
Abstract
Multipotent bone marrow-derived mesenchymal stem/stromal cells (BMSCs) exhibit a finite life span after ex vivo expansion leading to cellular senescence. Many factors can contribute to this. Recently, our group has identified for the first time expression of the chemokine-like factor superfamily 8 (CMTM8) gene in cultured human BMSCs. In this study, we examine the role of CMTM8 in BMSC proliferation, migration, and differentiation. Functional studies using siRNA-mediated knockdown of CMTM8 in human BMSCs resulted in decreased capacity to undergo proliferation and migration and an increased capacity for osteogenic differentiation in vitro. Furthermore, reduced CMTM8 levels led to a decrease in the epidermal growth factor receptor (EGFR) signaling pathway during BMSC proliferation and migration, respectively. Supportive studies using retroviral mediated enforced expression of CMTM8 in BMSC resulted in an increased capacity for proliferation and migration but a decreased osteogenic differentiation potential. Collectively, these data suggest that CMTM8 promotes BMSC proliferation and BMSC migration through the EGFR/ERK1/2 pathway. This study provides insight into novel regulatory mechanisms of human BMSC growth and cell fate determination.
Introduction
Bone marrow-derived mesenchymal stem/stromal cells (BMSCs) are a heterogeneous population of stem and progenitor cells with different proliferation and differentiation capacities. This is attributed to the nature of a stromal cellular maturation hierarchy, comprised mostly of committed self-renewing progenitor cells and a small population of multipotent stromal cells capable of differentiating into adipocytes, osteoblast, chondrocytes, and smooth muscle cells [1 –7]. However, many of the molecular mechanisms mediating BMSC growth and differentiation are yet to be discovered.
Using microarray analyses of human BMSC lines grown in culture media and osteogenic inducing media, our group recently identified the chemokine-like factor superfamily 8 (CLKLSF8) gene to be expressed in human BMSCs during proliferation and osteogenic differentiation in vitro. [8]. CLKLSF8 was renamed chemokine-like factor (CKLF)-like MARVEL transmembrane domain containing 8 (CMTM8) and belongs to the chemokine-like factor superfamily (CKLFSF). CKLFSK consists of nine members, CKLF and CMTM1–8 [9,10]. CMTM1 links the CKLFSF family with chemokines, and CMTM8 links it with members of the transmembrane 4 super family. The characteristics of CMTM2–7 are intermediate between CMTM1 and CMTM8 [10]. Therefore, CMTM8 protein contains four putative transmembrane regions; however due to the lack of the CCG motif, it is not a typical tetraspanin [11]. Moreover, CMTM8 has a MAL-related protein for vesicle trafficking and membrane link domain (MARVEL) domain, suggesting that CMTM8 might play a role in membrane protein trafficking and apposition events [12]. In addition, CMTM8 contains two tyrosine-based internalization consensus sequences, YXXØ, which can bind directly to the adaptor protein 2 that has a function in budding from the plasma membrane [13].
Other studies have demonstrated that CMTM8 enhances the ligand-induced internalization of epidermal growth factor receptor (EGFR) from the cell surface, attenuating EGFR-mediated signaling [14], and therefore leads to decreased proliferation. It has been reported that the CMTM8-induced absence of EGFR-mediated signaling triggers cells to undergo apoptosis through caspase-dependent and -independent pathways, as demonstrated by decreased levels of Bad-S112 phosphorylation [15]. In addition, CMTM8 may be unregulated in tumors and act as a tumor suppressor gene with important roles in the male reproductive, hematopoietic, and immune systems [16]. CMTM8 was found to be a suppressor of epithelial-to-mesenchymal transition (EMT)-like phenotypes through c-Met-ERK signaling [17], which could have an effect on BMSC migration.
Currently, no known function of CMTM8 has been described during BMSC growth, migration, and differentiation. This study aimed to determine whether CMTM8 is a potential mediator of BMSC proliferation, migration, and differentiation, using gene knockdown and gain-of-function studies.
