
Abstract
Fluorescent-activated cell sorting (FACS) remains a powerful tool to enrich blood-derived progenitor cells for the establishment of highly proliferative endothelial colony-forming cells (ECFC). Further investigation remains necessary to determine whether the retention of progenitor cell phenotypes after expansion can identify ECFC with enhanced proangiogenic and regenerative functions. This study employed FACS purification to segregate umbilical cord blood-derived ECFC using conserved provascular progenitor cell markers CD34 or aldehyde dehydrogenase (ALDH) activity. ECFC FACS purified for high versus low ALDH activity formed single cell-derived colonies and demonstrated tubule formation in Matrigel at comparable rates. Surprisingly, FACS purification of ECFC for CD34 enriched cells with enhanced colony-forming capabilities and tubule formation within the CD34− population. CD34 expression was enriched on early ECFC populations; however, steady-state expression of CD34 rapidly declined and stabilized on expanded ECFC after serial passage. CD34 expression on ECFC was shown to be cell density dependent and coincided with a loss of progenitor cell characteristics in vitro. Silica-bead surface membrane capture followed by proteomic analysis by label-free liquid chromatography tandem mass spectrometry (LC-MS/MS) identified >100 distinctions (P < 0.05) associated with the plasma membrane of CD34− versus CD34+ ECFC, including a significant enrichment of CD143 (angiotensinogen converting enzyme) on CD34+ cells. Despite an enrichment for traditional endothelial cell markers on the CD34+ ECFC in vitro, implantation of both CD34+ and CD34− ECFC within Matrigel plugs in immunodeficient
Introduction
From adaptable high-volume arteriole systems to fenestrated microvascular networks, endothelial cells serve as building blocks to form the intraluminal lining of blood vessels and provide a highly regulated interface between circulating blood and vascularized tissue compartments [1,2]. The dynamic array of endothelial cell phenotypes is governed by tissue-specific microenvironments that provide bioactive cues and an architectural matrix to direct endothelial specification during organ development [3,4]. To illustrate this concept, grafts of non-neural somite tissue transplanted within a neural microenvironment formed the organization of the blood–brain barrier observed in adjacent host tissues [5]. On the other hand, transplanted brain tissues developed a fenestrated interface within coelomic tissue beds, supportive of the notion that endothelial specification occurs in association with the microenvironment [5].
Contrary to tissue-resident primary endothelial cells, endothelial cell lines often demonstrate a plastic and variable phenotype in vitro, although the functional relevance of this dynamic surface marker expression is poorly understood [6 –11]. Specifically, it appears that the removal of endothelial cells from their native environment and subcultured in vitro can induce loss of organ-specific phenotype [12,13]. It has been previously demonstrated that phenotypic expression profiles across several progenitor cells lineages, such as hematopoietic and mesenchymal progenitor cells, can rapidly change based on microenvironmental cue experience during ex vivo expansion [14 –18]. Thus, the classification of endothelial cell subsets within expanding endothelial progenitor/precursor cells (EPC) in vitro remains a challenge.
The conceptual framework for EPC arose from seminal experiments by Asahara defining the ability of CD34+ peripheral blood mononuclear cells (MNC) to home sites of ischemia and promote vascularization after transplantation [10]. This work inspired three distinct culture techniques to isolate and propagate EPC-like colonies from human blood or bone marrow sources. As a result, three inter-related cell populations were established: colony-forming unit–endothelial cells (CFU-EC) [10,19], circulating angiogenic cells (CACs) [20 –22], and endothelial colony-forming cells (ECFC) [23 –25]. It was later uncovered that the early outgrowth populations (CFU-EC and CAC) comprised primarily myeloid hematopoietic cells with a proangiogenic secretory phenotype [15,20,26 –28].
In contrast, late outgrowth ECFC derived from plastic-adherent MNC gave rise to highly proliferative endothelial cells able to form vascular networks in vivo [6]. Although it is agreed that only ECFC give rise to pure endothelial cells in vitro, a lack of consensus surrounding the term “EPC” to describe freshly isolated versus cells expanded in culture remains [6,9]. Several surface markers or levels of enzymatic activity have been explored to further define and enrich EPC before colony establishment [9]. For example, freshly isolated CD34+/CD45−, CD133, CD146, or ALDHhi cells can enrich ECFC [29 –32]. However, it is poorly understood whether these same phenotypes can reselect and purify for a subpopulation of ECFC that retain progenitor-like characteristics following expansion in vitro.
The detection of circulating endothelial cell phenotypes in peripheral blood has been used as clinical diagnostic to noninvasively predict cardiovascular disease risk [19] and recovery rates [33] in patients with cardiovascular complications. Preclinical studies have demonstrated the therapeutic relevance of freshly isolated EPC; in addition, the purification of EPC has uncovered molecular mechanisms underlying tissue homeostasis or disease [34].
It remains precedent to further our understanding of the distinctions between EPC harvested from fresh and/or expanded ECFC sources. Specifically, this knowledge is crucial for accurately comparing results obtained during clinical trials using endothelial cells in applications of regenerative medicine [34]. There are currently over 350 clinical trials initiated, which indicate the use of EPC or endothelial populations (
The clinical application of ECFC will likely require extensive ex vivo expansion to obtain large numbers of ECFC required for cell therapies. Thus, it remains critical to elucidate maturation-induced phenotype changes for expanded ECFC to better direct the utility of therapeutic ECFC subsets [39,40]. We hypothesized that ECFC cultures contain a hierarchy of rare, proliferative progenitor cells that drive expansion and subsequently generate more differentiated endothelial cells that demonstrate dampened proliferative rates and blunted vascular regenerative function.
