
Abstract
Differentiation of trophoblast stem (TS) cells into various cell lineages of the placenta during mammalian development is accompanied by dynamic changes in its proteome for exerting the highly specialized functions of various cell subtypes. In the present study, we demonstrate that the autophagic machinery, which includes proteins for initiation, vesicle nucleation, and autophagosome maturation are robustly upregulated during differentiation of TS cells. Interestingly, basal levels of autophagy were detectable in the developing mouse placenta as well as TS cells. However, autophagic flux was actively triggered by induction of differentiation evident from LC3 maturation. Formation of Beclin1, Vps34, and PIK3R4 ternary complex at the phagophore assembly site that is typically known to induce autophagy was also enhanced during differentiation. Degradation of the p62/SQSTM1 cargo protein and its colocalization with LC3, a mature autophagosome marker, was most prevalent in the trophoblast giant cells (TGCs) and negligible in other trophoblast cells at day 6 of differentiation. Furthermore, disruption of autophagy by impairing lysosomal fusion in TS cells before induction of differentiation led to a decrease in the giant cell and spongiotrophoblast cell markers Prl3d1, Prl2c2, Prl4a1, and Tpbpα upon differentiation. In addition, inhibition of autophagy was associated with a decrease in nuclear size of TGCs. Taken together, these data highlight that autophagy is a necessary prelude in commitment of trophoblast differentiation from the multipotent TS cells probably by regulating protein turnover at the onset of differentiation.
Introduction
Trophoblast stem (TS) cells are multipotent, self-renewing stem cell population that are derived from trophectoderm lineage, similar to embryonic stem (ES) cells that are derived from inner cell mass (ICM) of the blastocyst [1 –3]. TS cells differentiate into various trophoblast cell types [4 –7] that form the structural and functional component of the highly specialized organ, placenta, which in effect is the “lifeline” of the developing embryo in utero. Inadequate trophoblast differentiation leading to compromised placental development results in adverse pregnancy outcome that is threatening to prenatal, postnatal survival of the fetus as well as long-term health and disease predisposition for both mother and offspring. Despite being recognized as a functionally important cell lineage for successful embryo development, molecular regulation of TS cell differentiation is not as extensively studied as ES cells. Nonetheless, several recent reports on global transcriptional and epigenomic regulation of rodent TS cell differentiation [8 –12] have substantially added to our understanding of TS cell differentiation.
During the course of differentiation of TS cells into various lineages, the cell fate depends on radical changes in expression of transcription factors and progenitor markers that are dynamically remodeled to exert its specific function. Since protein turnover and homeostasis are tightly regulated during development and differentiation, a coordinated balance in autophagic machinery in the trophoblast cells of the placenta might be crucial for maintenance of pregnancy. Autophagy has been demonstrated in villous cytotrophoblasts and syncytiotrophoblast layer of the human placenta [13,14] and also in human placenta with compromised pregnancies such as fetal growth restriction and pre-eclampsia [15 –18]. However, the role of autophagic network during trophoblast differentiation and placental development still remains elusive.
Autophagy is a highly conserved catabolic process, primarily involved in bulk lysosomal degradation of proteins and organelles that have been mostly attributed to nutrient deprivation, growth factor depletion, infection, hypoxia, and other forms of physiological stress1. The cytoplasmic components are sequestered into double-membrane vesicles called autophagosomes, which then fuse with lysosomes leading to degradation and release of the recycled products. Autophagosome formation is regulated by a series of protein complexes acting sequentially, for autophagy induction, initiation of autophagosome formation followed by extension and closure of the autophagosome double membranes. Induction of autophagy is associated with formation of ULK1, Atg13, and FIP200 complex that regulates autophagosome biogenesis, and interaction between Atg101 and Atg13 leads to the stability and basal phosphorylation of Atg13 and ULK1. Induction is followed by vesicle nucleation, which involves formation of a termolecular complex of Beclin-1, Vps34, and PIK3R4 [19]. Bif-1 directly binds to UVRAG, forming a complex with Beclin-1, resulting in increased PI3-kinase classIII/Vps34 activity required for autophagosome maturation [20]. Atg9A is an integral membrane protein that is required for both the initiation and the expansion of the autophagosome [21]. Formation of the autophagosome involves a ubiquitin-like conjugation system in which Atg12 is covalently bound to Atg5 and targeted to autophagosome vesicles [22]. This conjugation reaction is mediated by the ubiquitin E1-like enzyme Atg7. Atg16L1 binds Atg5 of the Atg12-Atg5 conjugate forming an 800 kDa multimeric complex [23]. The Atg12-Atg-5-Atg16L1 complex localizes to pre-autophagosomal membranes where it determines the site of LC3 lipidation and catalyzes the reaction required for the formation of mature autophagosomes [23,24]. SQSTM1 (Sequestosome1, p62) binds autophagosomal membrane protein LC3/Atg8, bringing SQSTM1-containing protein aggregates to the autophagosome [25]. Lysosomal degradation of autophagosomes leads to a decrease in SQSTM1 levels during autophagy.
Studies conducted in recent years provide increasing evidence about the role of autophagy during development and differentiation [26 –29]. In mammals, autophagy is induced in early fertilized oocytes and is essential for pre-implantation development [30]. The next phases of autophagy occur throughout mammalian embryogenesis and are primarily required for tissue remodeling. Previous reports provide evidence about the role of autophagy during differentiation of erythrocytes [31], lymphocytes [32,33], adipocytes [34], hepatocytes [35], neurons [36], and so on. Deletion of several autophagy-related genes (Atg) leads to either embryonic lethality or developmental abnormalities [37 –41]. Emerging role of autophagy in development and disease has generated a growing need to accurately identify and quantify autophagy in live cells, animals, and patients.
In this study, we investigated the involvement of autophagic machinery during TS cell self-renewal and differentiation. We have also analyzed the autophagic network in developing murine placenta. We have further demonstrated that inhibition of autophagy in TS cells specifically detains its ability to form differentiated trophoblast cells.
Materials and Methods
Animals and tissue collection
Mouse placental tissues were collected from pregnant female Swiss albino mice obtained from the Indian Institute of Chemical Biology (IICB) animal house. Sexually mature females were caged overnight with fertile males, and day 0.5 of pregnancy was designated by the presence of copulatory plug in the vagina of the female mice. Uteroplacental tissues were dissected out from timed pregnant females on different days of gestation and snap frozen in liquid nitrogen for protein collection. All tissue samples were stored at −80°C until use. IICB Animal Care and Ethics Committee approved all procedures for handling and experimentation with animals as per guidelines set forward by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India (
TS cell culture and differentiation
Mouse TS cells, a kind gift from Prof. Janet Rossant, Sick Kids Research Institute, University of Toronto, Canada, were cultured in the presence of 70% mouse embryonic fibroblast-conditioned medium and 30% media, containing RPMI-1640 (Sigma–Aldrich, St. Louis, MO) supplemented with 20% fetal bovine serum, 1% Penicillin–Streptomycin, 1% Glutamax (Invitrogen), 1 mM sodium pyruvate, 100 μM β-mercaptoethanol (Sigma–Aldrich), 25 ng/mL FGF4 (R&D Systems), and 1 mg/mL heparin. TS cells were seeded at 2.4 × 105 cells per 35 mm culture dish and were maintained for 48 h following which differentiation was induced. Differentiation of the TS cells was induced by withdrawal of FGF4, heparin, and conditioned medium [3,42]. Culture medium was changed after every 24 h to prevent starvation-induced autophagy. Mouse embryonic conditioned medium was collected by harvesting day 13.5 mouse embryos, as described previously [8].