Materials and Methods
Isolation of BMSCs
Human BMSCs were isolated from bone marrow aspirates of normal adult volunteers (18–35 years of age) with informed consent, approved by the Human Ethics Committee of the Royal Adelaide Hospital (protocol number 940911a). Magnetic activated cell sorting (MACS) isolated STRO-1 positive marrow cells were cultured in normal growth media (alpha modification of Eagle's media [αMEM] supplemented with 20% fetal calf serum, 2 mM
RNA extractions, cDNA synthesis, and real-time polymerase chain reaction analysis
Total RNA was isolated from culture-expanded human BMSCs (day 7–14 of osteogenic induction) using Trizol (Invitrogen) according to the manufacturer's instructions, then used for cDNA synthesis using Superscript® IV Reverse Transcriptase (Invitrogen LifeTechnologies, Carlsbad, CA,
Retroviral transduction
Full-length human sequence for CMTM8 (NCBI RefSeq: NM_001320308.1) was subcloned into the pRUF-IRES-GFP vector (Paul Moretti, Hanson Institute, SA, Australia). Retroviral transduction of CMTM8/pRUF-IRES-GFP or empty control pRUF-IRES-GFP into BMSC from three donors was performed as previously described [19]. Stably transduced high-expressing GFP BMSCs were selected by fluorescence-activated cell sorting (FACS), using a BD FACSAria™ Fusion flow cytometer (
siRNA knockdown transfections
Human BMSCs were seeded at 2.2 × 104 cells per well (24-well plate), then treated with sequence-specific siRNA targeting CMTM8 (ThermoFisher Scientific, Australia,
Bromodeoxyuridine proliferation assay
Cells were seeded into 96-well plates (2.5 × 103), and proliferating cells were assessed at the indicated time points using Cell proliferation Elisa, bromodeoxyuridine (BrdU) colometric kit (Catalog No. 11647229001; Roche Diagnostics Corporation, Indianapolis, IN) per manufacturer's instructions. Absorbance was read at 450 nm on an iMark microplate absorbance reader (Bio-Rad Laboratories, Hercules, CA).
Apoptosis assay and flow cytometry
For the quantitative evaluation of apoptosis, cells were collected and washed with cold phosphate-buffered saline (PBS). The ratio of apoptotic cells was detected by using an Annexin V-phycoerythrin (PE)/
Migration assays
In vitro scratch assay: 3 × 104 cells were seeded into a 24-well plate to create a confluent monolayer. Cell monolayer was scraped in a straight line to create a “scratch” with a p200 pipet tip. Debris was removed by washing the cells once with 1 mL of the 1 × PBS, and then replaced with 1 mL of normal growth medium. To obtain the same field during the image acquisition, markings were created to be used as reference points close to the scratch on the outer bottom of the plate with an ultrafine tip marker. T0 was obtained when images were taken using phase contrast microscope and after initial scratch. Cells were then incubated at 37°C in 5% CO2 for 16 h. Images were taken again at T16 at the same spot using the reference point. The area and length of uninvaded space within each well were measured using ImageJ at T0 and T16.
Epidermal growth factor and Erlotinib treatments
BMSCs were seeded in triplicate in 96-well plates and serum deprived overnight. Culture cells were treated with 20 ng/mL recombinant human epidermal growth factor (EGF; Peprotech, Rocky Hill, NJ) or specific concentrations of Erlotinib (Cell Signalling Technology, Danvers, MA) for the indicated times.
Western blot analysis
Whole cell lysates were prepared from MSC lines cultured in 9.5 cm2 wells in triplicate in a total of 100 μL of lysis buffer (50 mM Tris/HCL pH 7.4; 1 mM ethylenediaminetetraaceticacid pH 8.0; 100 mM NaCl; 0.25% sodium deoxycholate; 1% NP-40; 10 mM NaF; 0.5 mM phenylmethylsulfonyl fluoride; 5 mM sodium vanadate; 10 mM sodium pyrophosphate; 1 × complete protease inhibitor) (Roche Applied Science Roche Diagnostics GmbH, Mannheim, Germany,
Osteogenic differentiation assay
In vitro mineralization assays were performed as previously described [4,20]. In brief, BMSCs were seeded at 8° × 103 cells/cm3 in 24-well plates and cultured in mineralization media for up to 28 days with media changes twice weekly. At 18–28 days, the wells were washed thrice in 1 × PBS and fixed with 10% neutral buffered formalin (Thermo Fisher; AJA2518-5L) for 1 h. Mineralized cultures were stained with 2% Alizarin Red S (Sigma Aldrich, Inc., St Louis, MO) in reverse osmosis water. For quantitative assessment of mineral deposition, cells were seeded at 8 × 103 cells/cm2 in 96-well plates and cultured in mineralization induction media for up to 28 days with media changes twice weekly. At 18–28 days, the wells were washed thrice in 1 × PBS, and the mineralized matrix dissolved in 100 μL of 0.6 M HCl (Merck, Kilsyth, VIC, Australia) for 1 h at room temperature. The dissolved mineral solution was then transferred to 96-well microtiter plates, and calcium levels were quantitated using the Arsenazo III (C7529-500 PT; PM Separations). In brief, 4 μL of each supernatant was transferred to a single well of a fresh microtiter plate. A calcium chloride (calcium/phosphorous combined standard; Sigma Aldrich) standard curve was also established in triplicate. Arsenazo was added to each well at 200 μL per well. The plates were shook to mix and incubated at room temperature for 1 min, and the absorbance was read at 650nm on a microplate reader (iMark™ Microplate Absorbance Reader; BIO-RAD). After dissolution of the mineral with HCl, the wells were washed with 1 × PBS, and the cells were digested with 100 μL of proteinase K (100 μg/mL) (Invitrogen, Carlsbad, Canada) for 2–4 h at 55°C, then analyzed on white 96-well microtiter plate (Costar, Corning, New York, NY), and normalized to DNA content per well.