Herein, we demonstrate that umbilical cord blood (UCB)-derived ECFC expressed CD34 in a density-dependent manner and the expression of CD34 coincided with increased expression of mature endothelial cell surface antigens, but decreased clonogenicity, growth kinetics, and tube-forming capacity compared to CD34− ECFC in vitro. In addition, CD34+/− ECFC subsets were equally able to form vessel-like structures within Matrigel plugs after subcutaneous transplantation into the flank of
Materials and Methods
Isolation and culture of ECFC
Human UCB samples were obtained with informed consent by venipuncture of the umbilical vein following Ceasarian section at Victoria Hospital Birthing Centre, London, ON, Canada (REB No. 12934). The Human Studies Research Ethics Board (HSREB) at Western University approved all procedures. Human ECFC were established from UCB-derived MNC, as previously described [25]. Briefly, MNC were seeded on tissue culture plastic at 70,000 cells/cm2 in complete endothelial growth media-2 (EGM-2) + 2% fetal bovine serum + cytokines (Lonza, Basel, Switzerland). Media were replaced every 3 days for up to 14 days when colonies became confluent (Supplementary Fig. S1A). Colonies were lifted with TrypLE™ express (ThermoFisher, Waltham, MA) and cells were replated at 4,000 cells/cm2. ECFC monolayers were subsequently expanded in EGM-2 and passaged at ≈80% confluency. Cells were cryopreserved at passage 3 to be used for subsequent experiments.
Flow cytometric characterization of ECFC
To prepare ECFC for flow cytometric analyses or fluorescence-activated cell sorting (FACS), ECFC were dissociated with trypsin and treated with Aldefluor™ reagent (Stem Cell Technologies, Vancouver, Canada) to measure aldehyde dehydrogenase (ALDH) activity. Surface marker expression was also analyzed by co-staining with conjugated anti-human antibodies for CD34, CD31, CD45, VE-Cadherin, CD105, VEGFR2, TIE-2, and CXCR4. Data were acquired using an LSR II flow cytometer (BD Biosciences, San Jose, CA) analyzed using FloJo software v10.6 at the London Regional Flow Cytometry Facility.
Single-cell sorting and colony-forming assays
An automated cell deposition unit on a FACSAria III cell sorter (BD Biosciences) was used to plate single cells into each well of a 96-well plate containing EGM-2. ECFC were initially selected using forward and side scatter profiles to obtain single unpurified cells at random. In subsequent experiments, ECFC subsets were selected based on high versus low ALDH activity and CXCR4, CD34, or CD143 expression. Single-cell deposition was visually checked by light microscopy after plating. Media were replaced after 6 days and colonies were enumerated using an inverted light microscope on day 12 of culture. Enumerated colonies were defined as cell clusters containing >50 cells.
Tubule forming assay
To assess the tubule-forming function of ECFC subsets in vitro, 120,000 ALDHhi/lo or CD34+/− FACS purified ECFC were cultured on growth factor-reduced Geltrex matrices (ThermoFisher) in endothelial basal media without serum or cytokines (EBM-2) or in complete EGM-2. After ≈20 h, photomicrographs were taken by light microscopy, and tubule formation was quantified using manual counting of complete tubule number under light microscopy. Tube formation was enumerated using ImageJ software.
Growth kinetics and surface marker expression
FACS-purified CD34+/− ECFC subsets were plated at 4,000 cells/cm2 in seven different tissue culture flasks. Manual hemocytometer counts and flow cytometry analyses were performed every 24 h for 7 days to assess ECFC expansion kinetics and CD34 expression patterns. The media were changed on days 3, 5, and 6 as the near-confluent monolayer required more frequent medium changes to maintain cell growth.
Silica-bead isolation of plasma membrane proteins
Initially, 106 FACS-purified CD34+/− ECFC subsets were plated on tissue culture plastic for 8 h to permit adherence, before plasma membrane isolation and analyses of surface proteins by liquid chromatography tandem mass spectrometry (LC-MS/MS). For cell surface protein isolation, a 10% solution of colloidal silica beads (Sigma Aldrich, St. Louis, MO) was added to ECFC cultures to permit colloid silica bead/membrane-bound protein interactions, as previously described [41]. A polyacrylamide catalyst (Sigma) was used to stabilize protein/colloidal bead interactions, cross-linking the silica beads to the cell membrane. Bead-bound ECFC lysates were manually collected using cell scrapers and homogenized by probe tip sonication before ultracentrifugation using a HistoDenz gradient (Sigma) to isolate bead/protein conjugates. The beads, containing membrane-bound proteins, were resuspended in 8 M urea, 50 mM ammonium bicarbonate, 2% SDS (sodium dodecyl sulfate), and 5 mM DTT to solubilize proteins off the beads in a sonication water bath for 30 min. Protein concentrations were determined using the Pierce 660 nm protein assay.
Mass spectrometry analyses of cell surface protein expression
Protein extracts were reduced in 10 mM DTT for 30 min in the dark and alkylated with 100 mM iodoacetamide for 45 min at room temperature in the dark. To facilitate the removal of incompatible detergents, reducing and alkylating reagents, proteins were precipitated using chloroform methanol. After protein precipitation, peptides were generated through digestion with trypsin. High pH fractionation was performed using a Waters XBridge BEH130 C18 5 μm column. After collection, the protein fractions were vacuum concentrated (SpeedVac™) and acidified before liquid chromatography coupled with tandem mass spectrometry [14,18,42,43]. Data analyses were performed with MaxQuant version 1.5.0.30 using the Andromeda search engine [44]. MS/MS spectra were searched against the Human Uniprot database with trypsin specificity (20,264 entries) [45].
Surface marker enrichment analyses
Bioinformatic analyses were performed using Perseus software version 1.6.7. Protein expression from the CD34+ ECFC was directly compared with protein expression from the CD34− ECFC using LFQ intensity values. Datasets were filtered for proteins exclusive to either CD34− or CD34+ ECFC. Alternatively, nonexclusive proteins were filtered for detection in >50% of samples in each ECFC subset, value imputation was performed on missing values (width: 0.3, downshift: 1.8), and differential protein expression was determined with a cutoff of ≥2-fold difference and Student's t-test with permutation-based false detection rate (FDR) P < 0.05. Differently expressed proteins were compared to GO Biological Processes and Reactome database using open-source Metascape (
In vivo implantation of purified ECFC in Matrigel
All animal procedures were approved by the Animal Care Committee at Western University Canada (AUP 2015-012). The ability of CD34-selected ECFC subsets to form functional vasculature in vivo was assessed after implantation into immunodeficient
Results
ECFC can be segregated based on CD34 expression and/or ALDH activity
To validate the purity of ECFC cultures, flow cytometry was used to assess both endothelial and hematopoietic cell surface marker expression. To confirm endothelial identity, cultured UCB-derived ECFC highly expressed CD31, VEGFR2, VE-Cadherin, and CD105 (>97%) with <5% hematopoietic contamination measured using CD45 (Supplementary Fig. S1B–F). Analyses of markers commonly used to assess progenitor cell status (ALDH activity, CD34) or migratory competence (CXCR4) after ex vivo expansion are shown in Fig. 1A. Expanded ECFC were 64.5% ± 17.9% ALDHhi and 28.0% ± 14.7% ALDHlo (Fig. 1B). In contrast, CD34 (3.3% ± 1.3%) and CXCR4 (1.4% ± 0.6%) expression on cultured ECFC were consistently low (Fig. 1B).