Autophagy inhibition
For determination of autophagic flux, differentiated cells (on day 3 or day 6) were treated with 100 nM of Bafilomycin (Sigma–Aldrich) or 50 μM of Chloroquine (Sigma Aldrich) in respective complete growth medium. Control cells were treated with equivalent amount of DMSO. Cell lysates from Control, Bafilomycin-treated, or Chloroquine-treated cells were subsequently collected after 0, 1, 2.5, and 6 h of treatment.
To assess the effect of autophagy inhibition on differentiation using marker gene expression analysis, TS cells were treated with 100 nM Bafilomycin (Sigma–Aldrich) for 6 h at 60% confluence before onset of differentiation. Control cells were treated with DMSO. Cells were then replenished with differentiation medium and were allowed to differentiate till day 3 and harvested for RNA isolation.
Western blot analysis
Mouse placental tissues were homogenized in RIPA buffer (20 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1 mM Na2 EDTA, 1 mM EGTA, 1% NP40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 0.2 mM PMSF, and 1 mM sodium orthovanadate) supplemented with protease inhibitor cocktail (Sigma–Aldrich). For protein extraction from TS cells, cells were trypsinized with 0.05% Trypsin to ensure isolation of a pure population of stem cells. Cell pellet was washed in Dulbecco's phosphate-buffered saline (DPBS) (Gibco) and lysed in RIPA buffer. For protein extraction from differentiated trophoblast cells, RIPA buffer was directly added to the flasks and cells were scraped off. Cell lysates were sonicated (30 s per pulse, three pulses per sample at 10 MHz). Samples were then centrifuged and supernatants were collected.
Protein concentration was determined using Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad, Hercules, CA). The cell lysates were fractionated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a poly-vinylidene di-fluoride (PVDF) membrane. For LC3 blots, a 15% SDS-PAGE and low cutoff PVDF membrane was used. Blots were incubated for 1 h in blocking solution and overnight with primary antibodies. After washing, secondary antibody incubation was performed for 1.5 h at room temperature. An ECL reagent, Luminata Forte (Millipore, St. Charles, MO), was used for chemiluminescence signal detection. Images were acquired with the Chemidoc Imaging System (UVP LLC, Upland, CA), and band intensities were quantified with NIH ImageJ software.
Immunoprecipitation
Cell lysates (250 μg) were incubated overnight with either anti-Beclin antibody or control isotype-matched IgG at 4°C to allow formation of the antigen–antibody complex. PureProteome Protein A/G Mix Magnetic Beads (Millipore) were used to capture this complex at room temperature for 1 h. Magnetic beads bound to the antigen–antibody complex were washed thrice and eluted under denaturing conditions as per the manufacturer's protocol.
RNA isolation and quantitative real-time PCR analysis
Total RNA was isolated from differentiated cells, on day 3 after bafilomycin treatment using TRIzol reagent (Invitrogen, Carlsbad, CA). Cells were washed with DPBS (Gibco) and lysed using TRIzol reagent. Five micrograms of isolated total RNA was reverse transcribed using RevertAid Reverse Transcription Kit (Thermo Scientific) according to the manufacturer's instructions. Real-time PCR analysis was performed following the previously described protocol [43]. Briefly, 10-fold dilution of cDNAs and Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City) was used for each real-time PCR reaction. Reactions were run in a 7500 Real-Time PCR System (Applied Biosystems). Conditions used included initial holding stage (95°C for 10 min) and 40 cycles (95°C for 15 s and 60°C for 1 min) followed by a dissociation stage (95°C for 15 s, 60°C for 1 min, and then 95°C for 30 s). Primers used for real-time PCR are enlisted in Table 1. Samples were normalized to housekeeping gene, rpL7 for each of the genes. Three different biological replicates were used.
Primers Used for Real-Time PCR Analysis
Antibodies
A list of antibodies used for western blotting and immunoprecipitation experiments are enlisted in Table 2.
List of Antibodies Used for Western Blotting and Immunoprecipitation
Immunofluorescence
Undifferentiated TS cells were counted and plated on coverslips and/or induced for differentiation till day 6. Both TS cells and differentiated cells were treated with 50 μM of Chloroquine (Sigma–Aldrich) in their respective medium for 8 h. Cells were then harvested and fixed using 4% paraformaldehyde, washed with 1 × DPBS (pH = 7.4), and blocked with blocking buffer containing 5% goat serum and 0.3% Triton X-100. This was followed by incubation with anti-LC3 (12741; Cell Signaling Technology) antibody (1:100 dilution) and anti-p62 (ab56416; Abcam) antibody (1:50 dilution) in phosphate-buffered saline containing 1% bovine serum albumin and 0.3% Triton X-100 for 2 h. Cells were washed with 1 × DPBS three times and then incubated with Alexa Fluor-conjugated anti-rabbit IgG (4412; Cell Signaling Technology) and TRITC-conjugated anti-mouse IgG (T5393; Sigma–Aldrich) for 1.5 h at room temperature. Cells were again washed with 1 × DPBS three times followed by nuclear staining with Hoechst at a concentration of 2 μg/mL. Cells were washed again for five times, coverslips were mounted in grease-free slides using Fluoroshield solution (Sigma–Aldrich) and imaged at 630 × magnification using a Confocal microscope, TCS SP8 (Leica, Germany).
Hoechst and phalloidin staining
To observe morphology, size, and frequency of giant cell formation, TS cells were counted and plated over coverslips as per previously published protocol [44]. After 36 h, cells were treated with either 100 nM Bafilomycin (Sigma–Aldrich) or DMSO (vehicle control) for 6 h before onset of differentiation. Cells were then washed and allowed to differentiate for 72 h. Confluent differentiated cells were washed with 1 × DPBS and fixed with 4% paraformaldehyde. Cells were then stained with Phalloidin (13054; Cell Signaling Technology) dissolved in methanol at working dilution 1:200. After washing three times, cells were further incubated with Hoechst at a final concentration of 2 μg/mL for 20 mins. Cells were washed for five times before imaging with FV10i Confocal Laser scanning microscope (Olympus, Japan).