Statistics
Data analysis was carried out using Microsoft GraphPad Prism 7 (GraphPad Software, La Jolla, CA,
Results
CMTM8 is a promoter of BMSC proliferation
The role of CMTM8 during BMSC proliferation was assessed by performing knockdown and overexpression studies followed by staining cells for BrdU incorporation. The data showed that after siRNA-mediated knockdown of CMTM8 gene expression (Fig. 1A), BMSC proliferation rates were greatly reduced with no effect on cell morphology (Fig. 1B, C). The notion that CMTM8 is a promoter of BMSC proliferation was supported in experiments, demonstrating that CMTM8 overexpressing BMSCs (Fig. 1D) exhibited an increased proliferation rate with no observable effects on cell morphology (Fig. 1E, F).
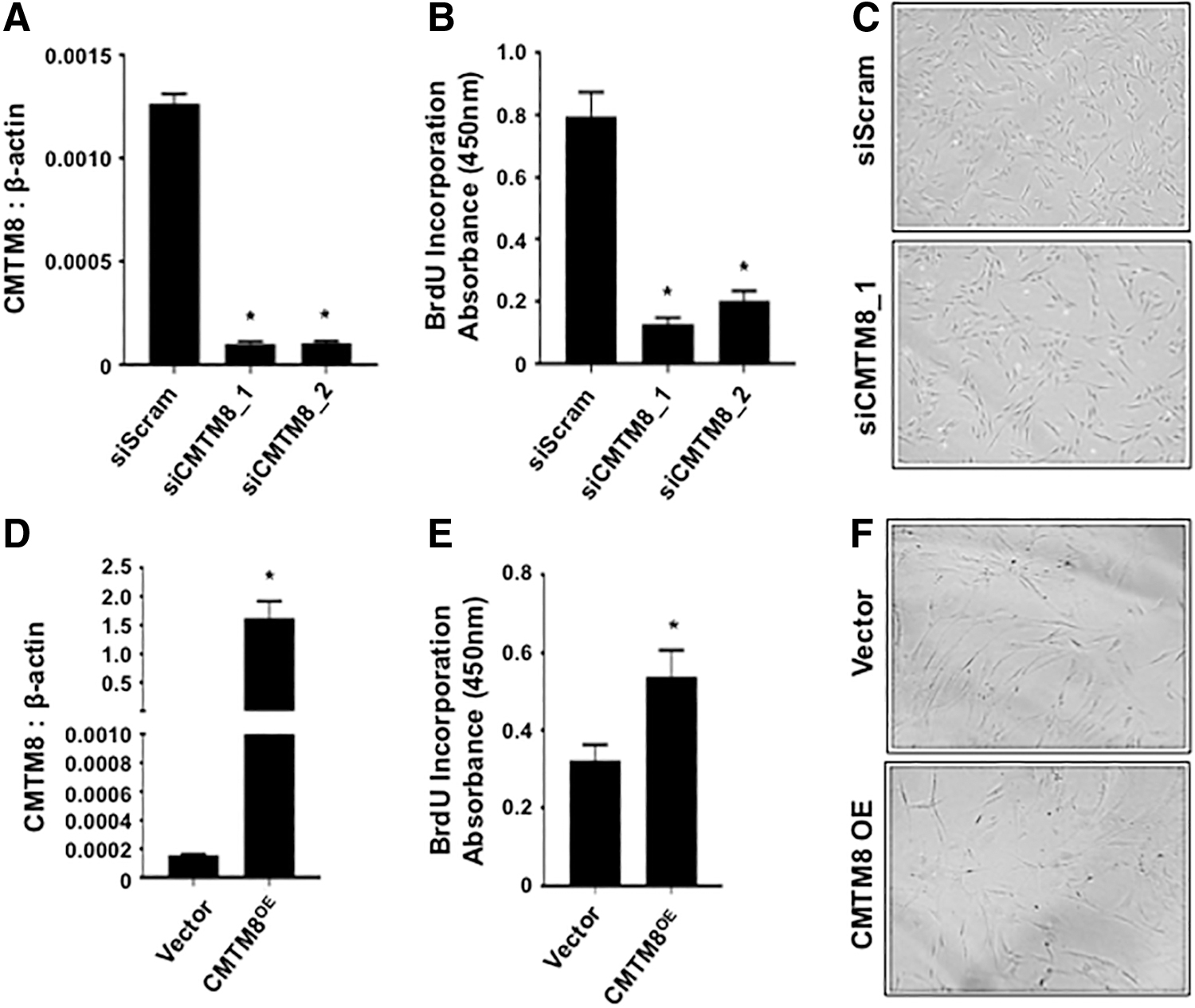
CMTM8 promotes BMSC proliferation.
