engineering has been identified as a promising alternative for dentin-pulp regeneration. The restoration of the pulpal functional must been considered from a clinical perspective, including vascularized pulp that supports nourishment, newly differentiated odontoblast that can produce new dentin, and nerve that restores the sensation of occlusal pressure and pain [1,4].
DPCs are heterogeneous mixtures of progenitor mesenchymal stem cells (MSCs) and connective tissue cell populations. Dental pulp stem cells (DPSCs) are regarded as reliable seeding cells for pulp engineering. They are capable of differentiating into odontoblasts, nerve cells, and endothelial cells [5 –7], and have the capacity to form complete dental pulp tissue containing an odontoblast layer, blood vessels, and nerve cells [1,4]. The fate of DPSCs depends on intrinsic genetic programs regulated by surrounding growth factors, transcription factors, and extracellular matrix (ECM) proteins [8,9].
The microenvironment is an important factor in determining the behavior of cells. MSCs, growth factors and ECM in niche, and their multiple interactions determine the tooth development [10]. MSCs can secrete various soluble factors, including growth factors, cytokines, and ECM proteins, which contribute to their environment [11 –15].
Recently, it is reported that the differentiation of MSCs appears to be a rare event in vivo, and is only modestly enhanced, while paracrine activity plays a predominant role [12 –15]. In vitro, MSCs retain the capacity to regenerate unique microenvironments similar to those from which they are derived [5]. Conditioned medium (CM) released from bone marrow MSCs (BMSCs) could induce repair of tissue, including bone and nerves [16 –18]. In certain regenerative therapies using MSC, such as treatments for cerebral ischemia and myocardial infarction, the injection of MSC-CM alone could ameliorate the injury and MSC transplantation [14,15].
It has been known that dental pulp is evolved from dental papilla. As the root continues to develop after the bell stage, the location of the dental papilla becomes apical to the pulp tissue [4]. Stem cells derived from dental apical papilla (SCAPs) are found in the root apex of immature permanent teeth and contribute to the formation of dental pulp [4,19]. In a previous study, we profiled the secretome of human BMSCs and SCAPs in vitro [20]. SCAPs and BMSCs secreted large amounts of soluble factors involved in processes including odontogenesis, angiogenesis, neuron development, collagen organization, and immunity [20]. However, the secretome differs among cell sources. We hypothesized that the paracrine production of SCAPs may mimic the microenviroment of dentin-pulp formation to maintain the function of DPCs in vitro.
The aim of this study was to elucidate the role of SCAPs paracrine action on the function of DPCs. A previous study has reported that CM from BMSCs have osteo/ondontogenic inductive effect on DPCs [9]. To achieve insights into the function of secretions from BMSCs and SCAPs, in this study, we compared the osteo/odontogenic, angiogenic, and neurogenic effects of soluble factors released from SCAPs and BMSCs in vitro. CM was collected from human SCAPs and BMSCs cultures, and their effects on human DPCs proliferation and differentiation were determined.
Materials and Methods
Sample collection and cell culture
Normal human impacted third molars with immature roots (n = 5) and normal human bone marrow obtained from the mandibular bone (n = 5) were collected from healthy patients (aged 16–30 years). Sample collection was in accordance with the guidelines of the Ethical Committee of the health science center of Peking University (Beijing, China; IRB00001052-11060 and PKUSSIRB-201734036). Human SCAPs, DPCs, and BMSCs were isolated, cultured, and identified as previously described [4,5,9]. The cells used in experiments were of passages 3–5. SCAPs and BMSCs were characterized by flow cytometry with antibodies for CD73, CD90, CD105, CD146, and CD34 (BD Biosciences) as previously described [20].
Preparation of CM
SCAPs and BMSCs were seeded on 100 mm plates at a density of 20,000 cells/cm2. When MSCs reached 70% −80% confluency, they were washed twice with PBS and cultured in serum-free medium (minimum essential medium α [α-MEM] or Dulbecco's modified Eagle's medium/nutrient mixture F-12 [DMEM/F12]). CM in DMEM/F12 was collected for neurogenic differentiation of DPCs, and CM in a-MEM was collected for the others. Subsequently, after incubation period of 24, 48, or 72 h, CM were collected. The CM collected at 24 and 72 h were used only for mineralization of DPCs. Then MSCs-CM were centrifuged at 130 g for 10 min to remove the cellular debris, passed through a 0.22-μm filter, and stored at −80°C until use.
Cell proliferation assay
DPCs were seeded at a density of 1.0 × 103 cells/well into 96-well plates with α-MEM containing 10% fetal bovine serum (FBS). Twenty-four hours later, the cells were divided into four groups. The medium in the control group was replaced with fresh medium containing 10% FBS, and the medium of the treated group was replaced with fresh medium containing different concentrations of CM from MSCs (25%, 50%, or 100%) and 10% FBS. Cell counting kit 8 (CCK-8; Dojindo, Japan) solution was added to each well of the plate and the absorbance was measured at 450 nm, 1, 3, 5, and 7 days after seeding, according to the manufacturer's protocol.
DPCs were grown in three different culture media at a density of 1.0 × 104 cells/well in six-well plates: (1) α-MEM plus 10% FBS medium as a control, (2) SCAPs-CM plus 10% FBS, (3) BMSCs-CM plus 10% FBS. CM was not diluted in this experiment. Cells were counted 1, 3, 5, and 7 days after seeding.