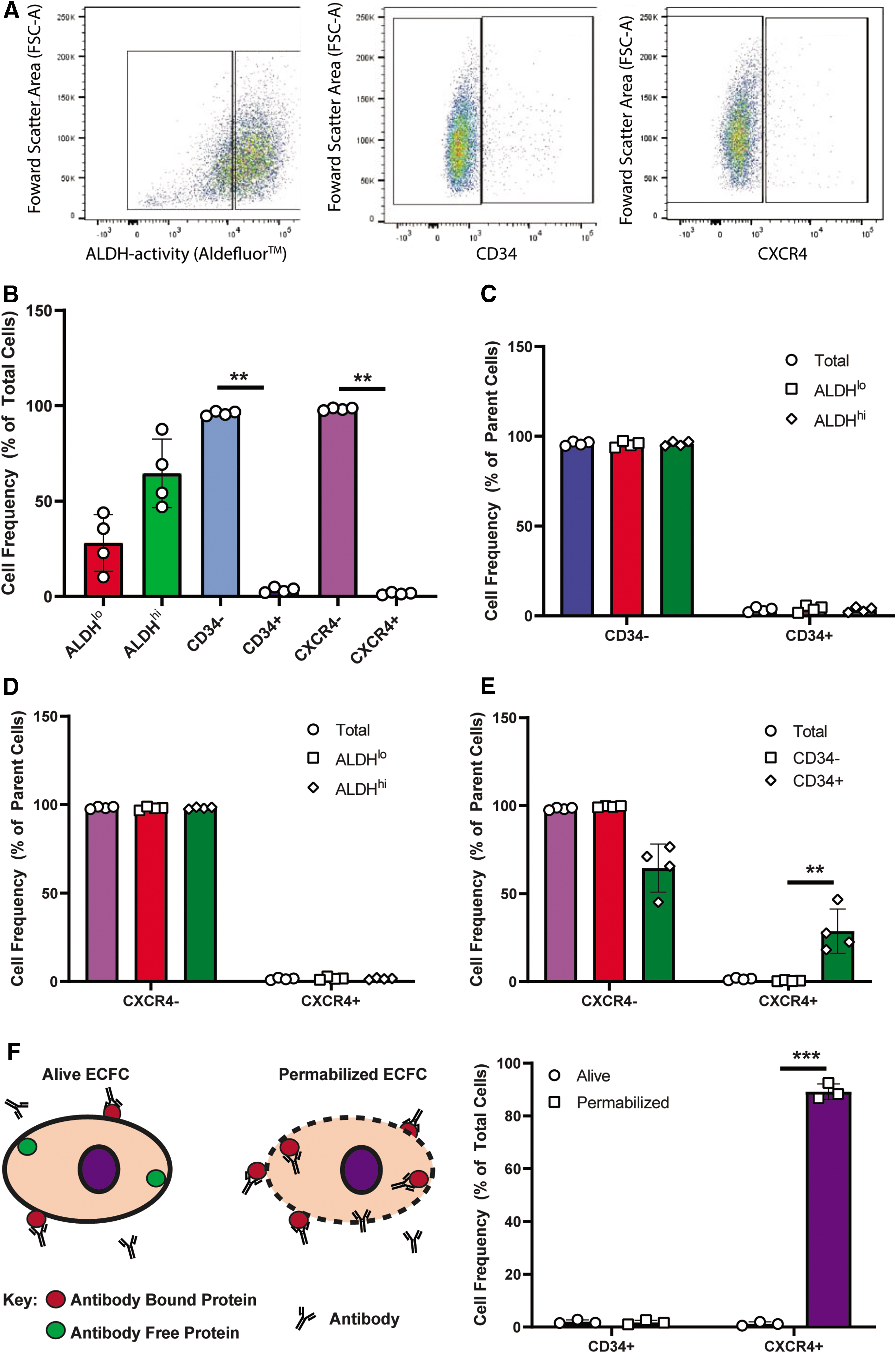
Putative progenitor marker expression assessment by flow cytometry on expanded UCB-ECFC. Expanded UCB-ECFC (at passage 4) were assessed for the expression of ALDH, CD34, and CXCR4 by flow cytometry to identify progenitor cell populations. Representative flow plots of ECFC expressing
UCB-derived hematopoietic progenitor cells with high ALDH activity often co-express CD34 [18,40,48,49]; however, CD34 or CXCR4 expression was not enriched within either ALDHlo or ALDHhi ECFC subset (Fig. 1C, D). In contrast, CXCR4 expression was significantly enriched within the CD34+ ECFC subset, compared to CD34− ECFC (Fig. 1E). Several reports have described dynamic internalization and externalization patterns of both CXCR4 and CD34; therefore, we determined whether ECFC possessed intracellular stores of CXCR4 or CD34 following fixation and permeabilization before flow cytometry. Notably, the vast majority of ECFC contained intracellular stores of CXCR4 (Fig. 1F). On the other hand, CD34 expression in ECFC appeared to be exclusive to the outer plasma membrane leaflet in ECFC (Fig. 1F).
Although low expression of CD34 was observed on culture-expanded ECFC, the CD34+ ECFC demonstrated increased CXCR4 expression, which may represent increased migratory capacity previously demonstrated for CXCR4+/CD34+ SCID-repopulating hematopoietic progenitor cells [50], and for putative circulating endothelial progenitor cells [51].