To investigate whether the reduced proliferation rate observed in CMTM8 knockdown BMSC was attributed to apoptosis, BMSCs were stained with AnnexinV and 7AAD (Fig. 2). Flow cytometric analysis found that CMTM8 knockdown resulted in no difference in the number of live AnnexinV−/7AAD− cells, early apoptotic AnnexinV+/7AAD−, or late apoptotic AnnexinV+/7AAD+ cells (Fig. 2A, B).
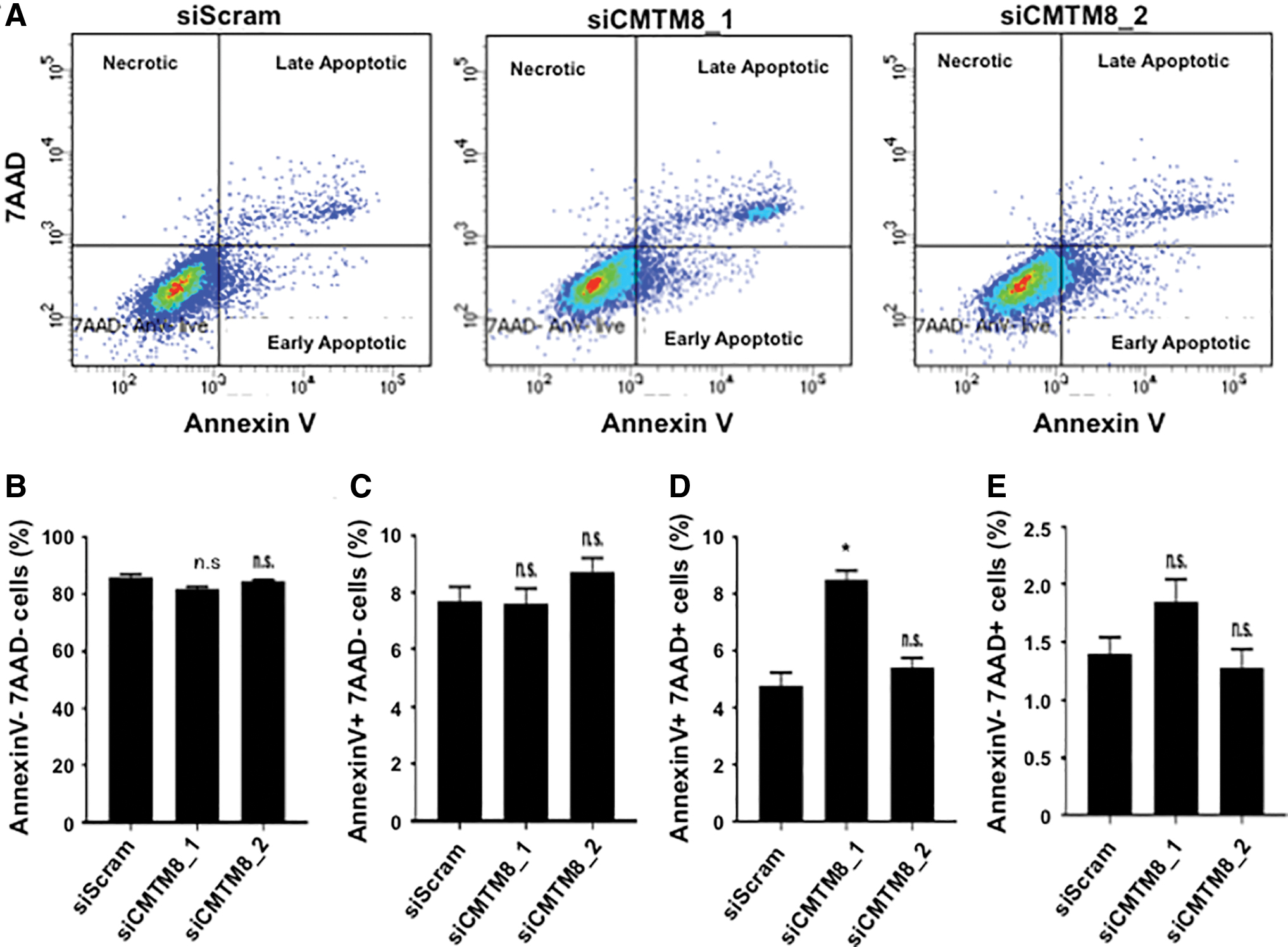
CMTM8 does not affect BMSC apoptosis. siScram- and siCMTM8-treated BMSCs were stained with AnnexinV and 7AAD and put through flow cytometry.
