
Abstract
Mesenchymal stromal cells (MSCs) have become a promising treatment for inflammation-related diseases, and their therapeutic efficacy mainly depends on crosstalk between MSCs and inflammation. However, methods to improve the immunosuppressive efficiency of MSCs in different diseases still need to be developed. In this study, we investigated whether preconditioning MSCs with a disease-related inflammatory cytokine could increase their immunosuppressive properties and improve therapeutic efficacy. In a contact hypersensitivity (CHS) mouse model, inflammatory profile screening revealed that among all tested cytokines, monocyte chemotactic protein-1 (MCP-1) exhibited the most significantly increased level in the local microenvironment. As expected, MSCs preconditioned with MCP-1 (P-MSCs) exhibited an enhanced ability to downregulate proinflammatory cytokine secretion, induce regulatory T cells, inhibit T cell proliferation, and polarize M2-type macrophages. In vivo experiments showed that P-MSCs alleviated ear swelling and local proinflammatory cytokine production more effectively than control MSCs. Mechanistically, MCP-1 could significantly activate the signal transducer and activator of transcription 3 (STAT3) signaling pathway and induce the expression of cyclooxygenase-2 (COX2) and prostaglandin E2 (PGE2) in MSCs. STAT3 inhibitor reversed the MCP-1-mediated enhancing of their immunosuppressive ability. Collectively, our findings demonstrate that CHS-related MCP-1 preconditioning enhanced the immunomodulatory effects of MSCs and improved their therapeutic efficacy in CHS. Enhancing the immunosuppressive efficacy of MSCs by preconditioning with certain disease-related inflammatory cytokines may provide a new strategy for MSC-based therapies for inflammatory diseases.
Introduction
Mesenchymal stromal cells (MSCs) have been revealed to be safe and effective in the treatment of a variety of inflammatory and autoimmune diseases because of their distinct immunomodulatory properties [1,2]. They exert their activities by influencing the proliferation, recruitment, function, and fate of both innate and adaptive immune cells, including T cells [3,4], B cells [5], dendritic cells (DCs) [6], and natural killer (NK) cells [7]. However, although numerous preclinical and clinical studies have shown that MSCs can be therapeutically relevant in various inflammatory diseases, many obstacles still limit the application of stem cell therapy. The immunomodulatory capabilities of MSCs are usually mediated by inflammatory cytokines and may vary depending on the levels and types of inflammatory cytokines.
Allergic contact dermatitis (ACD), in which epidermal and dermal cells are exposed to exogenous haptens, is one of the most common antigen-specific inflammatory skin diseases [8,9]. ACD and the murine model of ACD, termed contact hypersensitivity (CHS), are typical T cell-mediated inflammatory disorders, and T lymphocytes are likely the predominant effector population [10]. Therefore, it should be possible to prevent ACD or CHS responses by mainly targeting T cells. Recent studies have also demonstrated that MSCs are becoming a promising therapeutic option for CHS treatment [11 –13].
Monocyte chemotactic protein-1 (MCP-1) is the initial chemokine produced by activated resident skin cells such as keratinocytes, fibroblasts, and endothelial cells, and its production precedes the infiltration of monocytes and lymphocytes during the elicitation phase of ACD or CHS [14]. Many studies have indicated that MCP-1 not only serves as the key inflammatory factor in CHS but also mediates the development of CHS. Transgenic mice with increased expression of MCP-1 exhibit enhanced inflammatory responses during CHS and relatively strong infiltration of DCs into the skin [15], and downregulation of MCP-1 expression using an RNA interference strategy attenuates hapten-induced CHS [16]. Moreover, CCR2−/− mice display attenuated CHS skin inflammation [17], and a CCR2 antagonist decreases site-directed itch- and pain-like behaviors [18].
In this study, MCP-1 expression was robustly increased in our CHS mouse model and was used to precondition MSCs. The aim of this study was to determine whether MCP-1-preconditioned MSCs (P-MSCs) exhibit improved therapeutic effects on MCP-1-related diseases, such as mouse models of CHS.
Materials and Methods
Cell isolation, purification, and culture
MSCs and peripheral blood mononuclear cells (PBMCs) were obtained from healthy volunteers after informed consent was obtained. The study protocol was approval by the institutional clinical ethics committee of the Third Affiliated Hospital, Sun Yat-sen University, in compliance with the provisions of the current Declaration of Helsinki Principles and Good Clinical Practice guidelines. MSCs were isolated from the mononuclear cell fraction of bone marrow aspirates by Ficoll–Paque density gradient centrifugation and maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U/mL penicillin, and 100 U/mL streptomycin. In all experiments, the MSCs used were at passages 5–8. PBMCs were isolated by Ficoll–Paque density gradient centrifugation.
Stimuli and inhibitors
MSCs were activated with 100 ng/mL human recombinant MCP-1 protein (PeproTech) (P-MSCs) for 48 h. The cells were washed three times to remove residual recombinant protein before use in subsequent experiments. Signal transducer and activator of transcription 3 (STAT3) signaling was inhibited by adding HO-3867 (10 nM; PeproTech) to the culture medium. A similar amount of DMSO was added to a control culture.
Coculture assays
CD3+ T cells were purified with human CD3 microbeads (Miltenyi Biotec). The purity of the isolated CD3+ T cells was >95%, as confirmed by flow cytometric analysis using an anti-CD3 antibody (Ab) (BD Biosciences). In all coculture experiments, allogeneic MSCs were used, and CD3+ T lymphocytes from one donor were cocultured with MSCs from three different donors. MSCs (1 × 105) were cultured in 24-well plates for 1 day before treatment with MCP-1. CD3+ T lymphocytes were added to P-MSCs or control MSCs (5:1) and incubated for 3 days. CD3+ T cells cultured alone were used as controls.