Alkaline phosphatase activity assay and Alizarin red staining
DPCs were seeded at a density of 2.0 × 105 cells/well into six-well plates. When cells reached 90% confluency, they were cultured in different culture media. The osteo/odontogenic inducing medium (OM) was a-MEM supplemented with 10% FBS (Invitrogen), 10 mM β-glycerophosphate (Sigma-Aldrich), 50 mM ascorbate-2-phosphate (Sigma-Aldrich), and 100 nM dexamethasone (Sigma-Aldrich). OM plus SCAPs-CM was SCAP-CM supplemented with 10% FBS, 10 mM β-glycerophosphate, 50 mM ascorbate-2-phosphate, and 100 nM dexamethasone. The 50% SCAPs-CM was prepared by mixing a-MEM and SCAPs-CM in a 1:1 ratio. Alkaline phosphatase (ALP) activity assay was performed with an ALP kit according to the manufacturer's protocol (Nanjing Jiancheng Bioengineering Institute, China) and normalized based on protein concentrations.
For detecting mineralization, DPCs were cultured in different medium for 2 weeks, fixed with 70% ethanol, and stained with 2% Alizarin red (
After 2 weeks of culture in the SCAPs-OM, BMSCs-OM, OM, and control media, the total RNA of DPCs was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions.
Transwell migration assay
Tissue culture inserts (8 μm pore size; Corning) were seeded with DPCs at 1 × 105 cells/cm2 in α-MEM containing 0.1% FBS, and placed above the wells. Media in the wells beneath were of three types: (1) 0.1% FBS plus α-MEM medium as a control, (2) SCAPs-CM, and (3) BMSCs-CM. After 16 h of culturing, transmigrated cells were fixed with 4% paraformaldehyde (PFA) at room temperature and stained with 0.4% trypan blue. The total number of migrated cells was counted using an inverted phase contrast microscope.
Tube formation assay
DPCs were seeded at a density of 2.0 × 105 cells/well into six-well plates. When cells reached 90% confluency, they were cultured in different culture media: (1) 10% FBS plus α-MEM medium as a control, (2) angiogenic-inducing medium (AM), (3) AM plus SCAPs-CM (SCAPs-AM), and (4) AM plus BMSCs-CM (BMSCs-AM). The AM was a-MEM supplemented with 10% FBS (Invitrogen), 50 ng/mL vascular endothelial growth factor (VEGF; Peprotech), and 10 ng/mL basic fibroblast growth factor (bFGF; Invitrogen). After 7 days of culture, the total RNA of DPCs was extracted using TRIzol reagent for real-time reverse transcription–polymerase chain reaction (RT-PCR) and DPCs were digested for tube formation assay. In this study, 1.0 × 105 cells were seeded on Matrigel matrix-coated plate (Gibco) in a-MEM. After 15 h, the optical images of the wells were acquired. Tube-like structures in each well were counted, and the average number of tube-like structures in triplicate wells was determined.
Neurogenic differentiation induction and immunofluorescence staining
DPCs were seeded at a density of 1.0 × 106 cells/well into low-attachment six-well plates (Corning) in a medium optimized for neural stem cells. The medium was DMEM/F12 supplemented with 2% B27 (Invitrogen), 20 ng/mL bFGF, and 20 ng/mL epidermal growth factor (Invitrogen). After 6–9 days of culture, cells were fixed in 4% PFA, incubated in 0.1% (w/v) Triton X-100, and blocked with 1% bovine serum albumin. Samples were then incubated with primary antibody (1:100 Nestin, MAB1259, R&D systems; 1:800/1:100 β-III tubulin, ab18207, Abcam, United Kingdom) at 4°C overnight followed by secondary antibody (1:200 goat anti-mouse Alexa 488, and 1:200 goat anti-rabbit Alexa 488; Invitrogen) for 1 h at room temperature in the dark. After nuclear staining using DAPI, slices were washed and mounted in antifade mounting medium and examined by fluorescence microscopy. Isotype-matched control antibodies were used as a control.
DPCs were seeded at a density of 1.0 × 106 cells/well in different culture media: (1) 10% FBS plus a-MEM medium as a control, (2) media optimized for neural stem cells (UM), (3) UM plus SCAPs-CM (SCAPs-UM), and (4) UM plus BMSCs-CM (BMSCs-UM). After 14 days of incubation, the total RNA and protein of DPCs were extracted for real-time RT-PCR and western blot analysis.
Real-time RT-PCR
RNA was subjected to reverse transcription with oligo(dT) primers by using a reverse transcriptase kit (Promega). The quantitative RT-PCR (qRT-PCR) was carried out in triplicate in 96-well plates using a 7900HT Fast Real-Time system (Applied Biosystems) with SYBR green (Roche, China). The reaction conditions comprised 70°C for 5 min, 42°C for 60 min, and 95°C for 10 min. GAPDH was used as an internal control. The primer sequences are shown in Supplementary Table S1.