CMTM8 promotes BMSC proliferation through activation of EGFR
Previous studies have reported the mitogenic effect of EGF on BMSC [21]. We next examined whether the effect of CMTM8 on BMSC proliferation was mediated through the EGF pathway. Studies revealed that the mitogenic effect of EGF on BMSC was suppressed in BMSCs, after siRNA-mediated knockdown of CMTM8 (Fig. 3A). In parallel studies, CMTM8 overexpressing BMSCs exhibited an enhanced proliferative response in the presence of EGF compared with vector-only control BMSCs (Fig. 3B).

CMTM8 promotes BMSC proliferation through activation of EGFR signaling.
Previous studies have reported that CMTM8 facilitates the internalization of EGFR into cytoplasm, where EGFR is phosphorylated [14]. To investigate whether the CMTM8 affects BMSC proliferation through EGFR signaling, si-RNA-CMTM8-transfected BMSC and control siScram-transfected BMSC from three donors were stimulated with EGF and assessed by western blot (Fig. 3C). The levels of total EGFR were not significantly different between CMTM8 knockdown and control BMSCs in the presence or absence of EGF (Fig. 3C[i]). However, when cells were treated with EGF, there was an increase in the levels of phosphorylated EGFR in all cells at 10 and 30 min post-EGF stimulation. Nevertheless, levels of phosphorylated EGFR were reduced in siCMTM8-transfected cells at 10 min post-EGF stimulation when compared with siScram control-transfected cells (Fig. 3C[ii], D–F). At 30 min post-EGF stimulation, reduced levels of phosphorylated EGFR in siCMTM8_1-transfected cells were still observed, compared with control, but only in cells from two of the three donors (Fig. 3C[ii], D–F). Assessment of ERK1/2, a major downstream effector of EGFR, found that total Erk1/2 levels were not significantly different between CMTM8 knockdown and control BMSC in the presence or absence of EGF (Fig. 3C[i]). When cells were treated with EGF, there was an increase in the levels of phosphorylated ERK1/2 post-EGF stimulation in all cells. Nevertheless, levels of phosphorylated ERK1/2 were reduced in siCMTM8-transfected cells at 10 min post-EGF stimulation when compared with siScram control-transfected cells (Fig. 3C[ii], G–I). At 30 min post-EGF stimulation, reduced levels of phosphorylated ERK1/2 in siCMTM8-transfected cells were still observed, compared with control, but only in cells from the two donors, which also demonstrated a decrease in protein levels of phosphorylated EGFR (Fig. 3C[ii], G–I).
To further investigate the effect of CMTM8 on EGF-mediated proliferation, we treated vector-only BMSC (Fig. 4A) and CMTM8 overexpressing BMSC (Fig. 4B) with the EGFR inhibitor, Erlotinib, for 3 days and assessed for BrdU incorporation. Our results showed that both populations treated with EGF increased their proliferation rates compared with control dimethyl sulfoxide (DMSO)-treated cells. However, both cell populations displayed a dose-dependent inhibition of cell proliferation in the presence of Erlotinib when compared with their respective DMSO control cells (Fig. 4A, B). Further comparisons found that CMTM8 overexpressing BMSCs showed more resistance to Erlotinib at higher drug concentrations at ≥2 μm (Fig. 4C). In addition, we observed that overexpression of CMTM8 did not result in changes in the expression of important EMT marker genes (Supplementary Fig. S1). These results suggest that CMTM8 promotes BMSC proliferation through the activation of EGFR signaling and phosphorylation of ERK1/2.