Cytokine assays
T cells were cultured with or without MSCs (5:1 ratio) for 3 days. During the last 6 h of incubation, PMA (50 ng/mL) and ionomycin (500 ng/mL) were added to the culture system, and brefeldin A (BFA; 10 μg/mL) was used to inhibit cytokine secretion (all from Sigma-Aldrich, St. Louis, MO). Interferon γ (IFN-γ) and tumor necrosis factor α (TNF-α; BD Biosciences) were analyzed by flow cytometry.
Treg assays
Purified CD4+ T cells were cultured with or without MSCs (5:1 ratio). At day 6 of coculture, CD25+FOXP3+ Treg cells were assessed by flow cytometry.
Proliferation assays
Purified CD3+ T cells were stained with 5 mM 5-(and-6)-carboxyfluorescein diacetate succinimidyl ester (CFSE, CellTrace; Invitrogen, Carlsbad,
Coculture of macrophages and MSCs
THP-1 cells (ATCC) were stimulated with PMA (10 ng/mL) (Sigma-Aldrich; P8139) for 48 h to induce their differentiation into macrophages. THP-1-derived macrophages were cocultured with MSCs and P-MSCs in the presence of 1 μg/mL LPS. At day 3 of coculture, the percentage of CD206+ macrophages was assessed by flow cytometry.
CHS model
BALB/c mice (female, 6–8 weeks old) were used. CHS reactions were induced in mice with dinitrofluorobenzene (DNFB). In brief, 25 μL of 0.5% DNFB (Sigma-Aldrich) in acetone/olive oil (4:1) was applied evenly to a shaved hind flank for 2 consecutive days. On day 5, the sensitized mice were challenged on the right ear by application of 10 μL of 0.25% DNFB in acetone/olive oil (4:1). An identical amount of acetone/olive oil (4:1) was administered to the left ear. MSCs (1 × 106) were intravenously injected through the tail vein on the day of the challenge. The control group received phosphate-buffered saline (PBS). Ear thickness was measured using the Soft Touch Micrometer (CLM1–15QM, Mitutoyo, Japan), and ear swelling was calculated as the difference in ear thickness before the first application and 24, 48, or 72 h after the last DNFB challenge. Sample size was calculated based on the resource equation approach, which is widely used to determine the sample size in animal studies by calculating the minimum and maximum numbers of animals required [19]. We set the sample size per group to 5 in this study. Five mice in each group were killed on day 2 after challenge. Ear samples were harvested; one was used for HE, and the other four samples were homogenized to detect cytokines. All experiments were carried out in compliance with the protocols approved by the Third Affiliated Hospital of Sun Yat-sen University Animal Ethics Committee.
Enzyme-linked immunosorbent assay
In total, 2 × 105 MSCs or 2 × 105 P-MSCs were seeded in one well of a six-well plate. After 24 h, the cells were rinsed with PBS, and 1 mL of serum-free minimum essential medium eagle - alpha modification (α-MEM), medium (Thermo Fisher Scientific, Waltham, MA) was added. The medium was collected 48 h later and used immediately or stored at −80°C. Ears were homogenized in PBS with 0.5% Triton X-100 (Sigma-Aldrich) and a protease inhibitor cocktail. Lysates were incubated at 4°C for 30 min, followed by centrifugation at 14,000 rpm for 10 min at 4°C. The supernatant was collected, and the protein concentration was measured with a Bradford protein assay system (Bio-Rad). The enzyme-linked immunosorbent assay (ELISA) kits used were mouse interleukin 6 (IL-6), prostaglandin E2 (PGE2), and tumor growth factor β (TGF-β) ELISA Kits (Thermo Fisher Scientific), and an ear homogenate inflammatory factor assay was performed using a BD cytometric bead array (CBA) mouse inflammation kit.
MSC proliferation assay
MSCs were resuspended in complete L-DMEM and seeded in a 96-well plate at 103 cells per well. The cells were evaluated using a CCK8 kit at each indicated time point over 7 days. MSC CFSE staining was performed according to the protocol of the Cell Trace™ CFSE kits (Thermo Fisher).
STAT3 and COX2 analysis by flow cytometry
MSCs and P-MSCs were fixed (BD Cytofix™ Fixation Buffer, Cat. No. 554655) for 10 min at 37°C, then permeabilized (BD Phosflow™ Perm Buffer III, Cat. No. 558050) on ice for 30 min, and then stained with either PE mouse IgG1, κ Isotype Control (Cat. No. 559320) or PE mouse anti-STAT3. For data analysis, MSCs were selected by their scatter profile. Flow cytometry was performed on a BD FACS LSRII flow cytometry system. Cyclooxygenase-2 (COX2) staining experiments were performed according to the intracellular staining protocol.
Statistical analysis
All results are expressed as the mean ± SD. Statistical comparisons were made using a two-tailed Student's t test (between two groups) or one-way analysis of variance (ANOVA; for multigroup comparisons). Changes in ear thickness were compared using a repeated-measure ANOVA. P < 0.05 was considered significant. Analysis and graphing were performed using the Prism 5.01 software package (GraphPad, San Diego, CA).
Results
MCP-1 expression significantly increased in inflamed ears after DNFB challenge
To analyze the alterations in local inflammatory factors in CHS mice, we induced CHS in mice using DNFB and monitored ear swelling at 0, 24, 48, and 72 h after elicitation. Compared with that in the control group, the ear thickness in the experimental group increased progressively, reaching a peak at 48 h postchallenge before decreasing slightly at 72 h (Fig. 1A). Histopathological examination revealed ear swelling and remarkable cellular infiltration at 48 h postchallenge in the DNFB-treated mouse ears (Fig. 1B). Next, we detected the levels of cytokines in inflamed ear homogenates collected at 48 h postchallenge using a BD™ CBA inflammation kit. As given in Fig. 1C, MCP-1, IFN-γ, TNF-α, IL-6, and IL-10 levels were very low in control ears but significantly increased in DNFB-treated ears. The MCP-1 level was almost 100 times higher in the inflamed ears than in the control ears and exhibited the most significant alteration among the tested proinflammatory cytokines. Because of the relatively abundant expression of MCP-1 in the inflamed ear in CHS mice, we investigated the potential role of MCP-1 in preconditioned MSCs and its effects on MSC-mediated immunomodulation.