Western blot analysis
Thirty micrograms of protein were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene fluoride membrane, which was blocked with 5% (w/v) nonfat dried milk, incubated overnight at 4°C with primary antibodies (VEGFA, 46 kDa, 66828, Proteintech; CD31, 130 kDa, GB11063, Servicebio, China; nestin, 207 kDa, 33475, Cell Signaling Technology; neural cell adhesion molecule, NCAM, 95 kDa, 60238, Proteintech; glial fibrillary acidic protein, GFAP, 49 kDa, GB11096, Servicebio, China; GAPDH, 37 kDa, GB12002, Servicebio, China) and reacted with horseradish peroxidase-conjugated secondary antibodies (Origene, China). Immunoreactive bands were visualized by enhanced chemiluminescence (Cwbiotech, China) at room temperature and digitized using the Fusion FX image analyzer (Viber Loumat, Germany).
Statistical analysis
Statistical calculations were performed using IBM SPSS Statistics 25.0 (IBM, Inc.) statistical software. All experiments were repeated at least three times and data were expressed as mean ± standard deviation. One-way analysis of variance was used to assess the significance of differences between groups. A P value ≤0.05 was considered significant.
Results
Effects of CM on DPC proliferation
Cell proliferation was monitored for 7 days postseeding by using CCK-8 and cell count. The CCK-8 results showed no difference in cell proliferation rates in the SCAPs-CM and control groups (Fig. 1A). None of the SCAPs-CM concentrations (25%, 50%, and 100%) had an effect on DPCs proliferation (Fig. 1A). CM from BMSCs at a concentration of 25% and 50% had no significant effect on the proliferation of DPCs compared with the control condition (Fig. 1B). CM from BMSCs (100%) markedly inhibited proliferation compared with control group at 7 days in culture (Fig. 1B).
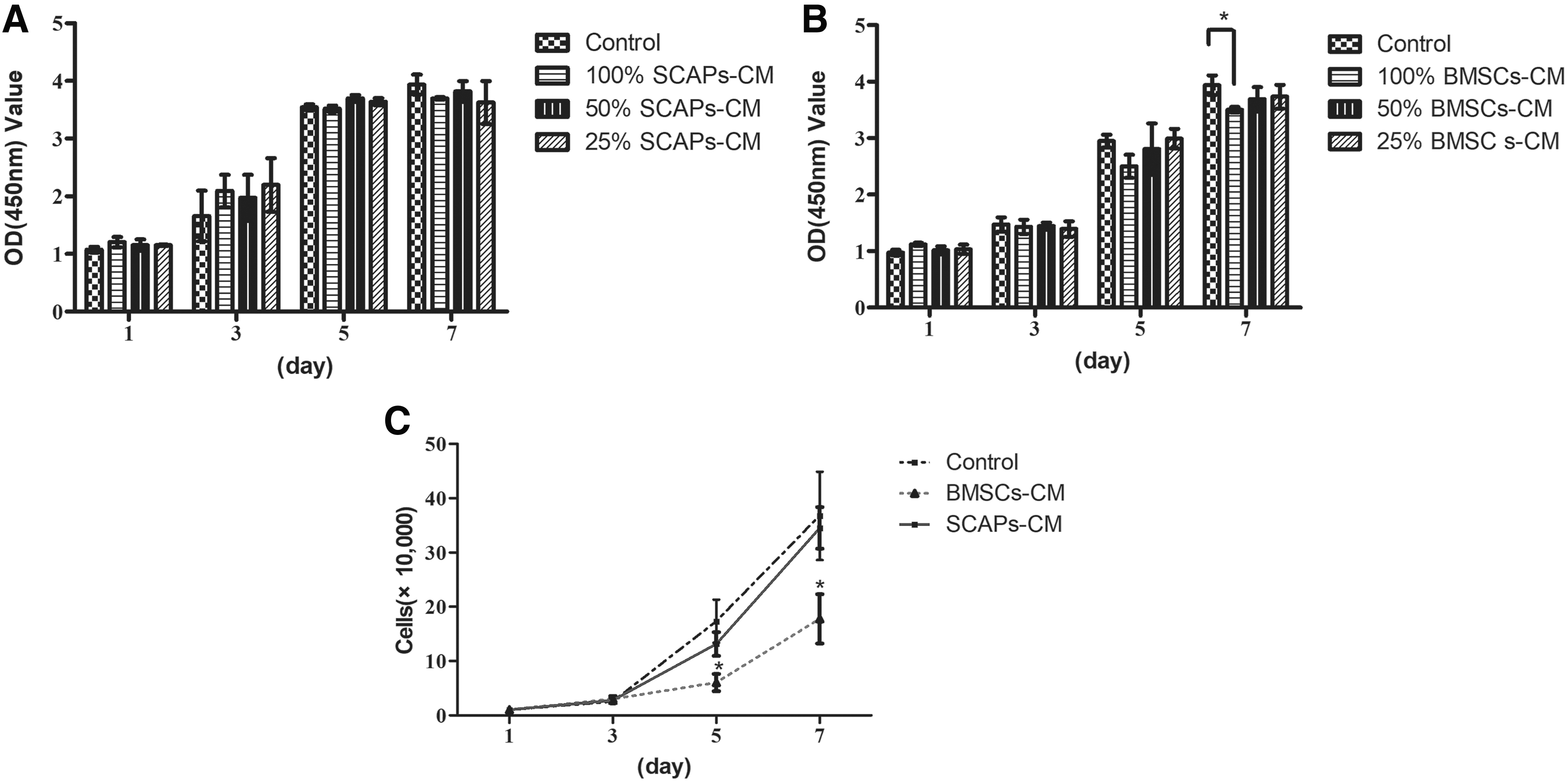
SCAPs-CM had no effect on DPCs proliferation, which was inhibited by BMSCs-CM.