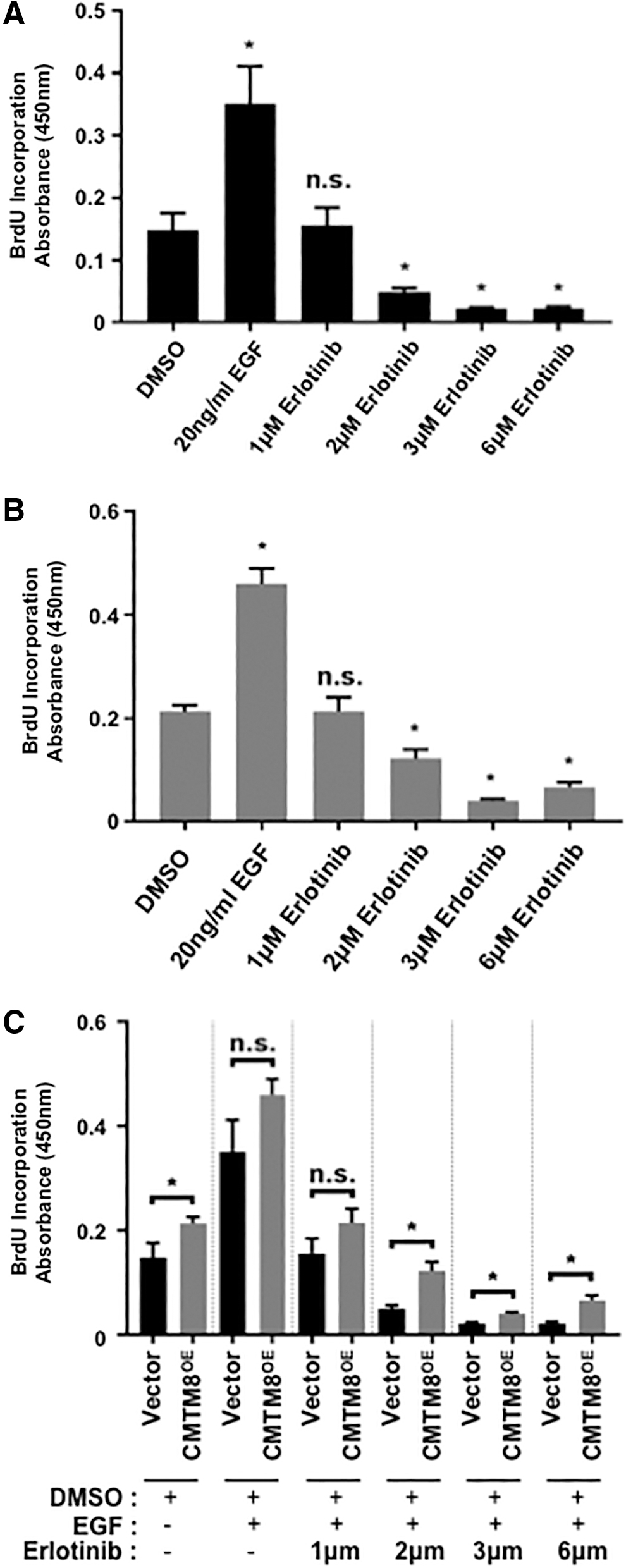
CMTM8 resists antiproliferation effect of Erlotinib.
CMTM8 promotes BMSC migration
Studies have shown that CMTM8 can inhibit EMT-like changes in HepG2 hepatocellular carcinoma cells [17], where EMT is a hallmark of cancer cell metastasis and cell migration. In this study, experiments examined whether CMTM8 played a role in BMSC migration, using an in vitro scratch assay. Images of migrating CMTM8 knockdown and control BMSC were compared between 0 and 16 h (Fig. 5A), and quantitated for area of occupation and migrating length. The data showed that CMTM8 knockdown BMSC had a decrease in length (Fig. 5B) and area (Fig. 5C) invaded after 16 h of incubation, compared with siRNA scramble control BMSC. To confirm this finding, scratch assays were performed using CMTM8 overexpressing and vector-only BMSCs (Fig. 5D). The CMTM8 overexpressing BMSC exhibited an increase in the length (Fig. 5E) and area (Fig. 5F) invaded after 16 h of incubation, compared with vector-only BMSCs. These data suggest that CMTM8 is a promoter of cell migration.

CMTM8 promotes BMSC migration. BMSCs were seeded into a 24-well plate, and in vitro scratch assays were performed. Images were taken, and area and length invaded by cells were measured at 0 and 16 h postincubation. Measurements were analyzed relative to the value obtained at 0 h; area measured is shown by red outline.
CMTM8 inhibits BMSC osteogenic differentiation
We next endeavored to establish the role of CMTM8 in human BMSC differentiation. Preliminary studies indicated that gene expression levels of CMTM8 were elevated in BMSCs cultured under osteogenic inductive conditions over time (Supplementary Fig. S2). Functional studies were performed using siRNA-mediated knockdown targeting CMTM8 transcripts in BMSC from two donors, cultured in osteogenic induction media (Fig. 6A). The data showed that CMTM8 knockdown resulted in an increase in Alizarin Red-stained mineral deposits (Fig. 6B) and extracellular calcium levels (Fig. 6C), after osteogenic induction, compared with BMSC transfected with control scramble siRNA. The CMTM8-mediated suppression of BMSC osteogenic differentiation was further supported by the increased expression of the osteogenic markers runt-related transcription factor 2 (RUNX2) and OSTEOPONTIN (OPN), in siRNA-CMTM8-transfected BMSCs compared with siScram-transfected cells (Fig. 6D, E).