CD34−/CXCR4− ECFC demonstrated increased colony-forming capacity
To assess progenitor-like functions in vitro, ECFC at passage 4 were sorted and plated as single cells based on either ALDH activity or CD34 or CXCR4 expression (Fig. 2), and assessed for clonal colony formation. When acquired without selection using forward and side scatter properties, 1 in 3 (34%) of ECFC established colonies (Fig. 2). After single-cell deposition of ECFC based on low versus high ALDH activity, the clonogenic capacity of ALDHhi ECFC was equal to ALDHlo ECFC (Fig. 2A, B). In contrast, CD34− ECFC efficiently formed colonies (51.3% ± 5.85%), while CD34+ ECFC did not (16.3 ± 4.7) (Fig. 2C, D). Similarly, CXCR4− ECFC formed significantly more colonies than CXCR4+ ECFC (Fig. 2E, F). Although ALDH activity did not select for increased clonogenic capacity, the common CD34− and CXCR4− populations retained the potential for clonal expansion.
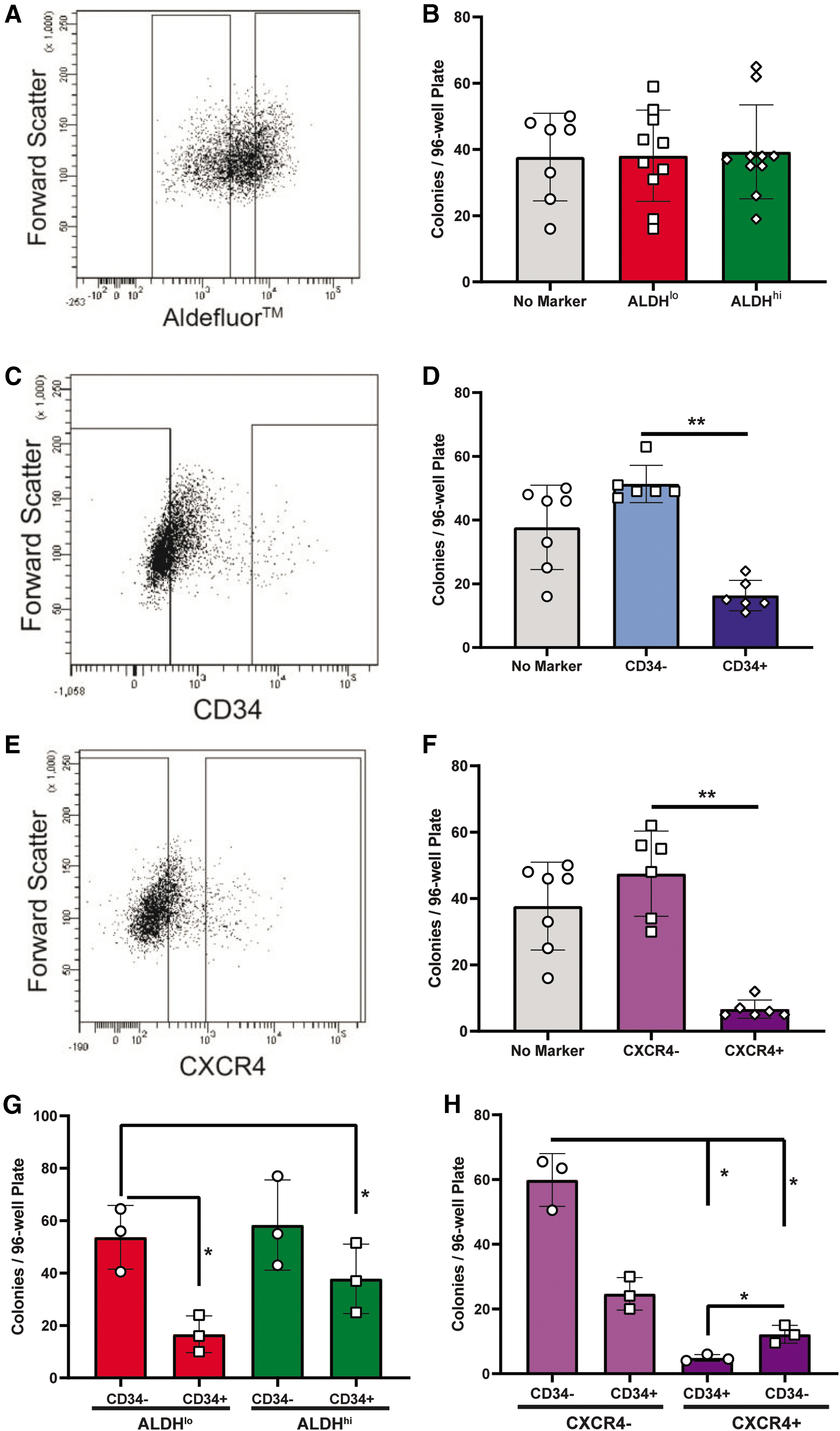
The absence of CD34 expression marked increased colony-forming capacity in UCB-ECFC. Colony-forming capacity of single ECFC purified by expression of
Next, we performed FACS using combinations of these aforementioned progenitor cell markers. Single-cell purification of ALDHloCD34+ and ALDHhiCD34+ ECFC consistently established less colonies than CD34− counterparts (Fig. 2G). Interestingly, CD34−/CXCR4− cells demonstrated the highest colony formation compared to any other combination selected for CD34 and CXCR4 expression (Fig. 2H). Collectively, these data suggest that ECFC expressing CD34 or CXCR4 have a reduced ability to establish colonies from single cells in vitro. Considering the notable stores of CXCR4 within ECFC compared to ECFC CD34 expression (Fig. 1F), we focused subsequent analyses on ECFC with or without CD34 expression.
Tube-forming capacity was increased by CD34− ECFC
To assess the relationship between progenitor marker expression and endothelial cell function in vitro, we assessed the tubule-forming capacity of ECFC subsets purified for ALDH activity or CD34 expression. Briefly, 120,000 purified CD34+/− or ALDHhi/lo ECFC were seeded in growth factor-reduced Geltrex in EBM or fully supplemented EGM-2. Similar to our previous results with clonogenic capacity, selection for low versus high ALDH activity did not alter the tubule-forming capacity of ECFC (Fig. 3A, B). In contrast, CD34− ECFC demonstrated increased tubule formation under minimal EBM or fully supplemented EGM conditions, compared to CD34+ ECFC (Fig. 3C, D).