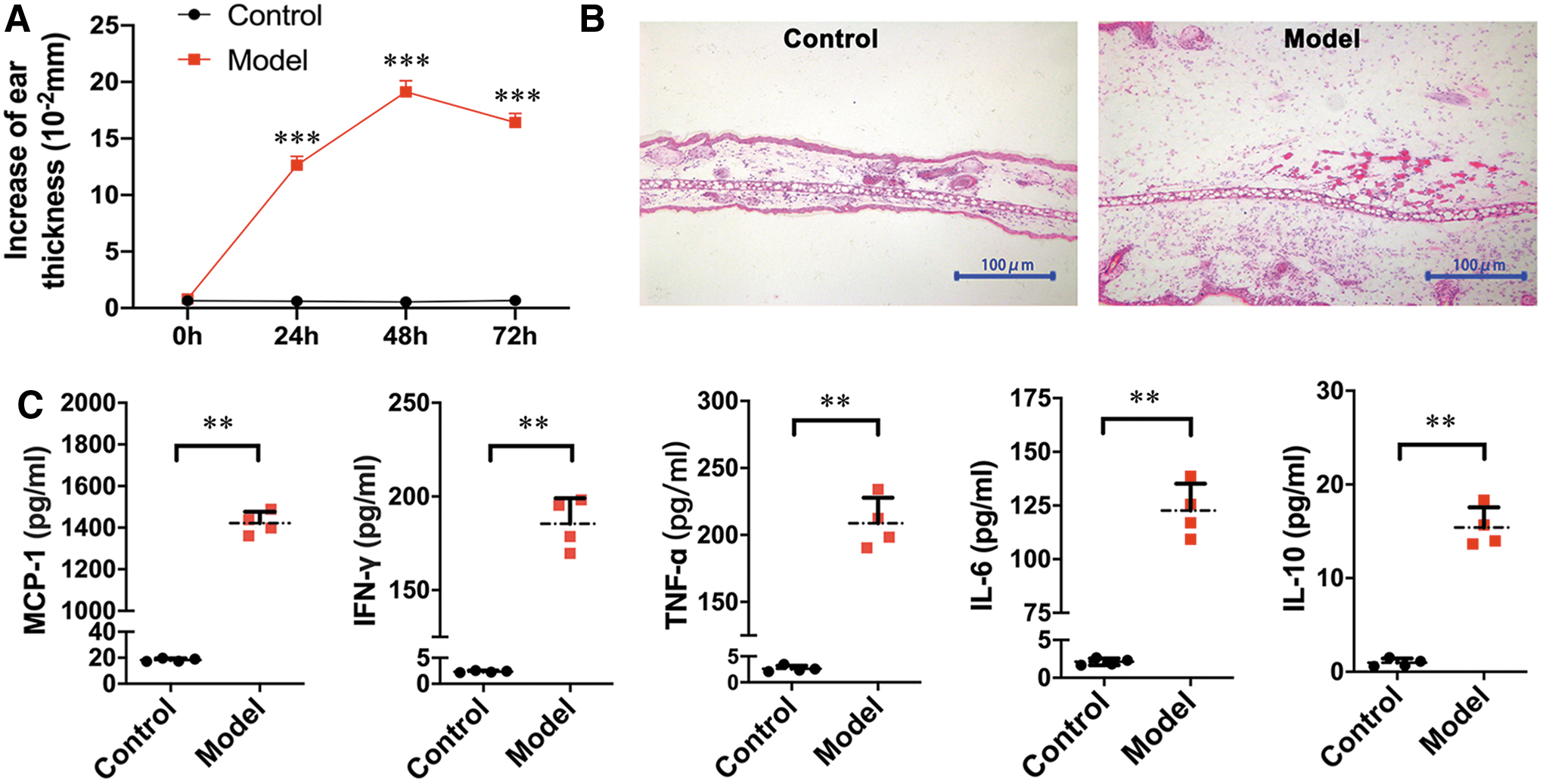
Inflammatory profiles changed in swollen ear tissue after DNFB challenge.
MCP-1 had no effects on the intrinsic characteristics of MSCs
First, we investigated the effects of MCP-1 on the characteristics of MSCs. We primed MSCs with MCP-1 (100 ng/mL, the indicated concentration), and then MSC phenotypic markers and differentiation potential were evaluated. As given in Supplementary Fig. S1, P-MSCs expressed the same panel of surface markers, including CD73, CD105, CD90, and HLA-ABC, as control MSCs and did not express CD34, CD45, HLA-DR, CD80, and CD86. In addition, we also investigated MCP-1-mediated effects on MSC apoptosis and proliferation. Apoptosis staining analysis showed that few cells were apoptotic during culture in vitro, and pretreatment with MCP-1 did not affect apoptosis in MSCs (Fig. 2A, B). In addition, a proliferation array showed that MCP-1 had effects on the proliferative ability of MSCs, as the proliferative ability of treated cells differed from that of untreated MSCs (Fig. 2C–E).