Statistical analysis
Data were analyzed by Student's t-test for comparison of independent means when two experimental groups were compared. One-way analysis of variance followed by Newman–Keuls multiple comparison test was used to compare multiple experimental groups as mentioned in figure legends. Each experiment was repeated three times using at least three different biological replicates.
Results
Autophagy induction was enhanced during differentiation of TS cells
To investigate whether autophagy is involved in differentiation of TS cells, protein components of the autophagy induction complex were analyzed in TS cells and differentiated cells on day 3 and 6 of differentiation by immunoblot assay (Fig. 1A). TS cells were cultured in the presence of factors derived from mouse embryonic fibroblasts (MEF) supplemented with FGF4 and heparin, and withdrawal of these mitogens led to differentiation into a mixed population of trophoblast cells that represent various cell types of the mature placenta. Spongiotrophoblast, trophoblast giant cells (TGCs), and syncytiotrophoblast cells are generally found on day 3 of differentiation, whereas TGCs are more prevalent on day 6 of differentiation as reported previously [42]. The Ser/Thr kinase ULK1 regulates autophagy initiation by forming a complex with FIP200 and Atg13. It was observed that induction of differentiation was associated with a stable and significant (P < 0.01) increase in ULK1 protein levels (Fig. 1B). Interestingly, phosphorylation of Ser (555) of ULK1 increased significantly upon differentiation by day 3, but it did not increase further as days of differentiation progressed. Phospho-ULK1 is known to stabilize the initiation complex. FIP-200 and Atg13, which are required for targeting the ULK1 complex to sites of autophagosome formation, and for ULK1 kinase activity, increased gradually with differentiation. In line with this, Atg101 was also upregulated gradually upon differentiation (Fig. 1A, B). Atg101 is recruited to the autophagy initiation complex by binding with Atg13. These data suggest that basal autophagy induction is active in self-renewing TS cells, but it gradually increases during differentiation.
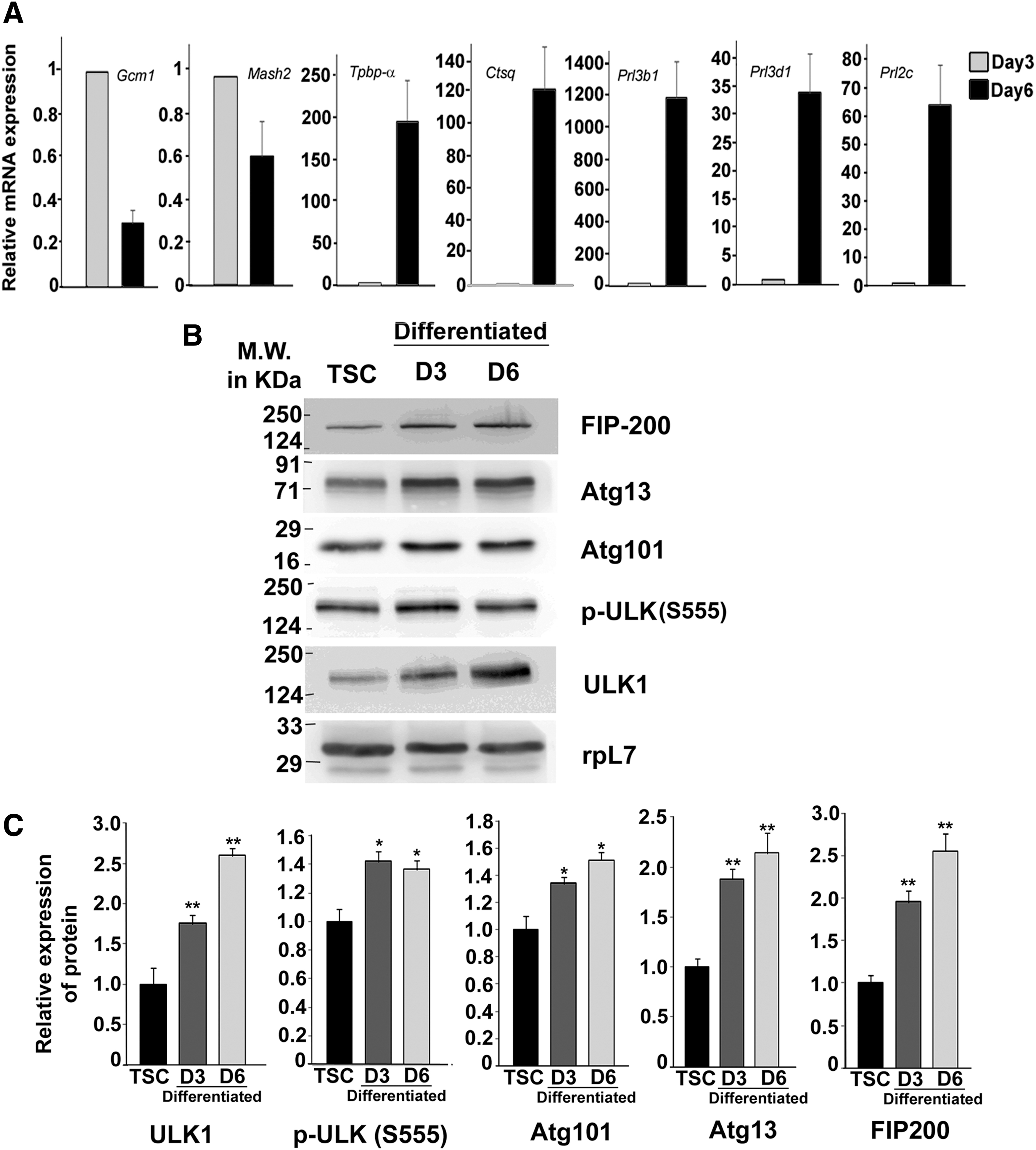
Induction of autophagy during differentiation of murine TS cell.
Vesicle nucleation required for autophagosome formation was augmented during differentiation of TS cells
Since autophagy induction is often terminated by negative feedback loops or other upstream signaling regulators, we investigated protein components required for vesicle nucleation by western blotting (Fig. 2A). For progression of autophagy, an isolation membrane is formed, which encloses cytoplasmic contents to be degraded. This isolation membrane, also called phagophore, is formed with the aid of the class III phosphoinositol-3 kinase/vacuolar protein sorting 34 (Vps34). Following ULK1 activation, Vps34 forms a complex with Beclin1 and PIK3R4. It was observed that Beclin1, the core component of the Vps34 complex, was significantly upregulated during trophoblast differentiation (Fig. 2A, C). Formation of the ternary complex of Beclin1 with PIK3R4 and Vps34, required for phagophore assembly, was enhanced during differentiation as shown by the co-immunoprecipitation assay (Fig. 2B). This suggests an increase in autophagosome formation by induction of differentiation. The UVRAJ protein binds to Beclin1; followed by Bif1 that ultimately enhances Vps34 complex activity. Remarkable increase in UVRAJ and Bif-1 protein levels during differentiation on day 3 and day 6 (Fig. 2A, C) was observed. Atg9A is the only known transmembrane protein required for vesicle nucleation since it delivers membranes to pre-autophagosomal structures called phagophores 25. Atg9A expression was negligible (almost undetectable) in TS cells and was significantly (P < 0.01) upregulated following differentiation (Fig. 2A, C). Taken together, assembly of autophagy inducers and phagophore formation at the phagophore assembly site (PAS) were enriched in differentiated trophoblast cells compared with TS cells.