ECFC tube-forming capacity was decreased in CD34+ ECFC. After ex vivo expansion, ECFC were sorted based on
CD34 expression on ECFC is reversible and elevated with increasing cell density
CD34 is enriched on founder ECFC populations; however, kinetics of CD34 expression during culture is less understood. Accordingly, we demonstrated that ECFC at passage 1 were highly enriched with CD34+ cells (Fig. 4A). However, CD34 expression rapidly decline during early passages and established a steady-state frequency during serial passaging (passage ≥3). We observed an initial trend toward accelerated growth kinetics within CD34− cells compared to CD34+ ECFC (Fig. 4B); however, equal cell numbers were obtained after 6 days of culture with both ECFC subsets. Cell density has been previously suggested to influence CD34 expression [7]. Consistent with these findings, we observed a 6.5-fold increase in CD34 expression when cells were cultured to high density (>90% confluent) before passage compared to when ECFC were harvested and passaged at <80% confluency (normal density) (Fig. 4C, D).
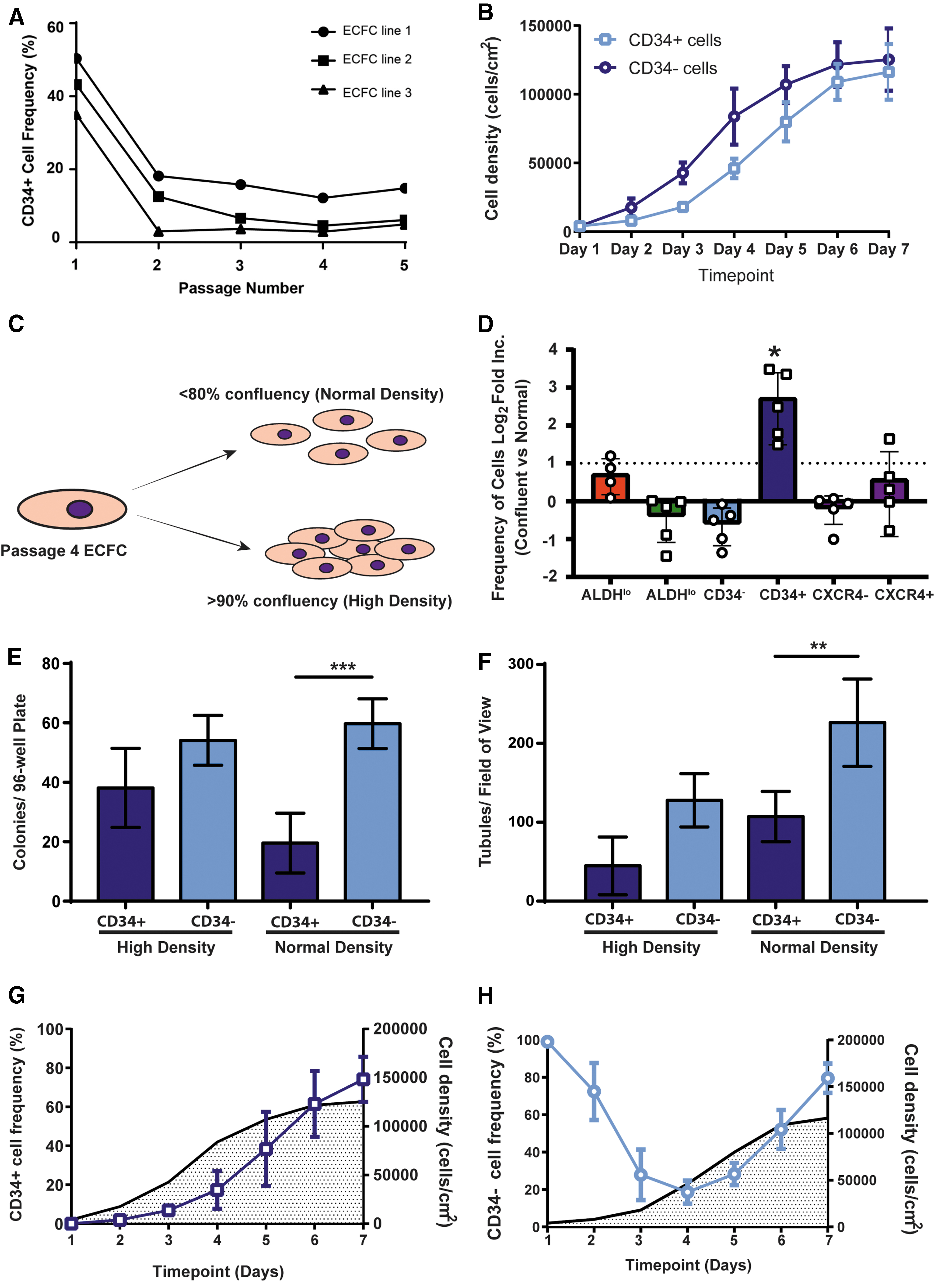
ECFC cultures showed temporal and cell density-dependent CD34 expression that impacted colony and tubule formation.
In contrast, other progenitor cell markers (ALDH and CXCR4) remained stable when ECFC reached confluence >90% (Fig. 4D). Next, we sought to assess the functional implications of increased CD34 expression with increased cell density. Interestingly, we did not observe differences in colony formation observed between CD34+ and CD34− ECFC after collection at high (>90%) cell density (Fig. 4E). Similarly, the ECFC subsets purified for CD34 after growth to high density did not demonstrate any difference in the ability to form tubes in vitro (Fig. 4F). These findings indicated that culture of ECFC to confluency increases CD34 expression and abrogated functional differences in colony and tubule formation in vitro.
It was unclear whether CD34 expression was turned on in CD34− cells under high cell density conditions. Accordingly, we investigated CD34 expression dynamics over 7 days alongside the expansion of purified CD34− (Fig. 4G) and CD34+ purified ECFC (Fig. 4H) at passage 4. Interestingly, CD34− cells consistently increased CD34 expression as cell density was increased, suggesting CD34− gave rise to CD34+ cells in vitro. The frequency of CD34+ ECFC gradually decreased to ∼20% over the first 96 h of culture (Fig. 4G), after which CD34 expression increased gradually as cell density increased. Collectively, these findings suggest that CD34 expression was reversible and cell density dependent; moreover, elevated CD34+ expression under high-density conditions does not impact endothelial functions in vitro.