Cell counts showed no statistically significant difference in cell numbers on days 1, 3, 5, and 7 in the SCAPs-CM and control groups. The BMSCs-CM group showed a lower proliferation rate than did the remaining groups on days 5 and 7 (Fig. 1C). Thus, SCAPs-CM had no effect on DPCs proliferation, which was inhibited by BMSCs-CM.
Effects of CM on DPCs osteo/odontogenic differentiation
ALP activity, an early marker for osteo/odontogenic differentiation, was more strongly induced in the SCAPs-OM (100%) group than in the OM group on days 3 and 5 (Fig. 2A). CM from SCAPs at a concentration of 100% enhanced DPCs ALP activity compared to a concentration 50% on days 3 and 5. There were no statistically significant difference among OM group, 50% and 25% SCAPs-OM groups on days 3. However, on day 5, ALP activity was significantly higher in the 50% and 25% SCAPs-OM groups than in OM. Thus, the optimal concentration for CM from SCAPs for the induction of osteo/odontogenic differentiation was 100%.
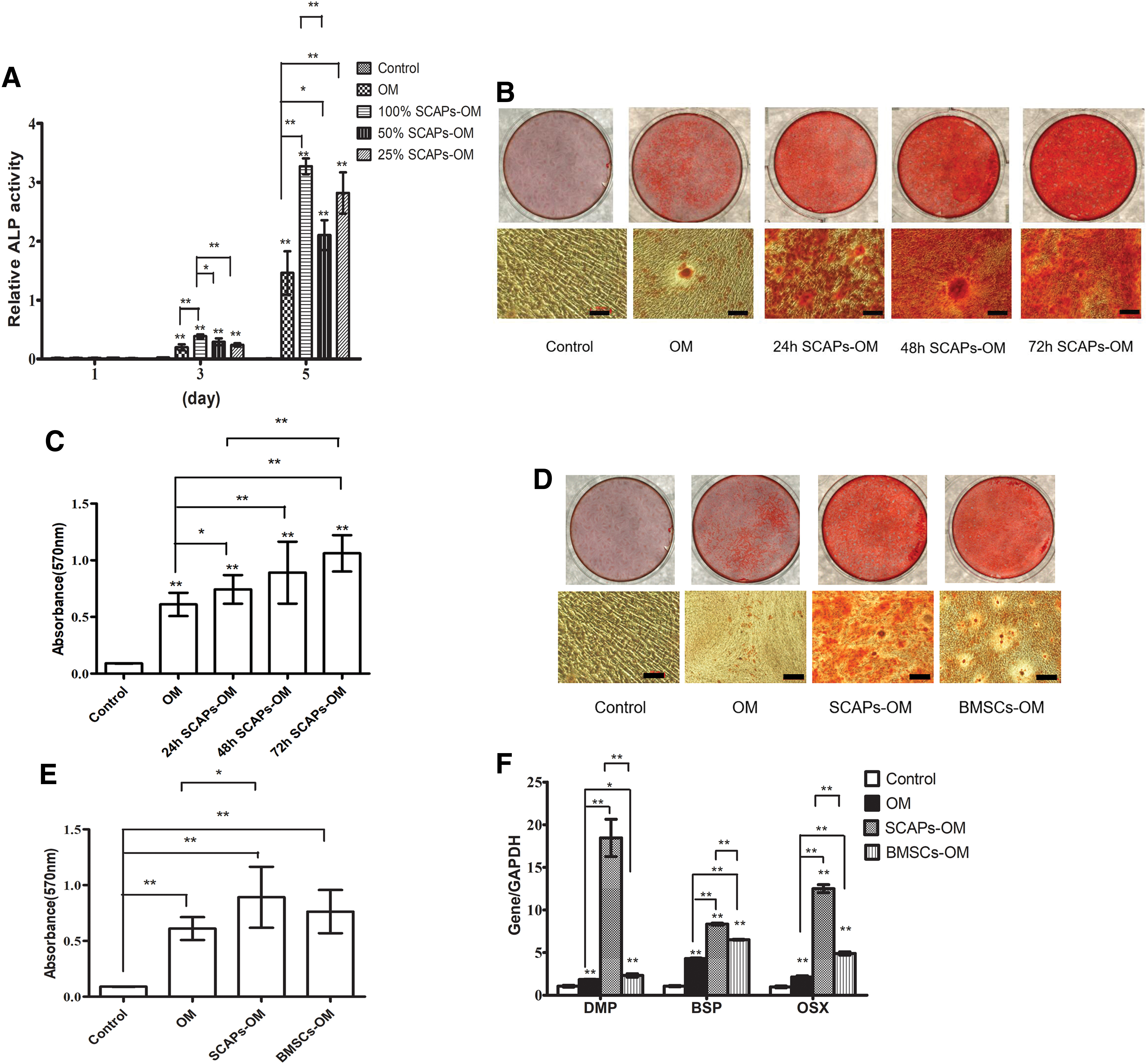
SCAPs-CM enhanced DPCs osteo/odontogenic differentiation.