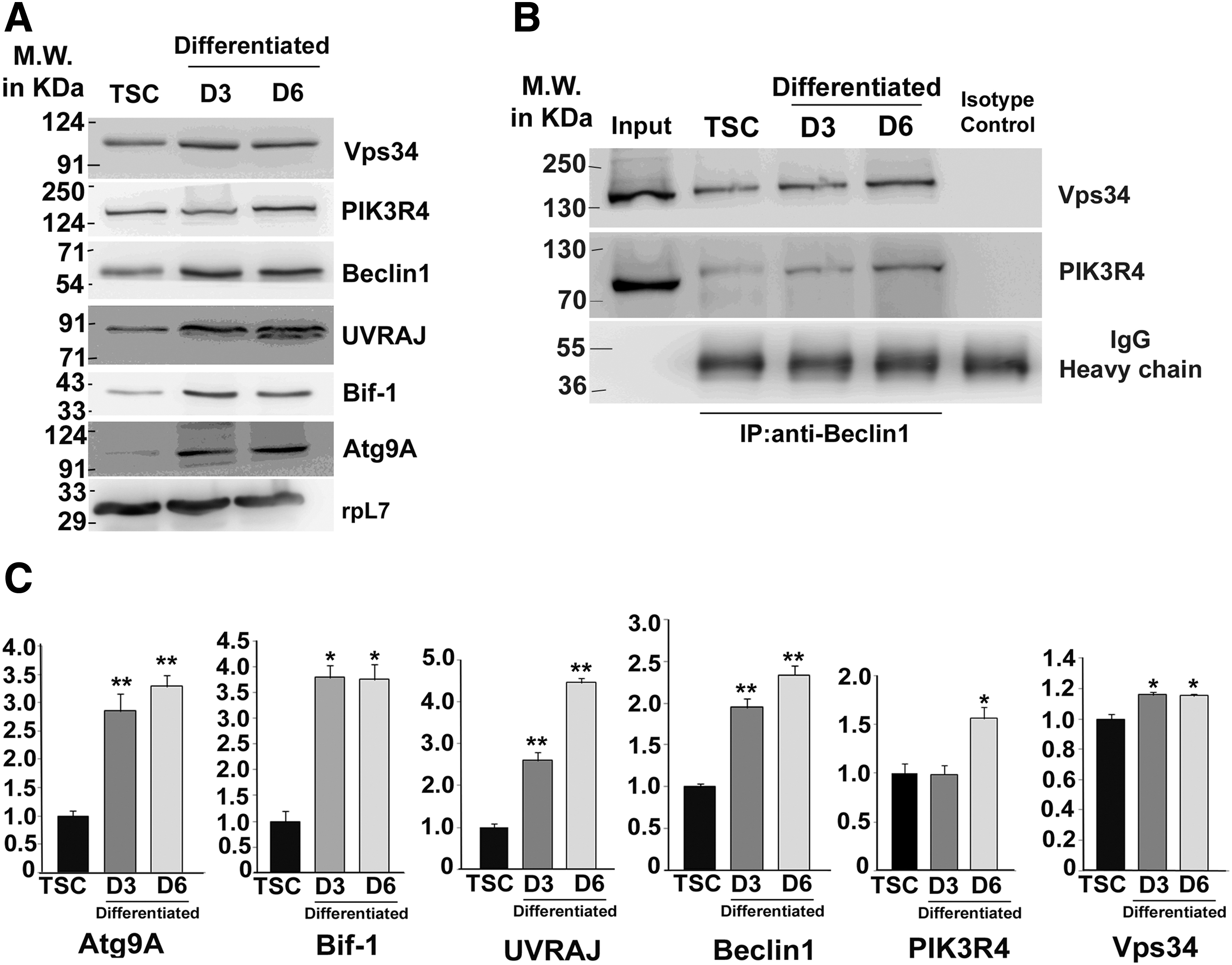
Phagophore assembly and vesicle nucleation for autophagosome formation are upregulated during TS cell differentiation.
Enhanced maturation of autophagosome marked differentiation of TS cells
Autophagy involves formation of double membrane-bound autophagosomes in a sequential ubiquitin-like conjugation system that involves collaborative activity of several Atg proteins. The key step that decides the stability of the autophagosome, which finally leads to fusion with lysosome, is conjugation of LC3 to phosphatidyl ethanolamine by the Vps34 complex formed at an earlier step. Atg5, Atg12, and Atg16L1 form a multimeric complex. Atg protein levels were analyzed by immunoblot assay (Fig. 3A) during differentiation. There was a significant increase (P < 0.01) in Atg5 as well as Atg12-bound Atg5 complex (Fig. 3A, B) during differentiation. Free Atg12 was not detectable in both TS cells and differentiated cells (data not shown). Atg12 conjugation is an essential step for elongation of the isolation membrane, which is mediated by Atg7 (E1-like enzyme) and Atg3 (E2-like enzyme) proteins. Expression of both Atg7 and Atg3 proteins were upregulated by induction of differentiation (Fig. 3A, B). Both α and β isoforms of Atg16L1 (visible as two bands in Fig. 3A) gradually yet substantially increased by day 6 of differentiation. Atg16L1 is known to interact with Atg5-Atg12 complex required for lipidation of LC3 (Atg8/Light chain 3). LC3-I is localized in the cytosol (16 kDa) and following lipidation forms LC3-II (14 kDa), which is incorporated into mature autophagosomes. It was observed that LC3-I expression was similar in TS cells and differentiated cells, but LC3-II formation was upregulated by differentiation (Fig. 3C). LC3-II/I ratio, which is a measure of autophagy, was significantly (P < 0.05) higher in differentiated cells (Fig. 3D). In addition, Atg14 that promotes membrane tethering and fusion of autophagosomes with endolysosomes was also upregulated on day 3 followed by decline in day 6 of differentiation (Fig. 3A, B).

Trophoblast differentiation is associated with enhanced maturation of autophagosome.
LC3-II persists on autophagosomes and colocalizes with the cargo protein p62/SQSTM1 to form aggregates in the cytoplasm before its fusion with the lysosomes leading to recycling of cellular components. Chloroquine treatment of cells prevents fusion of autophagosome with lysosomes and is used to visualize colocalized p62 and LC3 puncta. TS cells and day 6 differentiated trophoblast cells were treated with chloroquine for 8 h followed by double immunostaining with p62 and LC3 antibody (Fig. 3E). Interestingly, p62 and LC3 colocalization was highest in TGCs, found abundantly at day 6 of differentiation. Merge co-efficiency (data not shown, LAS-X software, Leica) was very low in both TS cells and other trophoblast cells (on day 6 of differentiation). Thus, these data demonstrate that although basal level of autophagy exists in TS cells, stability of mature autophagosomes increases by induction of differentiation in trophoblast cells.
Autophagy is an ongoing process during mouse placental development
Differentiation-dependent induction of autophagy led us to investigate whether autophagy continues to occur during mouse placental development throughout gestation. Protein levels of important markers of the mature autophagosomes were analyzed in developing placenta till late gestation. Interestingly, Atg5, Atg7, and Atg16L1, required for maturation of autophagosomes, increased as gestation progressed (Fig. 4A, B). However, LC3-II (lipidated form, 14 kDa) was almost undetectable in placenta. Thus, LC3-II/I ratio were also very low despite an increase in p62 levels. This might be due to lysosomal degradation and delipidation of LC3-II that is continuously recycled during autophagy. Assessment of autophagic flux is therefore required to conclude a real increase in autophagy during development and differentiation of trophoblast cells. As expected, Atg proteins required for maturation of autophagosome were localized in both labyrinth zone and junctional zone of mouse placenta by immunohistochemistry (Fig. 4C).
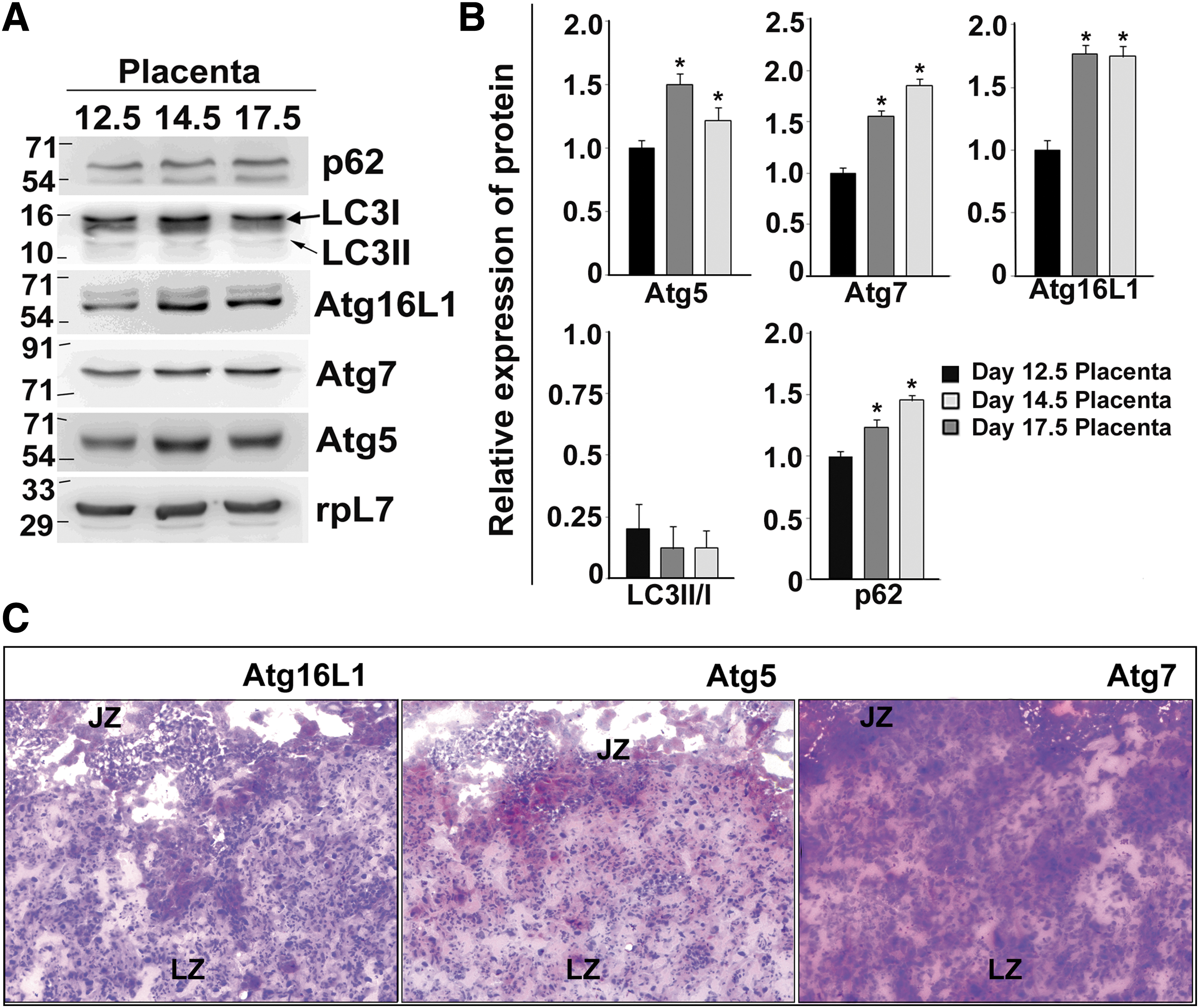
Autophagy continues to occur during mouse placentation.
Autophagic flux is active in trophoblast cells
To measure the autophagic flux, TS cells or differentiated trophoblast cells (day 3 and day 6) were treated with either bafilomycin or chloroquine or DMSO (vehicle control) over a time period of 1, 2.5, and 6 h to measure relative changes in p62 and lipidated form of LC3 (Fig. 5A). Amount of LC3-II reflects the number of mature autophagosomes. However, LC3-II can increase either by increase in autophagy or by inhibition of its degradation resulting from prevention of lysosomal fusion. Furthermore, LC3-II can also be found on non-autophagosomal structures that are not degraded in the lysosome. Therefore, to measure autophagic flux, it is important to measure degradation of p62. The cargo protein p62 binds to LC3-II in mature autophagosome and is degraded following autophagosome–lysosome fusion. It was observed that in control TS cells LC3-II levels were negligible, whereas bafilomycin treatment led to accumulation of its lipidated form along with increase in p62 levels. Chloroquine treatment of TS cells also increased LC3-II levels, but no change was observed in p62 level. To evaluate the autophagic flux, LC3-II/I ratio and p62 levels were also quantified (Fig. 5B). Since inhibition of lysosomal fusion resulted in accumulation of LC3-II along with either increased or constant levels of p62, therefore basal level of autophagy is also active in TS cells.