Surface proteomics on CD34 selected ECFC subsets revealed enrichment of mature endothelial cell pathways on CD34+ cells
In an effort to identify other surface markers that may correlate with an endothelial progenitor-like state in vitro, we employed silica-bead capture followed by LC-MS/MS proteomics analyses. We identified >4,800 proteins captured on silica-beads (Fig. 5A), however ∼25% of identified proteins were associated with cell surface or plasma membrane annotations, referred to as surfaceome. Specifically, we identified >800 proteins associated with the surfaceome of either CD34− or CD34+ ECFC subsets (Fig. 5B). The surfaceome signature in itself did not distinguish either ECFC subset (Fig. 5C), as 935 proteins were commonly expressed on both subsets. Nonetheless, 23 and 10 proteins were exclusively detected in the surfaceome of CD34− versus CD34+ ECFC, respectively (Fig. 5D and Table 1). This included the exclusive detection of CXCR4 in CD34+ ECFC, which corroborated our earlier flow cytometry analysis (Fig. 1).
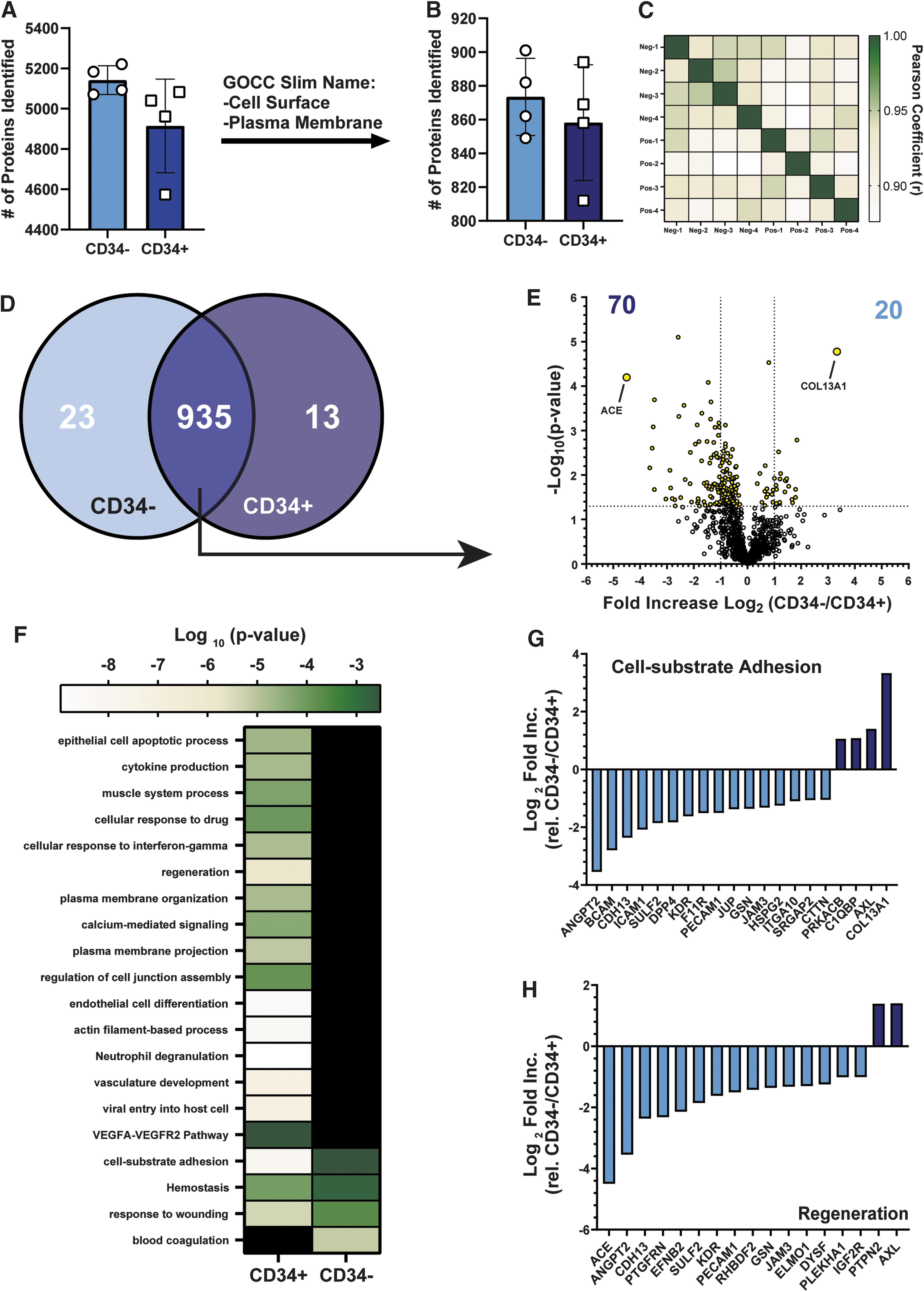
The surfaceome of CD34+ ECFC and CD34− ECFC. FACS-purified CD34− or CD34+ ECFC were temporarily seeded in EGM-2 to allow adherence.
Proteins Exclusively Identified on the Surfaceome of CD34− Versus CD34+ Endothelial Colony-Forming Cells
Furthermore, we identified 935 proteins that were detected in >50% of samples from each ECFC subset. Twenty and 70 proteins were differentially expressed twofold (P < 0.05) on either CD34− or CD34+ ECFC subsets, respectively (Fig. 5E). Comparison of these 90 proteins against the GO Biological Process and Reactome annotation databases identified an enrichment of proteins with functions toward endothelial cell development, vascular regeneration, VEGF-VEGFR2 signaling, and cell-substrate adhesion interactions on the CD34+ ECFC population (Fig. 5F).
AXL and COL13A1, associated with cell-substrate adhesion and/or regeneration, were elevated in the surfaceome of CD34− ECFC (Fig. 5G). In contrast, angiotensin-converting enzyme (ACE) and angiopoietin 2 (ANGPT2) were enriched within the surfaceome of CD34+ ECFC (Fig. 5H). Unfortunately, very few unfractioned ECFC demonstrated expression of COL13A1 or AXL on the external cell surface membrane, Cell fixation and permeabilization revealed that >90% of ECFC expressed these proteins on the inside of the cell (Supplementary Fig. S2). Further investigation is warranted to understand the relevance of exclusive or differentially expressed proteins between CD34− and CD34+ ECFC subsets.