Two weeks after culturing DPCs in different media, Alizarin red staining and calcium quantitation revealed that mineralization was significantly higher in the SCAPs-OM groups (24, 48, and 72 h) than in OM (Fig. 2B, C). The 24 h SCAPs-CM was collected after incubation period of 24 h in serum-free medium. More deposited mineralized matrix was observed in the 72 h SCAPs-OM group than in 24 h SCAPs-OM (Fig. 2B, C). There was no statistically significant difference between BMSCs-OM group and OM group with respect to the number of mineralized nodules (Fig. 2D, E). Thus, SCAPs-CM enhanced the mineralization of DPCs.
We examined the expression of dental dentin matrix protein (DMP), bone sialoprotein (BSP), and osterix (OSX), which are key transcription factors in osteo/odontogenic differentiation. RT-PCR showed stronger DMP, BSP, and OSX expression in SCAPs-OM and BMSCs-OM than in OM at 2 weeks (Fig. 2F). The RNA level of DMP, BSP, and OSX were higher in SCAPs-OM than in BMSCs-OM. SCAPs-OM had a significantly stronger effect on DPCs osteo/odontogenic differentiation than did BMSCs-OM.
Effects of CM on DPCs angiogenic differentiation
The chemotactic potential of DPCs was evaluated using a transwell migration system. SCAPs-CM and BMSCs-CM caused a significant increase in DPCs migration (Fig. 3A, B). Furthermore, SCAPs-CM group showed a stronger effect on DPCs migration than BMSCs-CM group.
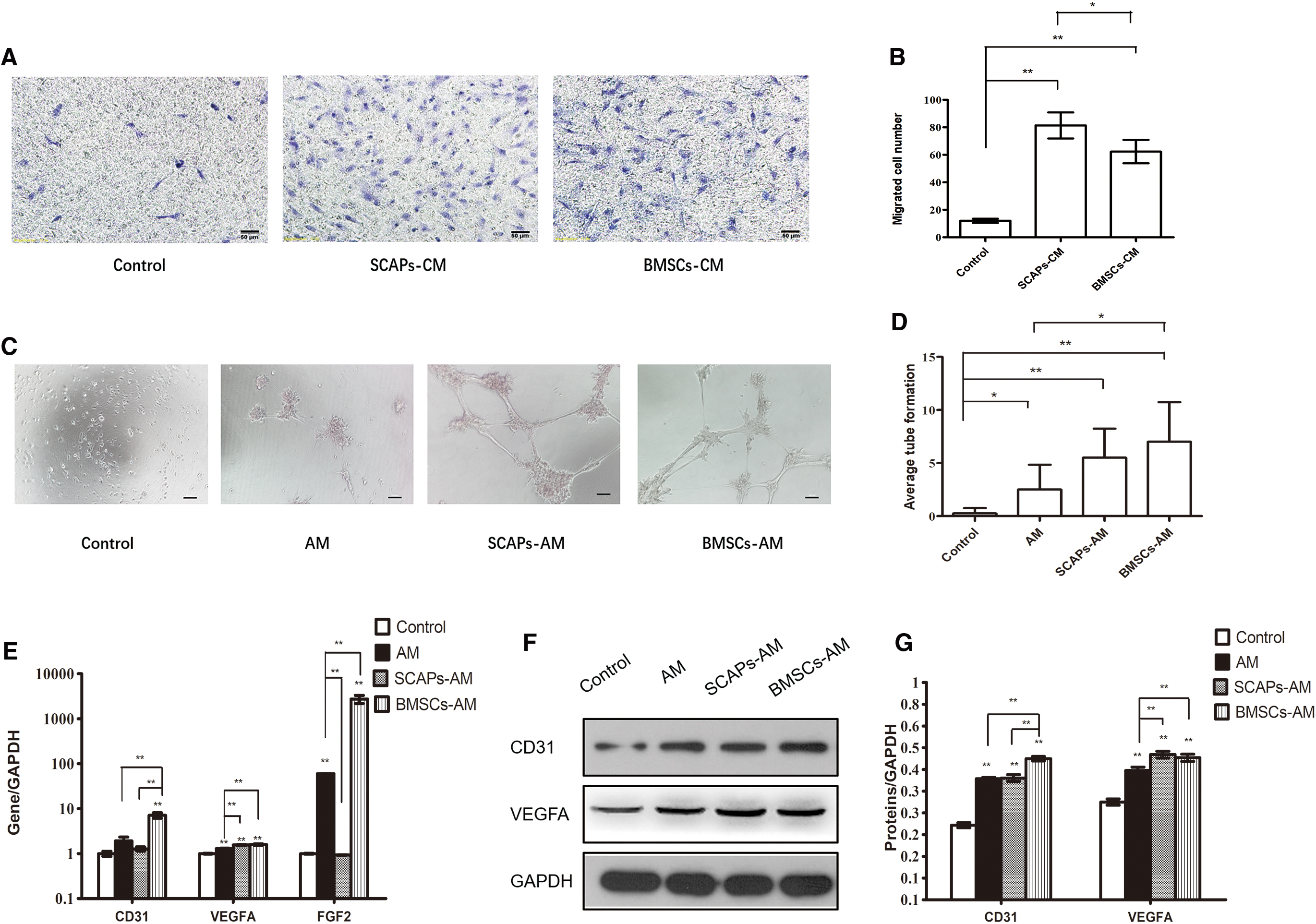
SCAPs-CM did not have any apparent effect on angiogenesis while BMSCs-CM had a pronounced effect.