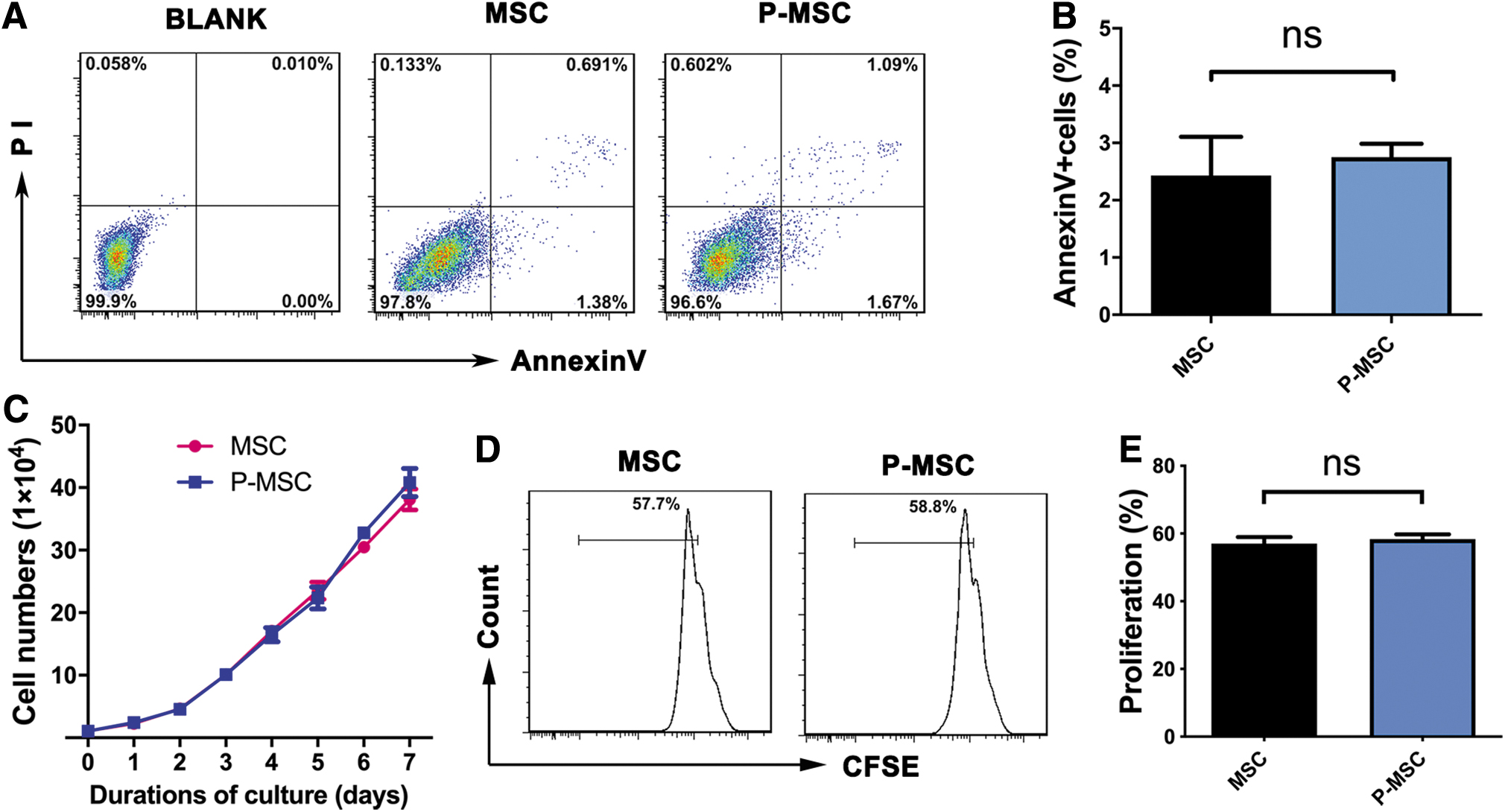
The viability of MSCs was not obviously influenced by preconditioning with MCP-1. Flow cytometry plots of Annexin V and PI staining of MSCs with or without preconditioning with MCP-1.
MCP-1 preconditioning enhanced the inhibitory effect of MSCs on T cell cytokine secretion
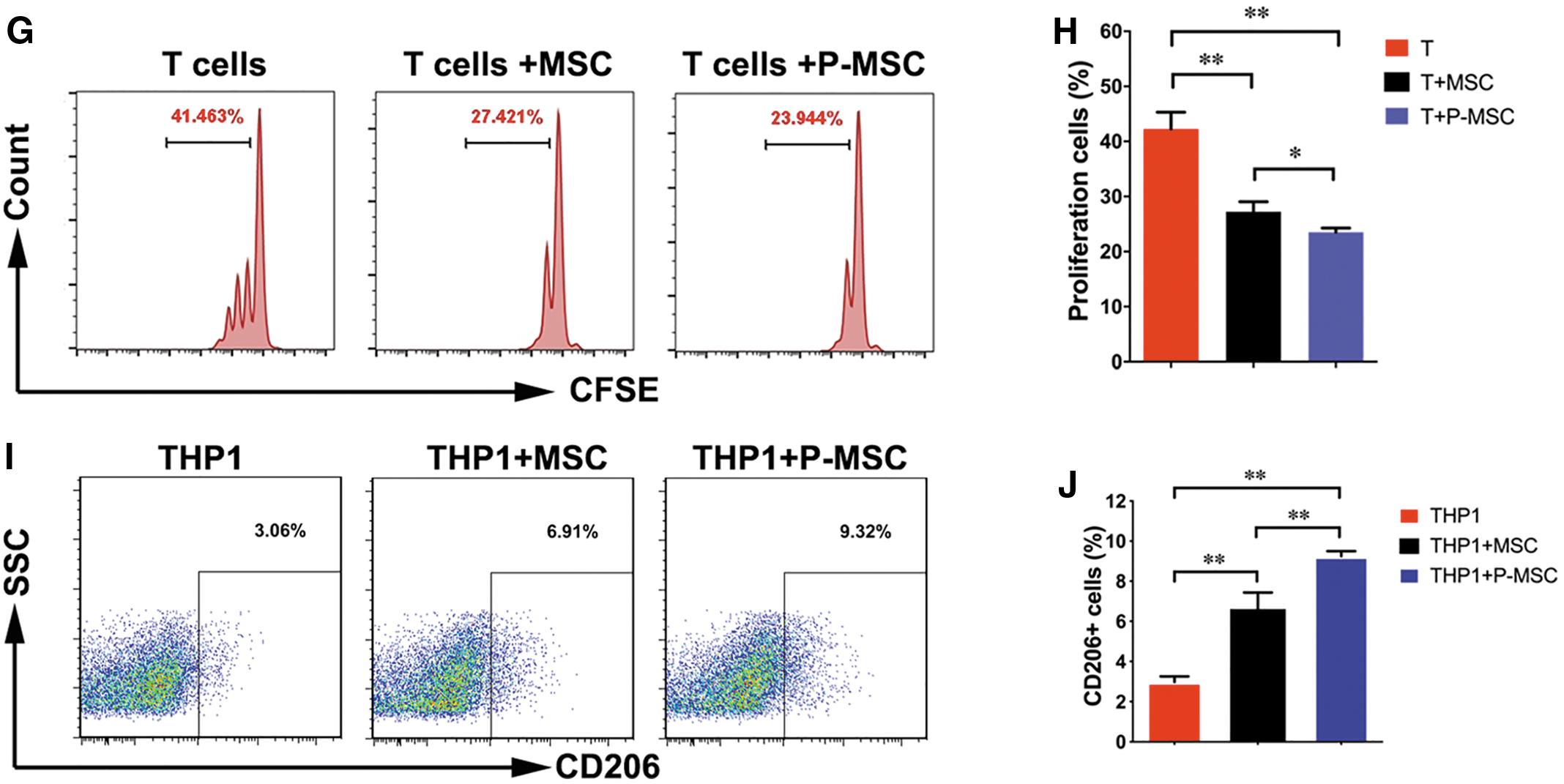
It is well known that MSCs can inhibit the T cell-mediated immune response. To evaluate the immunomodulatory ability of P-MSCs, we established an in vitro coculture system containing MSCs and T cells. When T cells were cultured alone, many IFN-γ-producing T cells and TNF-α-producing T cells were detected. Compared with the T cells cultured alone, T cells cocultured with MSCs showed significantly decreased frequencies of IFN-γ-producing T cells and TNF-α-producing T cells (Fig. 3A–D). Of importance, the secretion of IFN-γ and TNF-α by T cells was dramatically downregulated by P-MSCs compared with control MSCs (Fig. 3A–D). Next, we performed Treg induction and T cell proliferation experiments and demonstrated that P-MSCs have more potency than MSCs in inducing regulatory T cells (Fig. 3E, F) and inhibiting T cell proliferation (Fig. 3G, H). Macrophages are an important regulator of inflammation and the hypersensitivity reaction. We also found that P-MSCs polarized M2-type CD206+ macrophages more efficiently than MSCs (Fig. 3I–J). Overall, MCP-1 enhanced the immunomodulatory ability of MSCs to downregulate the T cell- and macrophage-mediated inflammatory response.
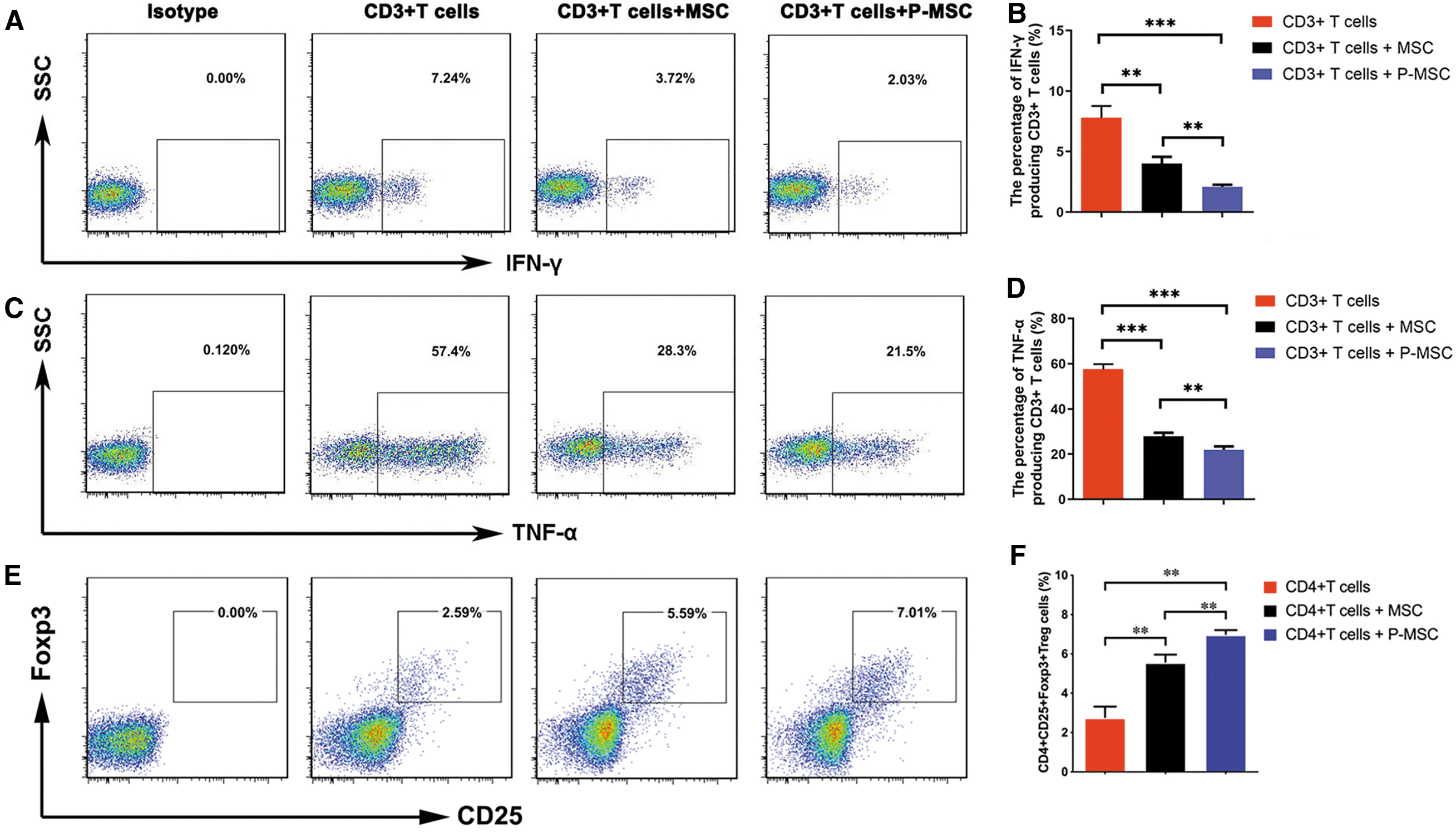
MCP-1 enhanced the ability of MSCs to inhibit T cell cytokine secretion. CD3+ T cells were cocultured with MSCs for 3 days, and flow cytometry was used to assess IFN-γ and TNF-α production.
