
Abstract
Congenital diaphragmatic hernia (CDH) leads to pathophysiologic pulmonary vasoreactivity. Previous studies show that mesenchymal stromal cell-derived extracellular vesicles (MSCEv) inhibit lung inflammation and vascular remodeling. We characterize MSCEv and human pulmonary artery endothelial cell (HPAEC) interaction, as well as the pulmonary artery (PA) response to MSCEv treatment. HPAECs were cultured with and without exposure to nitrofen (2,4-dichloro-phenyl-p-nitrophenylether) and treated with MSCEv. HPAEC viability, architecture, production of reactive oxygen species (ROS), endothelial dysfunction-associated protein levels (PPARγ, LOX-1, LOX-2, nuclear factor-κB [NF-κB], endothelial NO synthase [eNOS], ET-1 [endothelin 1]), and the nature of MSCEv-cellular interaction were assessed. Newborn rodents with and without CDH (nitrofen model and Sprague-Dawley) were treated with intravascular MSCEv or vehicle control, and their PAs were isolated. Contractility was assessed by wire myography. The contractile (KCL and ET-1) and relaxation (fasudil) responses were evaluated. HPAEC viability correlated inversely with nitrofen dose, while architectural compromise was directly proportional. There was a 2.1 × increase in ROS levels in nitrofen HPAECs (P < 0.001), and MSCEv treatment attenuated ROS levels by 1.5 × versus nitrofen HPAECs (P < 0.01). Nitrofen-induced alterations in endothelial dysfunction-associated proteins are shown, and exposure to MSCEv restored more physiologic expression. Nitrofen HPAEC displayed greater MSCEv uptake (80% increase, P < 0.05). Adenosine, a clathrin-mediated endocytosis inhibitor, decreased uptake by 46% (P < 0.05). CDH PA contraction was impaired with KCL (108.6% ± 1.4% vs. 112.0% ± 1.4%, P = 0.092) and ET-1 (121.7% ± 3.0% vs. 131.2% ± 1.8%, P < 0.01). CDH PA relaxation was impaired with fasudil (32.2% ± 1.9% vs. 42.1% ± 2.2%, P < 0.001). After MSCEv treatment, CDH PA contraction improved (125.9% ± 3.4% vs. 116.4 ± 3.5, P = 0.06), and relaxation was unchanged (32.5% ± 3.2% vs. 29.4% ± 3.1%, P = 0.496). HPAEC exposure to nitrofen led to changes consistent with vasculopathy in CDH, and MSCEv treatment led to a more physiologic cellular response. MSCEv were preferentially taken up by nitrofen-treated cells by clathrin-dependent endocytosis. In vivo, MSCEv exposure improved PA contractile response. These data reveal mechanisms of cellular and signaling alterations that characterize MSCEv-mediated attenuation of pulmonary vascular dysfunction in CDH-associated pulmonary hypertension.
Introduction
Congenital diaphragmatic hernia (CDH) is a disease with high mortality, in which malformation of the diaphragm and subsequent herniation of abdominal contents into the thoracic cavity are associated with hypoplastic pulmonary parenchyma and abnormal pulmonary vasculature, often leading to pulmonary hypertension (PH) [1 –3]. While the underlying cause in the human disease remains unknown, the animal model most frequently used to study CDH is generated by maternal ingestion of the teratogen nitrofen (2,4-dichloro-phenyl-p-nitrophenylether), leading to offspring with posterolateral diaphragmatic defects, pulmonary hypoplasia, and pulmonary vasculopathy [3,4]. Although this vasculopathy has been defined structurally and functionally, specific effects of nitrofen on the pulmonary vasculature and vascular cell subtypes remain poorly characterized.
Mesenchymal stromal or stem cells (MSCs) are progenitor cells with emerging clinical applicability and therapeutic potential [5 –7]. Broad translation and diverse tissue/disease targets highlight a spectrum of plausible mechanisms of action, including the release of myriad factors that have both paracrine and endocrine effects. MSCs and their released factors have shown the propensity to facilitate homeostasis in the setting of distress, minimizing or reversing cell injury and inflammation, while mediating and enhancing cellular preservation, repair, and/or recovery.
Extracellular vesicles (EV) are one family of factors released by some (if not all) cell types, which may mediate MSC effects. Mesenchymal stromal cell-derived EV (MSCEv) are subcellular, lipid bilayer-surrounded vesicles with surface markers/receptors that contain proteins, lipids, cytokines, chemokines, and nucleic acids [8,9]. Exosomes and microvesicles are specific subtypes of EV characterized by their size, surrounding lipid bilayer/receptor characteristics, and associated contents, although specific physical and biochemical criteria are neither definitive nor exclusive. EVs may serve an intermediary role in cellular communication after initial cell release through transmission of soluble materials, ligand binding, and/or internalization by target cells [7]. To initiate exploration into the therapeutic potential of MSCEv for the vasculopathy associated with CDH-associated PH, in vitro identification of the effects of nitrofen on endothelial cells, the subsequent mitigation of these effects with MSCEv, and the nature of endothelial cell-MSCEv interaction were undertaken.
In this study, we first investigated the cytotoxic effects of nitrofen on human pulmonary artery endothelial cells (HPAECs). Then, we examined the effects of human MSCEv therapy on the expression of endothelial dysfunction-associated proteins in nitrofen-exposed HPAECs. Furthermore, using HPAECs, we characterized the mechanism by which MSCEv interact with endothelial cells under normal and nitrofen-exposed conditions, both in vitro and in vivo. Finally, we evaluated the effects of MSCEv on pulmonary artery (PA) vasoreactivity in rodents with nitrofen-induced CDH.