We measured the angiogenic differentiation of DPCs cultured with SCAPs-CM or BMSCs-CM. After 7 days of culture, the BMSCs-AM group showed a pronounced increase in tubulogenesis compared to the AM group (Fig. 3C, D). The SCAPs-AM group showed no notable increase in DPCs tube formation (Fig. 3C, D).
Furthermore, at mRNA level, the angiogenic markers (CD31, VEGFA, and FGF2) expression was significantly higher in the BMSCs-AM group than in the AM group (Fig. 3E). VEGFA level was significantly higher and FGF2 level was significantly lower in SCAPs-AM group than in the AM group by the RT-PCR (Fig. 3E). There was no difference between SCAPs-AM group and AM group in the RNA level of CD31 (Fig. 3E). The analysis performed by western blot showed that protein (CD31 and VEGFA) and mRNA changed toward the same trend (Fig. 3F, G). Thus, DPCs angiogenic differentiation was not affected by SCAPs-CM but was significantly enhanced by BMSCs-CM.
Effects of CM on neurogenic differentiation of DPCs
DPCs formed sphere-like clusters when cultured in media optimized for neural stem cells, in a low-attachment plate. After 6–9 days of incubation, sphere-like clusters started to attach to the plates and cells were scrambled from the clusters (Fig. 4A). These cells were neuron-like and were positive for nestin and β-III tubulin.
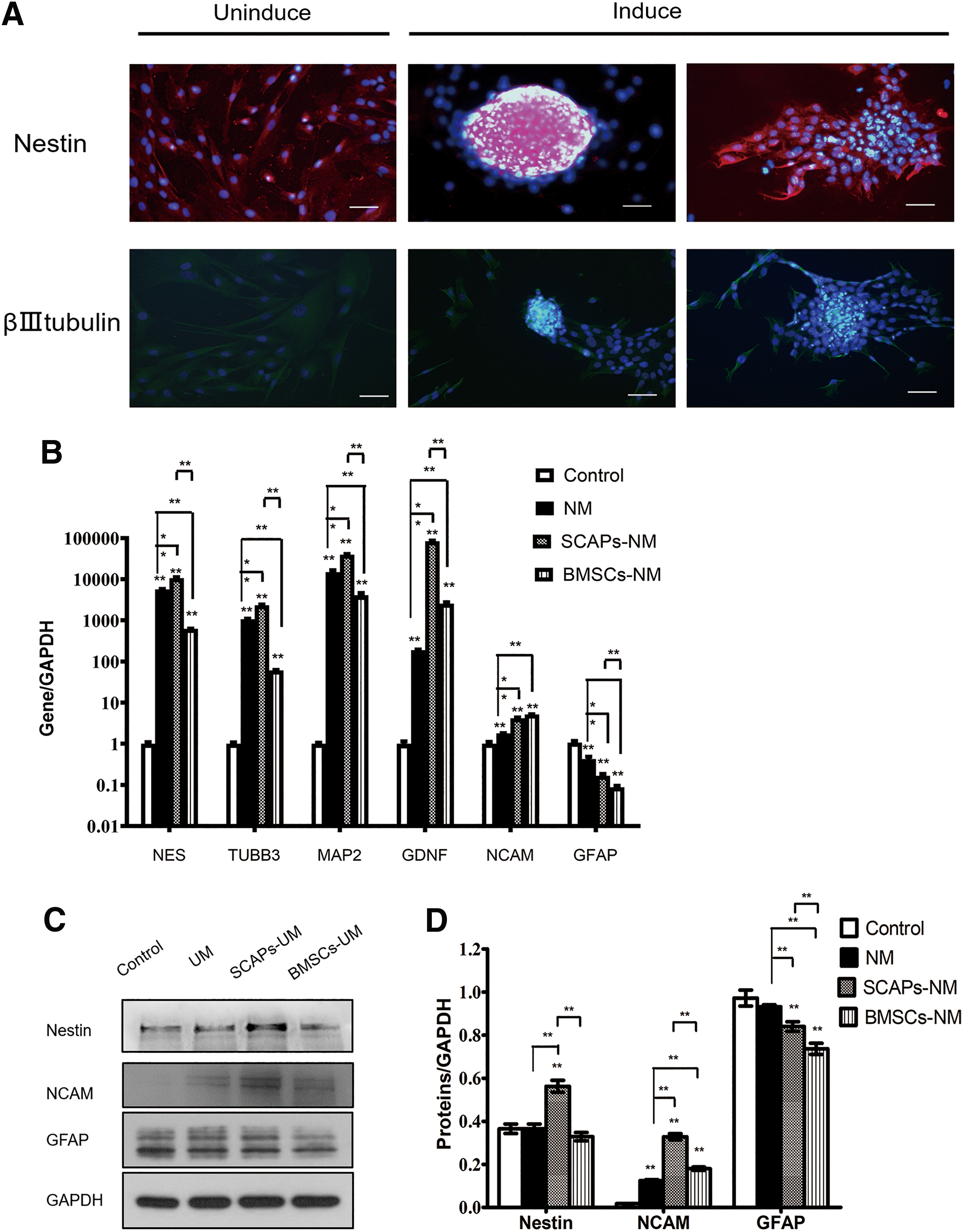
SCAPs-CM enhanced DPCs neurogenic differentiation.