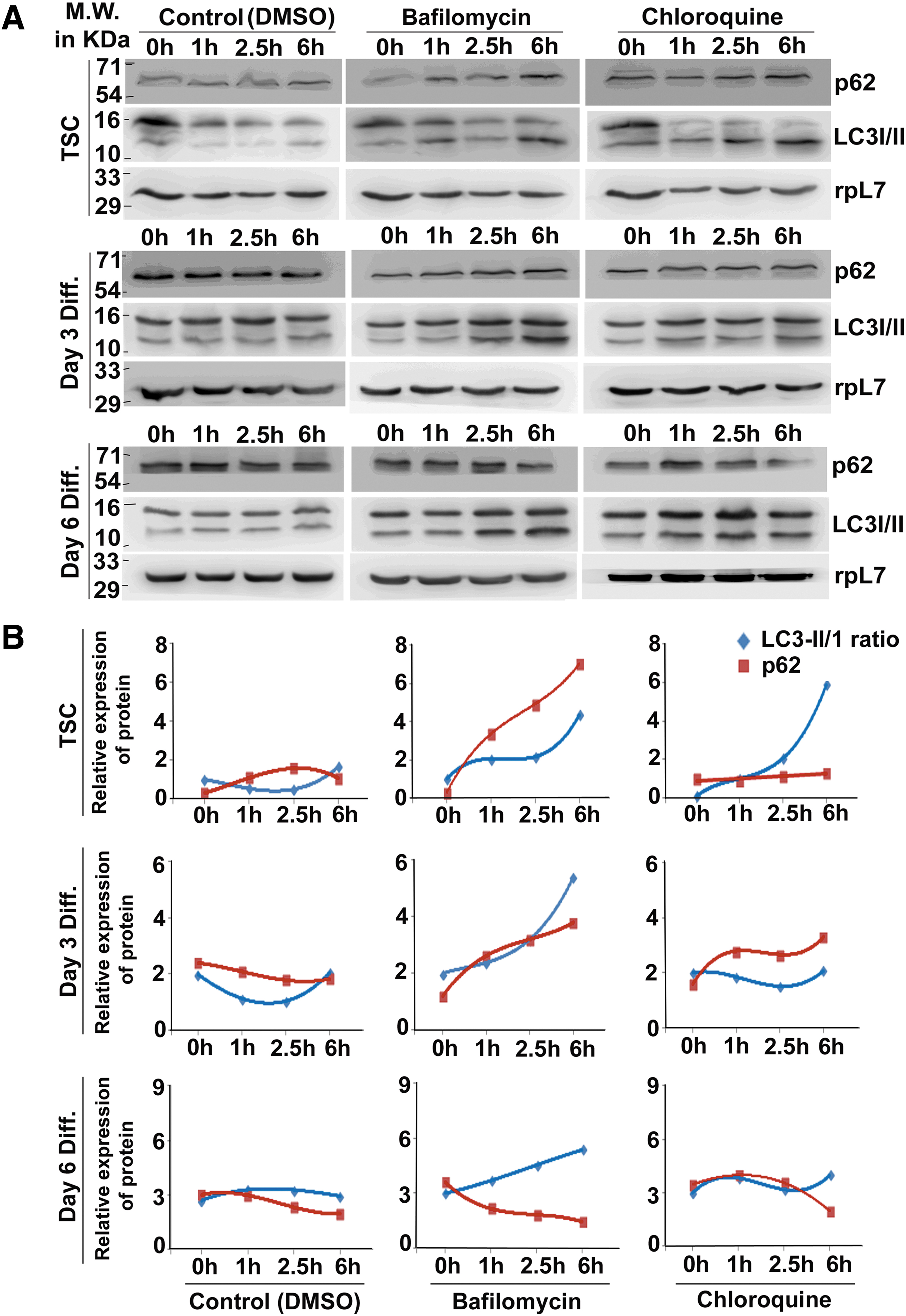
Presence of autophagic flux is evident in trophoblast cells.
Interestingly, in differentiated trophoblast cells, LC3-II accumulation was highest after 6 h of bafilomycin or chloroquine treatment compared with their respective controls. Bafilomycin treatment was more potent than chloroquine with respect to LC3-II accumulation on both day 3 and day 6 of differentiation. As expected, on day 3 of differentiation, inhibition of lysosomal fusion led to accumulation of p62 protein indicating a positive autophagic flux. On day 6 of differentiation, a gradual rise in LC3-II levels was accompanied by decrease in p62 protein despite bafilomycin or chloroquine treatment (Fig. 5B). This result indicates an overriding effect of other active protein turnover machinery leading to degradation of p62 in day 6 differentiated trophoblast cells.
Inhibition of autophagic flux leads to compromised trophoblast differentiation
To determine if autophagy is necessary for trophoblast differentiation, TS cells were treated with bafilomycin for 6 h before differentiation. Cells were harvested after 72 h of differentiation. TGC and spongiotrophoblast markers, which are known to be expressed by day 2 of differentiation, were then analyzed by real-time PCR. There was a substantial decrease in expression of Prl3d1, Prl2c2, Prl4a1 (TGC markers), and Tpbpα (spongiotrophoblast marker) in bafilomycin-treated cells compared with vehicle control (Fig. 6A).
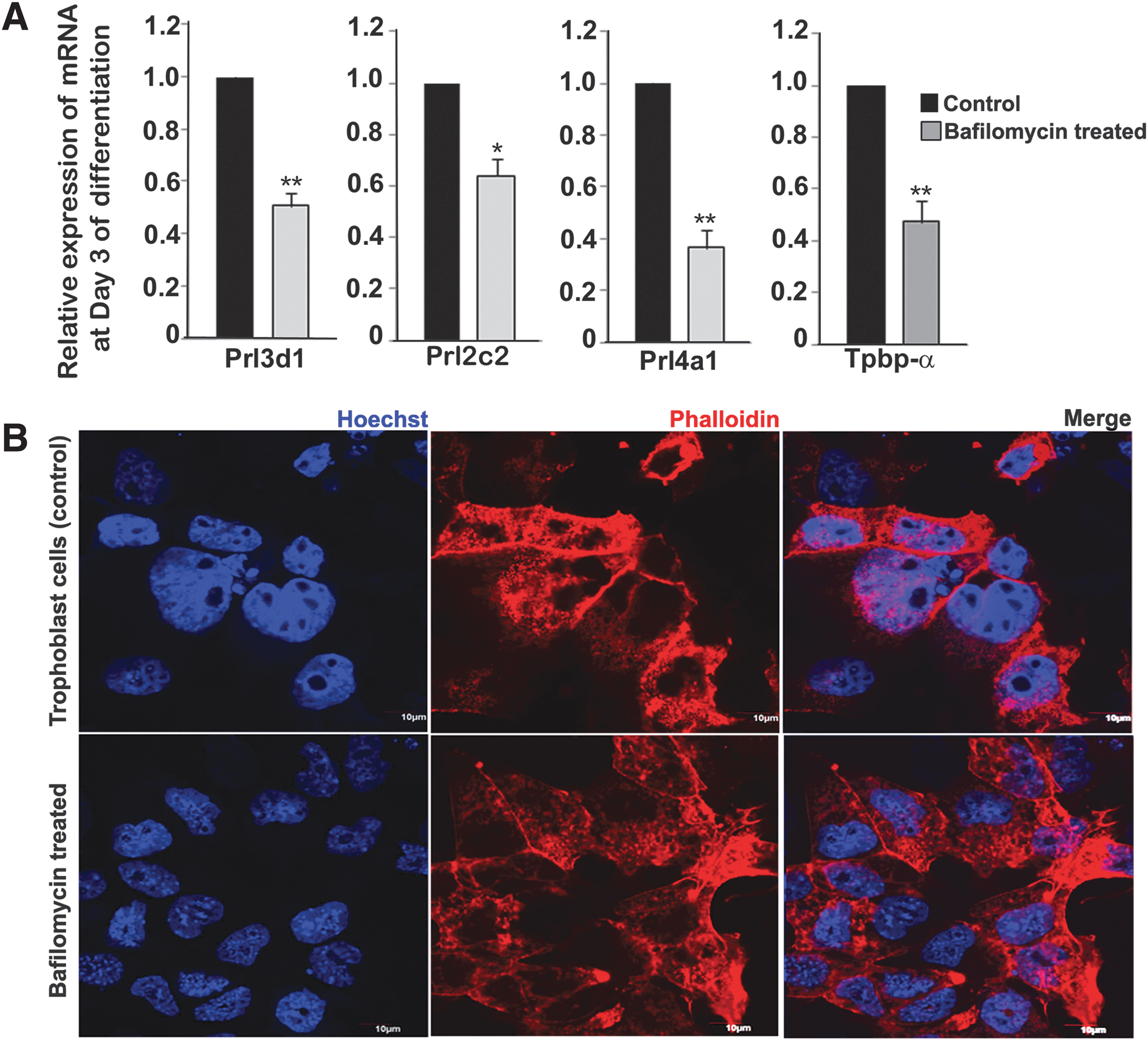
Inhibition of autophagy in self-renewing state impedes trophoblast differentiation.