P-MSCs dramatically alleviated CHS
To evaluate the roles of P-MSCs in the T cell-mediated inflammatory response in vivo, we constructed a DNFB-induced murine CHS model, which is a well-known T cell-mediated delayed-type hypersensitivity model. Next, control MSCs or P-MSCs were administered to CHS mice, and ear swelling was observed at 0, 24, 48, and 72 h postchallenge. As given in Fig. 4, the ear thickness of the CHS mice increased progressively, with the most obvious swelling occurring at 48 h. MSCs could reduce ear swelling (Fig. 4A, B). In addition, histopathological results showed that MSCs significantly reduced ear swelling and the infiltration of inflammatory cells into inflamed ears (Fig. 4C). Of importance, P-MSCs alleviated tissue swelling, and inflammatory cell infiltration was ameliorated with P-MSCs compared with control MSCs. Furthermore, we detected the levels of inflammatory cytokines within inflamed ear homogenates collected at 48 h postchallenge. We found that MSCs significantly reduced the levels of proinflammatory factors (MCP-1, IFN-γ, TNF-α, and IL-6) and increased the level of an inhibitory factor (IL-10). P-MSCs showed a stronger ability to downregulate inflammatory cytokine expression than did control MSCs (Fig. 4D). Overall, P-MSCs dramatically alleviated CHS, mainly owing to their outstanding ability to downregulate the inflammatory response.
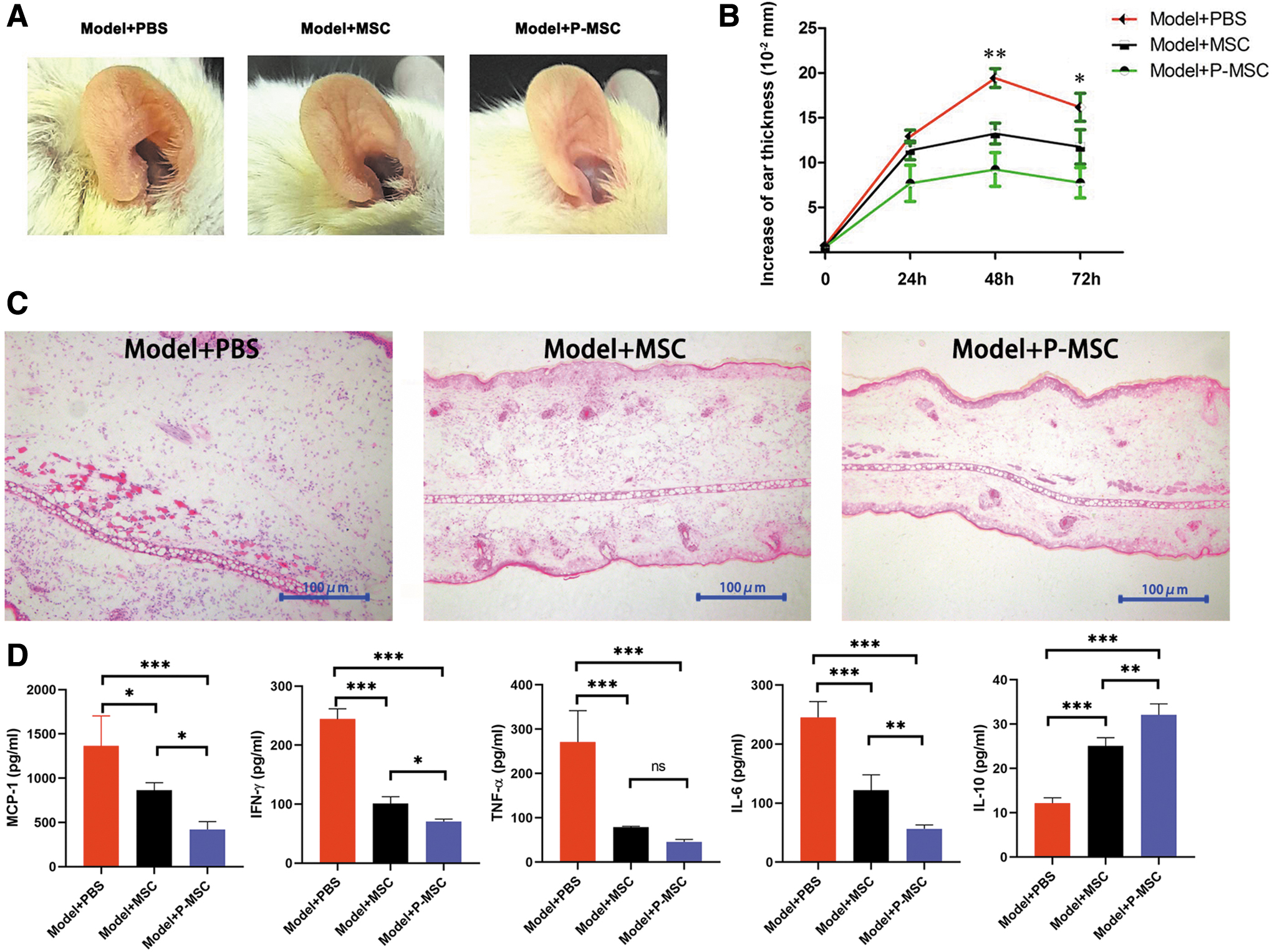
Preconditioning MSCs alleviated CHS.