To evaluate the effect of CM on neurogenic differentiation of DPCs, DPCs were cultured in different culture media. After 14 days of incubation, the mRNA level of neurogenic markers, including nestin (NES), β-III tubulin (TUBB3), microtubule-associated protein 2 (MAP2), NCAM, and glial cell-derived neurotrophic factor (GDNF), were significantly higher in the UM treated group than in the control (Fig. 4B). NES, TUBB3, MAP2, NCAM and GDNF were also significantly higher in the SCAPs-UM group than in the UM group (Fig. 4B). However, compared to the UM group, the BMSCs-UM group showed lower NES, TUBB3 and MAP2 expression and higher NCAM and GDNF expression (Fig. 4B).
In addition, the mRNA level of GFAP was significantly lower in the UM treated group than in the control. GFAP was also lower in the SCAPs-UM group and BMSCs-UM group than in the UM group. The result of nestin, NCAM, and GFAP were mostly consistent with our western blot analysis (Fig. 4C, D).
Discussion
The formation of dentin-pulp is a highly organized process involving odontogenesis, angiogenesis, and neurogenesis, wherein trophic factors play a crucial role. Trophic factors can improve the capacity of tissues to regenerate by improving cellular chemoattraction, differentiation, and proliferation. The dental pulp microenvironment of the root apex of immature permanent teeth, which is closely related to dental papilla, may play an important role in the formation of root dentin and dental pulp.
SCAPs were derived from dental apical papilla. The CM of SCAPs contains numerous trophic factors that participated in odontogenesis, angiogenesis, and neurogenesis [20 –24]. It is reported that in coculture system in vitro, dental papilla cell can stimulate osteogenesis and inhibit osteoclastogenesis in dental follicle cells (DFCs) [25]. The CM of DFCs is able to induce differentiation of SCAPs along the odontoblast lineage [26]. It is found that SCAPs-CM have a proangiogenic impact on endothelial migration and tube formation [27]. SCAPs-CM stimulated neurite outgrowth in vitro and SCAPs significantly enhanced axon regeneration in vivo [28]. It is hypothesized that the paracrine production of SCAPs may mimic the microenviroment of dentin-pulp formation. However, the role of SCAPs paracrine action on the function of DPCs is not clear. In this study, we investigated the function of SCAPs-CM on cell proliferation and directed differentiation potential of DPCs.
Despite largely similar phenotypic and cytological properties of MSCs, differences were observed in proteomic profile of MSCs derived from different tissues [29]. MSCs retain the capacity to generate structures resembling the microenvironments from which they are derived in vivo. Numerous studies have demonstrated that the secretome differs among cell sources [20,30 –32]. SCAPs secreted significantly larger amounts of chemokines and neurotrophins than BMSCs, whereas BMSCs secreted more ECM proteins and proangiogenic factors [20]. Osteogenic lineage related proteins were found to be higher in number in secretome of DPSCs and SCAPs when compared with BMSCs secretome [30]. However, MSCs can be seen to secrete both classes of stimulating and inhibitory factors. To achieve insights into the function MSCs, we compared the function of soluble factors released from SCAPs and BMSCs.
Trophic factor's effect on cell proliferation and differentiation will be various, depending on the cell species, culture conditions, and concentration of the trophic factors. We found that BMSCs-CM at a concentration of 100% had an inhibitory effect on DPCs proliferation, which was consistent with previous reports [9]. BMSCs-CM likely contains antimitotic signaling molecules that interfere with the cell cycle [9]. However, CM from BMSCs (25% and 50%) had no significant effect on the proliferation of DPCs. It is reported that DPSCs-CM could not enhance viability of DPSCs at 50% concentration and cause a significant decrease in cell viability at 100% concentration [32]. It is hypothesized that 100% has been less effective due to metabolic acidic by-products accumulated in the culture [32].
Furthermore, our data showed that SCAPs-CM at different concentrations had no effect on DPCs proliferation. It is likely because some of the factors in CM can increase the proliferation effect and some of the factors in CM may inhibit proliferation. Our secretome results indicated that SCAPs secrete less metalloproteinase inhibitor 1 (TIMP1), which promotes cell proliferation, and interleukin-6 (IL6) that inhibits cell proliferation, than do BMSCs [13].
Recent evidences suggest that stem cells paracrine action is regulated by environment condition. In some occasions, changes in culture conditions are intentionally introduced to enhance the production of certain growth factors in CM. For instance, the stress microenvironmental factors, including hypoxia, serum deprivation, and glucose deprivation, favored the production of angiogenic paracrine factors [24]. It is reported that serum deprivation selects for MSCs with a more pluripotent phenotype and enhances their proangiogenic and osteogenic capacities [33 –35]. We collected CM-MSC upon no serum in this study.
BMSCs-CM has osteo/odontogenic inductive effects on DPCs in vitro and induces bone regeneration in vivo [9,16]. Upon implanting BMSCs or BMSCs-CM in prepared bone defects in a rat calvarial model, it is found that the BMSCs-CM group had a greater area of newly regenerated bone than did the BMSCs group, and that BMSCs-CM regenerated bone mainly through mobilization of endogenous stem cells [16].