Next to analyze the morphology of the differentiated cells as a result of inhibition of autophagy, Hoechst and phalloidin staining was performed on day 3 differentiated cells. An overall reduction of nuclear size was observed in bafilomycin-treated group compared with control. Approximately 10 different fields were observed from 3 different biological replicates. A representative photomicrographic image is shown in Fig. 6B.
Activation state of AMP-activated protein kinase and mammalian target of rapamycin during trophoblast differentiation and their implication in trophoblast autophagy
Since the hierarchical steps of autophagy gradually increase during differentiation of TS cells, we wanted to exclude the possibility that growth factor inhibition during induction of differentiation might be the inducer. To test this hypothesis, we analyzed the phospho-AMP-activated protein kinase (AMPK) and phospho-mammalian target of rapamycin (mTOR) levels by immunoblot assay (Fig. 7). AMPK is activated by phosphorylation at Thr-172 due to an elevated AMP/ATP ratio induced by starvation or cellular stress (27). However, in the presence of sufficient nutrients, the mTOR kinase is autophosphorylated at Ser-2481(28). mTOR and AMPK signaling orchestrates and integrates nutritional status and stress signals and regulates autophagy. During induction of differentiation, there was a gradual decrease in phosphorylation of AMPK at Thr-172 (Fig. 7). This was associated with an increase in Ser-2481 phosphorylation of mTOR and its downstream effector p-70 S6 kinase (Fig. 7). Interestingly, inhibition of autophagy after induction of cell differentiation did not further affect differentiation (data not shown), whereas inhibition in self-renewing state before differentiation led to downregulation of differentiation markers (Fig. 6). These data clearly suggest that elaboration of equipoised autophagic network during TS cell self-renewal and differentiation was not a consequence of nutrient deprivation or cellular stress. A schematic representation of the key findings has been summarized in Fig. 8.

AMPK and mTOR signaling during trophoblast differentiation
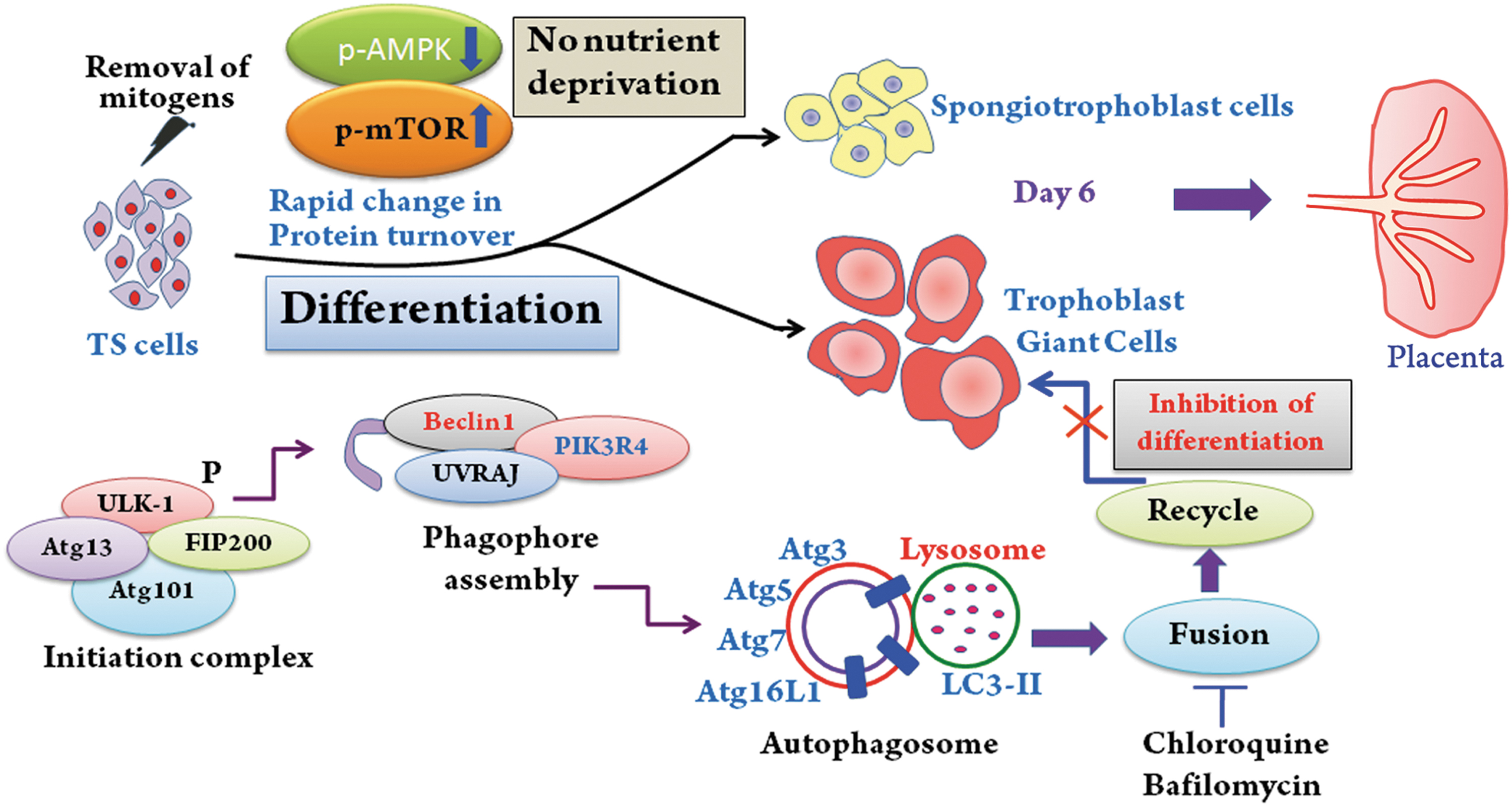
Autophagy is indispensable during TS cell differentiation. Schematic diagram that sums up the finding in this article. Differentiation of TS cells is associated with enhanced formation of initiation complex, phagophore assembly, and autophagosome formation. Inhibition of autophagy before induction of differentiation impedes differentiation. AMPK and mTOR phosphorylation fine-tunes the autophagic network during trophoblast differentiation and is not a consequence of nutrient deprivation. Autophagy is an ongoing process during placentation in mice.