CMTM8 inhibits BMSC osteogenesis.
To verify these findings, CMTM8 overexpressing BMSCs or empty vector BMSCs were cultured in osteogenic differentiation media (Fig. 6F). Overexpression of CMTM8 resulted in decreased Alizarin Red staining (Fig. 6G) and extracellular calcium levels (Fig. 6H), compared with empty vector control cells. Reduced BMSC osteogenic differentiation was further supported by the downregulation of osteogenic markers RUNX2 and OPN expression (Fig. 6I, J) compared with expression in vector-only infected BMSCs. Overall, these studies demonstrated that CMTM8 represses BMSC osteogenic differentiation.
Discussion
This study identified a new role for CMTM8 as a mediator of BMSC cellular proliferation and migration, and suppressor of BMSC osteogenic differentiation. To date, studies have focused on the functional role of CMTM8 in proliferation, migration, and apoptosis of different cancer lines [15,17,22 –28]. Reports in the literature have demonstrated the ability of CMTM8 to induce apoptosis through caspase-dependent and -independent pathways [15]. Furthermore, CMTM8 overexpression was reported to decrease levels of Bad-S112 phosphorylation and induced cancer cell lines such as human cervical carcinoma cell line (HeLa), human prostate carcinoma cell line (PC3), and human breast adenocarcinoma cell line (MCF-7) to undergo apoptosis through caspase-dependent and -independent pathway [15]. In our study, AnnexinV and 7AAD staining determined that the decrease in proliferation due to CMTM8 knockdown was not due to the cells undergoing apoptosis. However, CMTM8 knockdown by siCMTM8_1 did show an increased percentage of 4 in the number of late apoptotic cells compared with siScram-treated cells. This could be due to off target affects by siCMTM8_1 as this was not observed in siCMTM8_2-treated cells. In addition, the level of increase in the percentage of late apoptotic cells by siCMTM8_1 was relatively minor when compared with the large number of live cells, which accounted for up to 80% of the total cells. Overall, the effect of siRNA-mediated CMTM8 knockdown on the late apoptosis of BMSC was negligible, where CMTM8 appears to have a reverse function in promoting postnatal stem cell growth in contrast to cancer cell lines.
Other studies have demonstrated that enforced expression of CMTM8 facilitated ligand-induced internalization of EGFR from the cell surface, and therefore silencing EGFR-mediated signaling [14]. Corresponding to CMTM8, a typical tetraspanin, CD82, comprised of a YXXØ motif has been shown to regulate EGFR internalization in epithelial cells [29]. Therefore, with two YXXØ motifs and one MARVEL domain, CMTM8 has been shown to accelerate the internalization of EGFR [14]. It was also found that CMTM8 inhibited cell proliferation after EGF stimulation [14], and that EGF-induced activation of its receptor, EGFR, led to phosphorylation of its downstream component proteins, ERK1/2. However, after the initial intense ERK1/2 phosphorylation, phosphorylation decreased dramatically in CMTM8 overexpressing cancer cell lines after EGF treatment [14]. On the contrary, siCMTM8-treated cells showed a slow and steady decrease in ERK1/2 phosphorylation. These studies suggest a negative effect of CMTM8 on EGF-induced signaling [14]. Most of the internalized EGFRs are directed for lysosomal degradation and therefore desensitization of the EGFR signaling. However, there are also studies reporting that the endocytosed EGF–EGFR complex preserves its capability to generate signaling cascade from endosomes [30 –32]. Therefore, the biological significance of compartment-restricted signaling in the context of the EGFR system is unclear.