MCP-1 enhanced MSC-mediated immunomodulatory functions by activating the cyclooxygenase-2-prostaglandin E2/STAT3 pathway
Because the STAT3 pathway was previously reported to be one of the important downstream signal transduction pathways of MCP-1 and STAT3 has been shown to be involved in augmenting the ability of MSCs to inhibit the T cell response and control delayed-type hypersensitivity [20], we explored whether STAT3 participates in the ability of MCP-1 to affect the plasticity of MSCs. First, we analyzed the activity of the STAT3 pathway. We found that MCP-1 preconditioning could increase the level of STAT3 phosphorylation in MSCs (Fig. 5A, B) while enhancing the ability of MSCs to inhibit T cell secretion of IFN-γ and TNF-α (Fig. 5C, D). To further confirm the role of STAT3 in augmenting the immunomodulatory ability of MSCs, H0–3867, a selective STAT3 inhibitor, was added to the MCP-1 preconditioning system for MSCs. We found that STAT3 inhibitor treatment reversed the effects of MCP-1 preconditioning on MSCs (P-MSCs) (Fig. 5C, D), suggesting that STAT3 activation was important in the ability of MCP-1 to augment the immunomodulatory ability of MSCs. A previous study reported that STAT3 and COX2 can form a positive feedback loop [21]. Moreover, COX2 activation induces PGE2, which is one of the immunosuppressive mediators secreted by inflammatory stimulus-primed MSCs. Thus, we speculated that MCP-1 augments the immunomodulatory ability of MSCs by regulating PGE2. To test this hypothesis, we analyzed changes in COX2 and PGE2 expression in MSCs after MCP-1 treatment. The results showed that MCP-1 could significantly induce the expression of COX2 in MSCs (Fig. 5E–G). Of importance, ELISA results showed that the secretion of PGE2 was dramatically increased in P-MSCs (Fig. 5G). Overall, MCP-1 enhanced MSC immunomodulatory functions mainly by activating the COX2-PGE2/STAT3 pathway.
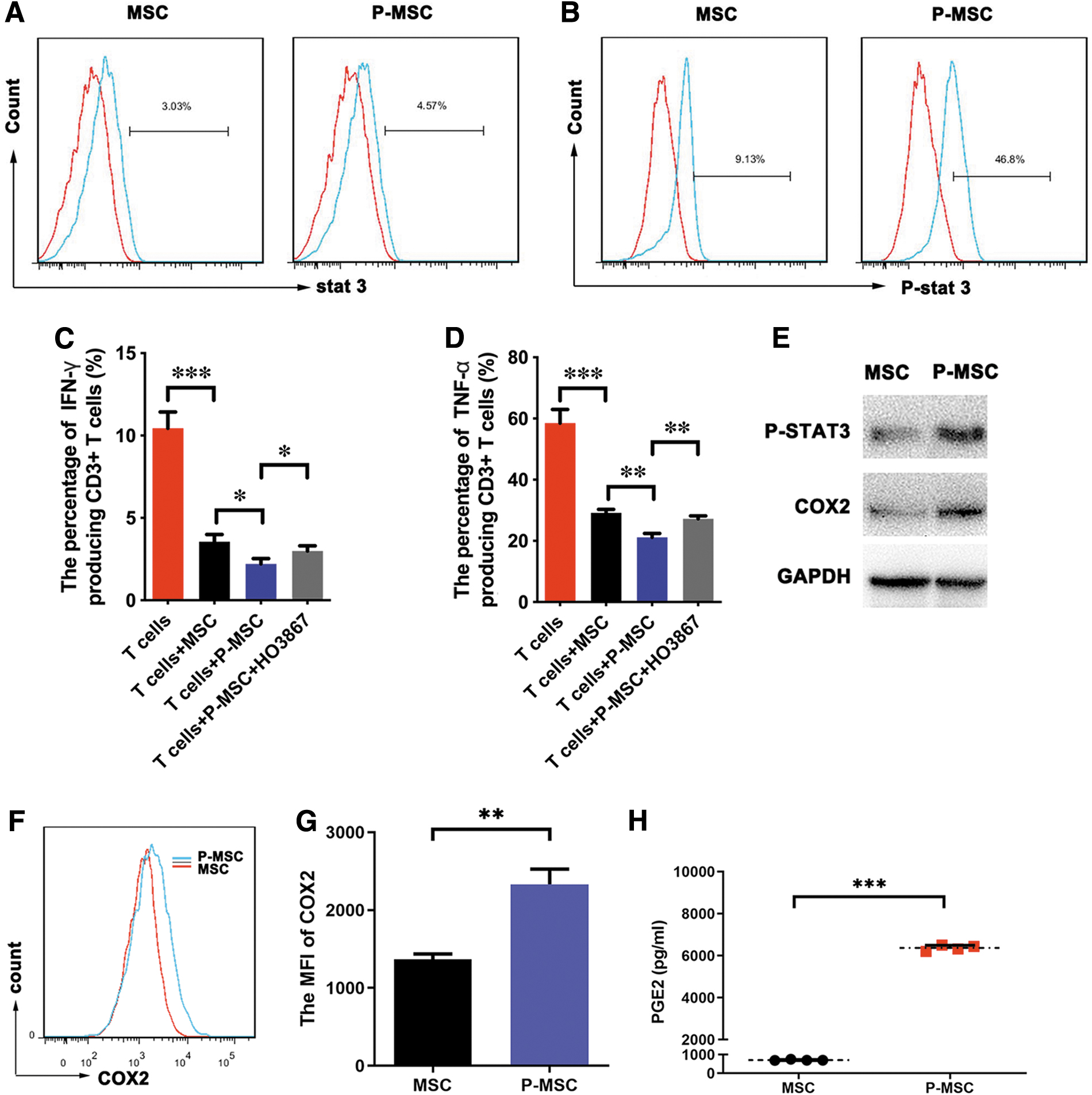
MCP-1 enhanced MSC immunomodulatory function by activating STAT3 signaling.