Angiotensin-converting enzyme expression was enriched on CD34+ ECFC, but did not impact colony-forming capacity
Angiotensin-converting enzyme (ACE/CD143) was shown to have 27-fold greater expression on CD34+ ECFC compared to CD34− ECFC by surfaceome analyses. Therefore, we sought to determine whether ACE was could be used as a potential biomarker to select for more mature endothelial cell subset in vitro. When validating the frequency of ACE expression on ECFC subsets, ≈88% of CD34+ cells expressed ACE, while only 12.6% of CD34− ECFC expressed ACE by flow cytometry (Supplementary Fig. S3A). Therefore, we purified ECFC subsets based on CD143 expression, and used the single-cell deposition to assess colony formation. Both CD143+ and CD143− subsets demonstrated low, but equal colony-forming capacity (Supplementary Fig. S3B). Of the validated hits from the proteomic screen, the expression of CD143, COL13A1, and AXL did not provide surface useable markers for the identification of ECFC after culture with differential colony-forming function.
In vivo transplantation of CD34+/− ECFC subsets in Matrigel enabled the formation of vessel-like structures
To assess the function of CD34+ versus CD34− ECFC subsets to form tubes in vivo, CD34+ or CD34− ECFC were purified and injected subcutaneously into the flank of immunodeficient

Purified CD34+/− ECFC demonstrated equal vascularization in Matrigel plugs subcutaneously injected into immunodeficient mice. CD34+/− ECFC were purified and combined with mouse SMC within Matrigel and subcutaneously implanted into
Discussion
ECFC remain the gold standard for progenitor-like endothelial cells that possess potent proangiogenic properties in vitro and in vivo. Initially, ECFC can be efficiently propagated in culture; however, the proliferative capacity of expanded progeny is dampened as phenotypic and molecular transformations occur during serial passages, ultimately resulting in culture-induced senescence [6]. The ability to distinguish highly proliferative progenitor cells within heterogeneous ECFC cultures would potentially enable the isolation of more potent provascular cell subsets for vascular therapies.
In this study, we assessed a combination of known progenitor cell functions and cell surface marker expression during culture [52,53] to identify a subpopulation of ECFC with enhanced progenitor cell characteristics. Specifically, we employed in vitro assays to assess putative progenitor cell functions, including growth kinetics, single-cell colony formation, and tubule formation. Our findings suggest CD34 expression was variable and cell density dependent and may not be a strong marker for the selection of culture-expanded ECFC with progenitor cell-like characteristics. Similarly, CD34 expression did not segregate ECFC with vessel-like forming capacity in vivo.
In an attempt to identify additional markers associated with progenitor-like function, we also employed silica-bead plasma membrane capture followed by label-free proteomics analysis to compare the surfaceome of CD34− ECFC versus CD34+ ECFC. Membrane proteins highly expressed on CD34+ ECFC suggested a mature endothelial cell surface phenotype; however, subcutaneous implantation of CD34+ ECFC into
Hematopoietic stem/progenitor cells (HSPC) and EPC originate from a common hemangioblast during fetal development [26,29,54]; thus, shared phenotypic markers have been used to enrich HSPC and EPC cells from several blood-derived human sources [6,26,29,53 –55]. CD34 has been used for over a decade to identify HSPC [29] and a considerable number of studies have selected CD34+ cells before the establishment of ECFC cultures [7,29,31,55]. In response to injury, EPC expressing CXCR4 will home sites of ischemia by the SDF-1/CXCR4 axis and contribute to angiogenesis and vasculogenic processes [50 –52,56]. It also is well established that progenitor cells will rapidly differentiate or undergo dynamic phenotypic changes in response to the “unnatural” microenvironment of culture. As a result, it remains poorly understood whether cell surface markers on de novo isolated or uncultured EPC are retained on ECFC expanded in vitro.
In addition to surface markers, high ALDH activity was enriched in de novo isolated HSPC and EPC populations and loss of high ALDH activity coincided with HSPC differentiation in vitro [14,40]. We determined that ALDH activity did not correlate with the expression of CD34 or CXCR4, as previously observed by Ferreras et al. [31]. Although similar isolation techniques were utilized in this study, the cell source being from peripheral blood of an adult versus UCB in our study may explain the differences observed across these experiments. It is also interesting to note that each outgrowth endothelial cell (OEC) line had different CD34 expression after isolation, where some OEC expressed CD34 to a high degree and other OEC minimally expressed CD34 [31]. Furthermore, selection of ECFC using high versus low ALDH activity did not segregate ECFC with enhanced colony- or tubule-forming capacity in vitro.
Alternatively, CXCR4 and/or CD34 expression on expanded ECFC was uncommon (<5%) and served as a negative selection maker for colony- or tubule-forming capacity in vitro. Previous reports have demonstrated expression of CXCR4 and CD34 on the plasma membrane of cells is dynamic and can rapidly transition between intracellular and extracellular compartments [55 –57]. Thus, we assessed ECFC for intracellular stores of CXCR4 or CD34 after cell permeabilization. Indeed, CXCR4 expression was located within the intracellular compartment of ECFC, thus was deemed unsuitable as a selection maker due to the possible capacity for rapid externalization.
Although we were unable to demonstrate a correlation between ALDH activity and CXCR4 expression on the surface of ECFC, Tu et al. observed higher CXCR4 transcription within the ALDHlow cells [58], and induced CXCR4 expression on ALDHhigh ECFC augmented endothelial cell homing to the site of in vivo injury. Notably, the only cytokine constituent in the culture media used by Tu et al. was b-FGF, and provides a possible explanation for the differential observations with other studies using complete EGM-2 culture media. Guan et al. have previously demonstrated endothelial phenotype and function were determined by the media used for endothelial cell culture [59]. This carries large implications regarding differences in surface marker expression when comparing different studies using divergent media constituents and tissue sources for ECFC isolation.
To consolidate our findings with Tu et al., we demonstrated that ALDH expression did not enrich for cells with elevated CXCR4 expression. This implies that the capacity to externalize CXCR4 in the ALDHlow cellular fraction upon exogenous stimulation is more potent. For example, stimulation of L-selectin recipient cells by cell-extrinsic ligands drives CXCR4 externalization [60]. An important point to note is many studies involving the profiling of CXCR4 expression on ECFC by flow cytometry fail to profile the CXCR4 content within the cell. Based on our results, ECFC contain internal CXCR4 stores; thus, studies demonstrating increased CXCR4 by flow cytometry may only provide a snapshot of a cell's capacity to respond to external cues leading to CXCR4 externalization.