Discussion
TS cells on induction of differentiation undergo substantial transformation with respect to morphology (cell size and shape), transcription factors, protein markers, and cellular properties. The catabolic degradation of cellular components by the well-conserved autophagy pathway is essential for both cell proliferation and remodeling. Although induction of autophagy has been primarily attributed to nutrient deprivation and stress, emerging evidences suggest its role during differentiation. In this report, we analyzed whether autophagic network enables cellular energy homeostasis required for TS cell self-renewal and differentiation. We have demonstrated that protein components that promote autophagy are sequentially and robustly upregulated during differentiation of trophoblast cells. We further tested whether enhanced autophagic machinery associated with differentiation is a consequence of growth factor depletion/stress or is a necessary component for trophoblast differentiation.
The AMPK and mTOR signaling are most commonly studied inducers and inhibitors of autophagy, respectively, that ultimately converges and regulates the activation of ULK1 complex that in turn promotes autophagosome nucleation. Our data demonstrating that p-AMPK levels were negligible in differentiated trophoblast cells and that p-mTOR gradually increases upon induction of differentiation rule out nutrient deprivation-induced autophagy in differentiated trophoblast cells. Increased levels of p-mTOR, an inhibitor of autophagy, might act as a negative feedback mechanism to balance the rates of autophagy during differentiation.
The protein components required for vesicle maturation increased during differentiation. Atg9A is required for expansion of the autophagosomes. Vesicle nucleation can occur by any autophagy stimulator, but to form an autophagosome, stability of the vesicles is extremely important. This is promoted by the Vps-34 complex, Beclin1, being one of the major components. By immunoprecipitation, we demonstrated stabilization of Vps-34, PIK3R4, and Beclin1 ternary complex in differentiated trophoblast cells.
The final steps of autophagosome maturation and stability also increased during differentiation of TS cells. This was evident from increase in Atg5-, Atg7-, Atg3-, Atg16L1-, and Atg12-bound Atg5 complex. The lipidated form of LC3 (LC3-II, 14 kDa) was induced only after differentiation supported by the increase in LC3-II/I ratio conventionally used to measure autophagy. Interestingly, Atg14 that aids in tethering of LC3-II to maturing autophagosomes increased on day 3 followed by a decline on day 6, which might be due to recycling of this protein during lysosomal degradation. Substantial colocalization of p62 and LC3-II was directly demonstrated in the TGCs on day 6 of differentiation (observed by formation of aggregates visible as yellow color of the merge). Unmerged p62 puncta was visible mostly in the TS cells and other trophoblast cells that accumulated due to chloroquine treatment.
Apart from our in vitro model of TS cell differentiation, we also investigated autophagy in vivo. The major protein components of a mature autophagosome were detectable in the developing mouse placenta. Although we observed an increase in Atg5, Atg7, and Atg16L1 during progressing days of gestation, lipidated form of LC3 was almost undetectable. However, this might be due to continuous degradation of LC3-II that is recycled to generate LC3-I after fusion of lysosomes. Variation in the expression levels of Atg proteins in mouse placental region might be attributed to real-time dynamics of autophagic events.
Measuring the amount of lipidated LC3-II in the presence of lysosomal inhibitors represents the amount of LC3 that is delivered to lysosomes for degradation and is used to measure the autophagic flux. p62 is incorporated into autophagosomes through direct binding to LC3 and is efficiently degraded during autophagy. Thus, expression of p62 is inversely correlated with autophagic flux. However, cases of starvation-induced autophagy without reduction in p62 levels have also been reported in autophagy-deficient cells, where p62 accumulation occurs [45]. To measure the autophagic flux, we treated TS cells and differentiated cells with pharmacological inhibitors of autophagy (Bafilomycin and Chloroquine) in a time-dependent experiment and found that there was gradual accumulation of both lipidated form of LC3 and p62, which are used as markers for autophagy [46]. LC3-II levels were very low in unstimulated cells, and in both stem cells and day 3 of differentiation, there was an accumulation of p62 protein by treatment of bafilomycin since the transit of LC3-II through the autophagic pathway was blocked. However, on day 6, there was a time-dependent decrease in p62 levels with increase in LC3-II, which clearly indicates a positive flux. Since chloroquine treatment was not so potent like bafilomycin in our cell culture model, p62 degradation was observed only after 6 h of treatment and not in the previous time points on day 6. p62 can function as a proteotoxic stress sensor and connects the ubiquitin-proteasome system with autophagy [47,48]. This also corroborates with our previous data where p62 and LC3 localization was predominant in the TGCs, which are found as majority on day 6 of differentiation.
Inhibition of autophagy led to a downregulation of TGC and spongiotrophoblast cell markers, with a direct functional consequence of decrease in size of polyploid differentiated cells. This indicates that autophagy plays a major role during endoreduplication of the TGCs. Furthermore, it also emphasizes that metabolic network modulation by autophagy can not only integrate cellular proteomic changes but also fine-tune transcriptional regulation. Most importantly, inhibition of autophagy in already committed cells, soon after induction of differentiation, does not affect differentiation markers but lead to cell death (data not shown). However, when autophagy is inhibited before differentiation, it can change cell fate and compromise differentiation. In conclusion, our data provide evidence that autophagic network and trophoblast differentiation are developmentally intertwined and that autophagy is an indispensable pre-requisite for trophoblast differentiation.
Footnotes
Acknowledgment
We thank Professor Janet Rossant, Hospital for Sick Children, Toronto, Canada, for providing the mouse TS cell line.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the Department of Biotechnology (BT/PR13997/MED/31/306/2015 to R.A.). S.C. was a recipient of Senior Research Fellowship from UGC, India. R.B. is a recipient of Shyama Prasad Mukherjee Junior Research Fellowship from CSIR, India.