The CM from SCAPs enhanced the osteo/odontogenic differentiation of DPCs. Our ALP activity results showed that 100% SCAPs-CM was the most conducive to the early osteo/odontogenic differentiation of DPCs, and enhanced differentiation in a dose-dependent manner at 3 days, but not at 5 days. This could be due to a complex effect of negative and positive mineralization-associated factors secreted by SCAPs. At 14 days, the 72 h SCAPs-OM group showed more calcium deposition than did 24 h OM group. It is hypothesized that MSCs cultured for longer time would lead to CM enriched in soluble factors. Furthermore, mineralization-associated genes were significantly more strongly induced in the SCAPs-OM group than in the BMSCs-OM group. Because RNA expression occurs before protein formation, SCAPs-CM could have a significantly higher effect on DPCs osteo/odontogenic differentiation than BMSCs-CM.
Our previous secretome analysis of SCAPs and BMSCs showed that both secreted bone morphogenetic protein 1 (BMP1), insulin-like growth factor 2 (IGF2), and transforming growth factor beta-2 (TGFβ2). BMP1 and TGFβ2 are shown to control dentin formation in vivo [36 –38]. It is demonstrated that osteogenic differentiation of stem cells can be promoted by IGF2 [39]. SCAPs secrete more TGFβ2 and IGF2 than do BMSCs, while BMSCs secrete more BMP1 [20].
SCAPs-CM and BMSCs-CM significantly increased DPCs migration. SCAPs-CM showed a stronger effect on DPCs migration than BMSCs-CM. Our results could be due to the secretion of factors, such as CXC chemokine ligand 12 (CXCL12) and VEGF, which promote cells chemotaxis [40 –43]. Further, SCAPs-CM had no effect on DPCs angiogenic differentiation, which was significantly enhanced by BMSCs-CM. MSCs secrete angiogenic (VEGF, bFGF, CXCL12, etc.) and antiangiogenic (TIMP1 and TIMP2) factors [21,27,44]. Our secretome showed that SCAPs secreted significantly larger amounts of CXCL12 than did BMSCs, and BMSCs secreted more angiogenic factors [20].
The in vitro characteristics of MSCs are associated with their in vivo function for therapeutic use [45]. Dental apical papilla appears to contain less blood vessels and cellular components than the dental pulp and the apical cell rich zone [4]. From our results, we speculate that SCAPs may offer no advantage over BMSCs in terms of inducing the angiogenic differentiation of DPCs.
Nestin is the marker of neural progenitor cells [46,47]. β-III tubulin is considered to be an early neuronal marker and is important for axonal growth during brain development [48,49]. DPCs expressed nestin and β-III tubulin probably due to the neural crest cell origins of dental pulp [50]. Immunofluorescence staining showed that β-III tubulin positive and nestin positive neurosphere-like cells and neuron-like cells were formed after culturing with neurogenic-inducing medium for 6–9 days.
Furthermore, after culturing for 14 days, our results showed that SCAPs-CM enhanced the expression of NES, TUBB3, MAP2, NCAM and GDNF in DPCs, whereas BMSCs-CM attenuated the expression of TUBB3 and MAP2 and enhanced the expression of NCAM and GDNF. MAP2 and NCAM are neuron-specific structural molecular, have been implicated as markers that are specific for neurogenic differentiation [51,52]. GDNF is a neurotrophic protein that promotes the survival and morphological differentiation of dopaminergic neurons [53]. Thus, SCAPs-CM likely enhances the neurodifferentiation of DPCs at neural stemness stage and maturation stage. BMSCs-CM offer no advantage on the neurodifferentiation of DPCs and may stimulate the neurotrophic action of DPCs.
It is reported that SCAPs releases brain-derived neurotrophic factor to regulate neurite outgrowth from cultured trigeminal neurons and trigger directed axonal targeting [54]. Our secretome showed that SCAPs secreted significantly larger amounts of neurotrophins than did BMSCs [20]. It is indicated that SCAPs-CM has a greater neurogenic inductive effect on DPCs than BMSCs-CM. In this study, glial cell marker GFAP was significantly lower in the UM treated group than in the control. It was also lower in the SCAPs-UM group and BMSCs-UM group than in the UM group. It is speculated that DPCs are not prone to differentiate into glial cells when culturing in the medium optimized for neural stem cells. Moreover, SCAPs-CM and BMSCs-CM might not induce the glial cell differentiation.
In conclusion, SCAPs-CM had no effect on DPCs proliferation and angiogenic differentiation, but promoted migration, osteo/odontogenic and neurogenic differentiation of DPCs. SCAPs-CM is more potent in enhancing DPCs osteo/odontogenic and neurogenic differentiation than BMSCs-CM. Our results could indicate the molecular mechanisms involved in dentin-pulp formation. It indicates that SCAPs-CM can serve as additive to improve pulp tissue repair and regeneration.
Footnotes
Author Disclosure Statement
The authors declare no potential conflicts of interest with respect to the authorship and/or publication of this article.
Funding Information
This work was supported by the Research Foundation of Peking University School and Hospital of Stomatology (no. PKUSS20170102), the National Natural Science Foundation of China (no. 303076129), and the Peking University Taisheng stomatology development foundation (no. 94000-6672-H78).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.