CD34 frequency was highest at passage 1 and decreased to a steady state by passage 3 for each ECFC line tested. Purified CD34+ ECFC at passage 4 formed significantly fewer colonies and tubules in vitro than CD34− ECFC, similar to studies by Ferreras et al. [31]. Conversely, other investigators have suggested that CD34+ ECFC have increased capacity to form tubule-like structures in vitro [7,8]. The divergence in these data from previous studies may be partially accounted for by the selection of tissue source (ie, placental vs. umbilical cord blood) and/or culture conditions. Specifically, it appears UCB-derived ECFC align better with peripheral blood-derived ECFC compared to placental-derived ECFC [7,31].
In agreement with Tasev et al., we demonstrate the expression of CD34 on ECFC was dependent on cell density and coincided with decreased tubule-forming capacity [7]. In contrast, we did not observe consistent changes to the expression of ALDH or CXCR4 after ECFC were grown to high-density confluency. To further demonstrate the dynamic nature of CD34 expression, we assessed ECFC density alongside the CD34+ cell frequency over the course of a 7-day culture using FACS-purified CD34− versus CD34+ ECFC. CD34− ECFC gradually increased CD34 expression over time as cell density increased. Similarly, FACS-purified CD34+ ECFC rapidly reduced CD34 expression early during culture and increased CD34 expression as cell density increased. We also observed that CD34− ECFC demonstrated accelerated growth kinetics compared to CD34+ ECFC.
Since CD34 expression on ECFC was dynamic and increased at high cell densities, we next assessed the impact of confluency on endothelial cell functions in vitro. Although CD34+ cell frequency increased at high cell densities, the statistical differences in clonogenicity and tubule forming observed between CD34+ and CD34− ECFC were abrogated when harvested at high-density conditions. Thus, increasing CD34 expression on ECFC appeared to be a response to cell-cell and/or cell-microenvironment cues that did not lead to functional changes in vitro. Collectively, these findings suggest a need to standardize culture conditions to prevent culture artifact, such as elevated CD34 expression. Moreover, we speculate CD34 expression reflects a transitional state of ECFC in vitro that warrants further investigation.
We employed silica-bead surface protein capture followed by label-free LC-MS/MS to identify prospective surface markers that correlate with progenitor functions. We identified >900 plasma membrane-associated proteins that were associated with cell-substrate adhesions, response to wounding, and regeneration. Specifically, we identified a collection of mature endothelial cell surface markers, such as CD31, ACE, VEGFR2, and ANGPT2, were significantly enriched on CD34+ ECFC. In contrast, proteins highly expressed on the surface of the CD34− ECFC did not possess obvious categorical association.
One protein of interest on CD34− ECFC was AXL, a protein tyrosine kinase that regulates cell proliferation and survival [61]. AXL is also postulated to attenuate immune responses [62], a function of progenitor cells in tissues with low turnover rates [63], a characteristic of endothelial cells in vivo [64]. Unfortunately, we were limited in this study by the lack of conjugated human-specific antibodies for flow cytometry on the outer leaflet of the membrane. For example, antibodies tested against AXL did not detect cell surface expression, instead considerable intracellular stores were detected. In addition, we validated the outer membrane expression of ACE/CD143 on ECFC was highly enriched within the CD34+ ECFC subpopulation. ACE is expressed on mature endothelial cells in vivo and acts as the rate-limiting enzyme in the renin-angiotensin pathway by processing angiotensinogen I to angiotensinogen II in blood plasma [65]. We speculated ACE represented a promising marker to predict why a large proportion of CD34+ cells was unable to make colonies or lacked tubule formation function in vitro. In addition, the expression of ACE has been previously demonstrated to be linked to cell density [66]. Unfortunately, sorting for ACE+ versus ACE− ECFC revealed no differences in the generation of colonies after single-cell deposition.
Our surfaceome analysis identified a significant enrichment of Mucolipin 1 (MCOLN1), a member of the transient receptor potential cation channel family, on CD34− ECFC. This membrane-associated protein primarily plays a role in calcium release from late endosomes and lysosome vesicles. Previous studies have implicated the mobilization of intracellular Ca2+ stores during CXCR4 signaling to enhance ECFC migration through PI3K/AKT signaling [67]. Moreover, endothelial cell proliferation has been linked to the mobilization of intracellular Ca2+ [68,69]. Indeed, our enrichment analyses indicated a significant enrichment of proteins involved with calcium-mediated signaling. Thus, it is tempting to speculate that the increased presence of MCOLN1 on CD34− ECFC could, in part, drive the clonogenic capacity and tube-forming functions of ECFC.
Following in vitro analyses, we began to speculate that CD34 expression may simply reflect the microenvironment of culture and cellular state during expansion. Therefore, CD34− and CD34+ ECFC were transplanted subcutaneously within Matrigel plugs to assess endothelial cell function in vivo. Surprisingly, CD34− and CD34+ ECFC generated vessel-like structures and occasionally appeared to integrate with murine endothelial cells. Thus, ECFC vessel-forming function retained vessel forming in vivo independent of CD34 expression.
In summary, our data suggest that CD34 expression on ECFC after culture expansion in vitro did not mark functionally enhanced EPC. Endothelial progenitor cell phenotype and functional capacity were altered after establishment in vitro and was dependent on culture density [11,70]. Furthermore, CD34+ and CD34− ECFC subsets were functionally equivalent in vivo, despite measurable functional differences in colony and tube formation in vitro. These data suggest that surrogate in vitro analyses need to be supported by in vivo studies to assess the function of endothelial progenitor cells relevant to applications of regenerative medicine. Collectively, standardization for the isolation, culture, and phenotype should be assessed before potential use in vascular regenerative therapies.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by an operating grant from the Canadian Institute of Health Research (CIHR) (MOP#378189) and Juvenile Diabetes Research Foundation (2-SRA-2015-60-Q-R) awarded to DAH, in addition to NSERC discovery grant and Canadian Foundation for innovation grant awarded to GAJ.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.