Our previous studies have demonstrated that EGF is a potent mitogen for human colony forming BMSCs [21]. In this study, our data showed that CMTM8 promotes BMSC proliferation through activation of EGFR signaling. Protein analysis demonstrated that EGF treatment did not alter the expression of total EGFR, but did increase the levels of phosphorylated EGFR after stimulation with EGF. Interestingly, siRNA-mediated knockdown of CMTM8 in BMSC resulted in lower levels of phosphorylated EGFR compared with control, suggesting that CMTM8 enhances EGFR phosphorylation. Assessment of ERK1/2, a major downstream effector of EGFR, found that total ERK1/2 levels were not altered but phosphorylated ERK1/2 levels were decreased after CMTM8 knockdown when compared with control. These data indicate that CMTM8 regulates total EGFR/phosphorylated EGFR ratios in BMSC, in the presence of EGF to promote cell proliferation. Confirmatory studies investigated the effect of CMTM8 on EGF-mediated proliferation, after treatment with the EGFR inhibitor, Erlotinib, in a dose-dependent manner. Interestingly, CMTM8 overexpressing BMSCs showed higher proliferation rates when treated with high levels of Erlotinib, suggesting that CMTM8 overexpression conveys a resistance to the antiproliferation effect of Erlotinib.
As a tumor suppressor gene, CMTM8 has previously been reported to inhibit the EMT potential of the HepG2 cells through c-Met signaling [17], which is an important feature of cell migration, metastasis, and invasion. The transmembrane c-Met tyrosine kinase receptor is activated by its ligand, hepatocyte growth factor (HGF) [33 –36], where downregulation of CMTM8 results in EMT-like morphological changes that can be blocked by suppressing MEK and ERK2 [17]. In addition, the protein expression levels of c-Met- and HGF-induced c-Met/ERK signaling were found to be increased when CMTM8 expression was downregulated. It was also confirmed by CMTM8 overexpressing cells where HGF-induced c-MET/ERK signaling was inhibited and cell migration rate was reduced [17]. Together, these findings suggest that CMTM8 functions as a negative regulator of HGF/c-MET signaling to ERK, which then leads to decreased EMT-like changes in cancer cells. While this study showed that CMTM8 promotes BMSC migration, examination of the expression levels of common EMT-associated markers failed to find significant differences (Supplementary Fig. S1), suggesting that CMTM8 does not promote BMSC migration through regulating the expression of EMT genes. As a transmembrane protein, CMTM8 could affect the cell–cell communication and the sensitivity toward a stimulant, which could then effect the migration of BMSCs.
To date, the effect of CMTM8 on BMSC cell fate determination has yet to be explored. Our study found that CMTM8 is an inhibitor of BMSC osteogenic potential through suppression of the osteogenic master regulatory gene, Runx2. These observations appeared to be independent of EGF signaling during differentiation, where the addition of EGF or EGF and inhibitor (Erlotinib) had no affect on the osteogenic potential of CMTM8 overexpressing BMSCs (Supplementary Fig. S3). Of note, we failed to detect any affect of CMTM8 on the adipogenic differentiation of BMSC (Supplementary Fig. S4), where adipogenesis is generally inversely affected by factors that promote bone formation and visa versa. Interestingly, our study showed that CMTM8 gene expression levels were upregulated during BMSC osteogenic induction over time, implying that CMTM8 may be part of a negative feedback loop, acting as a switch to block BMSC differentiation to allow for cellular proliferation and migration to occur. Collectively, these findings suggest that CMTM8 is a novel molecular inhibitor of BMSC osteogenic differentiation.
Conclusions
In this study, we have identified a previous unknown function for CMTM8 in human BMSC proliferation, migration, and differentiation. For the first time, CMTM8 has been shown to regulate human BMSC growth by promoting proliferation through EGFR signaling pathway and regulate cell fate by inhibiting osteogenesis (Fig. 7). This study contributes to development of new strategies in bone regeneration and tissue engineering by manipulating the expression level of CMTM8. Our future efforts will attempt to investigate the role of CMTM8 in vivo using CMTM8 knockout mouse model.
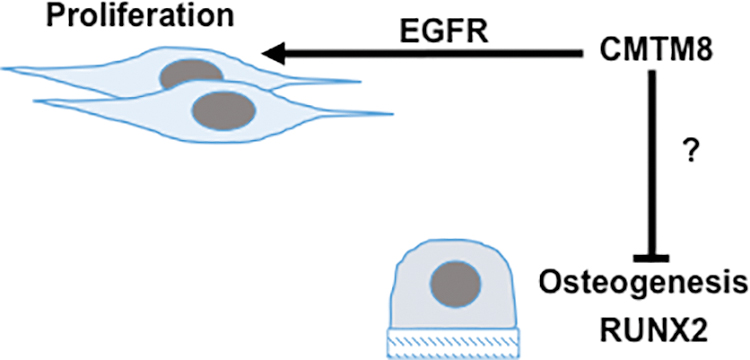
Model for CMTM8 modulation of BMSC functions. CMTM8 inhibits BMSC osteogenesis and promotes proliferation through EGFR signaling. Color images are available online.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported in part by the National Health and Medical Research Project Grant (APP1120989) and the Australian Cranio-Maxillo Facial Foundation.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.