Discussion
In this study, we identified MCP-1 as being highly expressed in inflamed ears in our CHS model and found that MCP-1 preconditioning of MSCs enhanced the ability of MSCs to inhibit T cell secretion of the cytokines IFN-γ and TNF-α. Moreover, MCP-1 enhanced MSC-mediated immunomodulation by activating the COX2-PGE2/STAT3 pathway. Indeed, we found that systemic infusion of P-MSCs dramatically attenuated CHS in our mouse model.
MCP-1 and its receptor CCR2 are involved in the pathophysiology of ACD. They are widely expressed in the immune system, and the synthesis of CCL2 and CCR2 can be significantly upregulated in primary sensory neurons under pathological conditions [18]. MCP-1 is the dominant monocyte/macrophage attractant expressed during the elicitation phase of CHS [14,22]. In this study, we found that the expression of MCP-1 was significantly increased (by >50-fold) in vivo, which is consistent with some previous evidence [14,18].
MSCs have the inherent function of repairing injured tissue and significantly suppress T cell proliferation. However, in patients with ACD or other inflammatory diseases, the levels of many inflammatory mediators are increased, which are common manifestations of the inflammatory response and potentially affect hazardous cell engraftment, viability, and the overall therapeutic response to MSC therapy [23]. It is important to enhance MSC functions and ultimately improve therapeutic outcomes by evaluating potential techniques. One approach that has been investigated for preparing cells to treat these physiological insults is in vitro preconditioning. Preconditioning approaches have been successfully used to augment the therapeutic function of MSCs.
There are no previous reports on P-MSC characterization or P-MSC immunomodulatory capacity, and it is unclear whether the therapeutic efficacy of MSCs during CHS is related to MCP-1. MCP-1-deficient MSCs do not inhibit IFN-γ production by T cells [24]. In this study, we demonstrated that MCP-1 preconditioning had no effects on MSC apoptosis or proliferation. Furthermore, MCP-1 enhanced the MSC-mediated inhibition of T cell cytokine secretion in vitro. These results suggest that activation of the MCP-1 signaling pathway in MSCs enhances their therapeutic efficacy in a CHS mouse model.
MSCs are thought to inhibit T cell functions through two different mechanisms: direct cell–cell interactions and paracrine activity, which means the production of soluble mediators. The soluble immunosuppressive factors produced by MSCs include IL-10, nitric oxide (NO), TGF-β, PGE2, and indoleamine 2,3-dioxygenase (IDO), all of which can inhibit the functions of major immune cell populations [25 –28]. We also found that MCP-1 preconditioning could significantly elevate COX2 and PGE2 expression in MSCs. COX2 is a key enzyme in the synthesis of PGE2, and MSC-derived PGE2 is involved in skewing the inflammatory environment toward an anti-inflammatory profile, altering the cytokine secretion profiles of T cell subsets (Th1 cells, Th2 cells, or Tregs) [29]. Altogether, these data suggest that MCP-1 signaling enhances the ability of MSCs to produce COX2 and PGE2 and enhanced their immunosuppressive function.
MCP-1 exerts its effects by binding to G-protein-coupled receptors, CCR 2 and CCR 11 receptors. These receptors, once activated, trigger a set of cellular reactions that result in inositol triphosphate formation, intracellular calcium release, and PKC activation [30,31]. The MAP kinases ERK1 and ERK2, Janus kinase JAK2, the stress-activated kinases JNK1 and p38, phospholipase C, and two isoforms of PI3-kinase (p85/p110 and C2α) have all been implicated in MCP-1 signal transduction [32 –34]. The Dai group has reported that the downstream of CCR2 activation involves STAT3 phosphorylation and IL-1β production [35]. It is unclear which signaling pathways are responsible for the enhanced therapeutic effects of P-MSCs. The STAT3 pathway is a relevant pathway involved in MSC-mediated immunomodulation, and blockade of STAT3 phosphorylation impairs the ability of MSCs to inhibit T cell proliferation [20]. Furthermore, measurement of STAT3 phosphorylation in MSCs as responder cells correlates with and predicts the ability to suppress T cells [36]. We revealed that the p-STAT3 level was upregulated in P-MSCs and that inhibiting STAT3 signaling reversed the enhanced inhibitory effects of P-MSCs on T cell cytokine secretion. P-MSCs can activate the STAT3 signaling pathway to exert immunosuppressive effects on T cells, which provides a new therapeutic strategy for MSC-based treatment of T cell-mediated inflammatory diseases.
Conclusions
In this study, our results demonstrated for the first time that preconditioning with MCP-1 enhanced the efficacy of transplanted MSCs in a CHS mouse model. Moreover, preconditioning MSCs with MCP-1 also enhanced their ability to inhibit T cell inflammatory factor secretion. We further demonstrated that MCP-1 preconditioning of MSCs enhances therapeutic outcomes through the COX2-PGE2/STAT3 signaling pathway. Our results uncover an enhanced the ability of P-MSCs to regulate T cell-mediated inflammatory diseases, especially MCP-1-related disorders.
Footnotes
Author Disclosure Statement
No competing financial interest exists.
Funding Information
This study was supported by the National Key Research and Development Program of China, Stem Cell and Translational Research (2017YFA010550), the National Natural Science Foundation of China (81971526, 81970109, 81803143, 81760112); the Key Scientific and Technological Projects of Guangdong Province (2019B020236004, 2017A020215193, 2017B020230004, 2016A030310131); the Guangdong Basic and Applied Basic Research Foundation (2019A1515012098, 2020A1515010272), the Key Scientific and Technological Program of Guangzhou City (201802020023), the Pearl River S&T Nova Program of Guangzhou (201906010095), the Fundamental Research Funds for the Central Universities (17ykpy50, 20ykpy149).
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.