Materials and Methods
Chemicals and cell culture
Nitrofen and amantadine, an independent inhibitor of clathrin-mediated endocytosis, were obtained from Sigma-Aldrich (St. Louis, MO).
HPAECs were purchased from Lonza Walkersville, Inc. (Walkersville, MD). Cells were cultured at 37°C under 5% CO2 in EGM-2 (Lonza Walkersville, Inc.) medium.
Isolation and characterization of human MSCs EVs
Human MSCs were isolated from commercially available fresh human bone marrow aspirates of a 34-year-old female (AllCells, Alameda, CA) using density centrifugation and plastic adherence as previously reported [7,10] and briefly described in the Supplementary Materials and Methods section in Supplementary Data and Supplementary Fig. S1. The allogeneic adult MSCs used fit the International Society for Cellular Therapy definition of “MSC,” have typical immunobiology for MSCs, and are among the most commonly available cellular therapy products worldwide [11]. MSCEv were isolated by sequential filtration assay and characterized using TEM, RNA, and protein assay, and counted by the IZON qNano system as described in the Supplementary Materials and Methods section in Supplementary Data and Supplementary Fig. S2.
Cell viability assay
MTT assays were performed to evaluate the in vitro cytotoxic activity of nitrofen in HPAECs. The cells were seeded in 96-well microassay culture plates (10,000 cells/well) and treated with varying concentrations of nitrofen (0, 0.05, 0.1, 0.5, and 1 mg/mL) for 24 h. Upon completion of the treatment, the cells were incubated in serum deprivation medium containing 0.5% MTT for 4 h. Subsequently, the medium was replaced with 100 μL of dimethyl sulfoxide (DMSO) to dissolve the formation. The absorbance was measured at 570 nm in a microplate spectrophotometer (Molecular Devices), and the experiments were repeated three times (n = 3 for all groups). For quantification of the viability of HPAECs, the following formula was used: Viability of cells (%) = (Absorbancesample/Absorbancecontrol) × 100. To assess the morphological changes of HPAECs induced by nitrofen, in a pilot study, cultured HPAECs were treated with nitrofen (0.1 mg/mL), either with or without MSCEv (1 × 1010/mL), for 24 h. Cell morphology was examined using a Leica DMIL LED microscope with a digital camera (DMC 2900). In addition, to evaluate the effects of MSCEv on nitrofen-induced HPAEC viability changes, cultured HPAECs were treated with nitrofen (0.1 mg/mL), either with or without MSCEv (1 × 1010/mL), for 24 h, and cell viability was analyzed using MTT assay as described above.
Reactive oxygen species detection
Determination of intracellular reactive oxygen species (ROS) levels in HPAECs was performed using the fluorescent 2′,7′-dichlorofluorescin diacetate (DCFDA)-cellular ROS detection assay kit (Abcam). Briefly, HPAECs were seeded into six-well plates at a density of 1 × 104cells/well and incubated for 24 h. The cells were then stained with 20 μM DCFDA in the dark for 30 min at 37°C. Subsequently, the extracellular dye was discarded, and the cells were exposed to medium only (control), nitrofen (0.1 mg/mL), or MSCEv (1 × 1010/mL) + nitrofen (0.1 mg/mL) for 4 h. Fluorescence was analyzed using a BD LSR II flow cytometer (BD Biosciences, Franklin Lakes, NJ) with maximum excitation and emission spectra of 495 and 529 nm, respectively. Automated compensation was performed by using FACSDiva Version 6.1.3 software (BD BioSciences, San Jose, CA). Each experiment was repeated on three separate occasions (n = 3 for all groups). The value for the untreated control was set to be 1, and the values for nitrofen only and MSCEv+nitrofen groups were normalized to the control.
Immunoblotting assay
Cultured HPAECs were treated with nitrofen (0.1 mg/mL), either with or without MSCEv (1 × 1010/mL), for 24 h. To prepare total cellular extracts, cells were isolated after treatment and sonicated in radioimmunoprecipitation assay (RIPA) solution (Thermo Scientific, Rockford, IL). Protein concentrations were measured by using the BCA protein assay reagent (Pierce, Rockford, IL). Proteins were separated by using 4%–15% sodium dodecyl sulfate polyacrylamide gel electropheresis (SDS-PAGE) (25 μg protein/lane) and transferred onto Immun-Blot PVDF membrane/filter paper (Bio-Rad, Hercules, CA). The PVDF membranes were then incubated with the indicated antibodies, including anti-p-PPARγ (1:1,000 dilution; Thermo Fisher Scientific), anti-p-eNOS (1:1,000 dilution; Cell Signaling Technology, Inc.), anti-nuclear factor-κB [NF-κB], anti-NOX-2 (NADPH oxidase 2), anti-LOX-1 and anti-endothelin 1 (ET-1; 1:1,000 dilution; Abcam). Subsequently, the membrane was incubated with goat anti-rabbit or anti-mouse HRP-labeled secondary antibodies. Proteins were detected by using Pierce enhanced chemiluminescence western blotting substrate. β-Actin (Sigma-Aldrich) was used as a loading control.
Small interfering RNA treatment
Both siControl (ON-Targetplus Non-Targeting Control Pool) and OnTarget siLOX-1 Smart Pool, containing four target sequences, were purchased from Dharmacon. HPAECs were seeded in six-well plates (50,000 cells/well). The transfection was performed the next day after seeding in 2% fetal bovine serum media with siControl or siLOX-1 diluted in antibiotic-free medium using the Lipofectamine RNAiMAX (Invitrogen) diluted in OPTI-MEN (Gibco). Small interfering RNA (siRNA) was added to the respective wells to a final dose of 50 nM. Media were changed the day after siRNA transfection, and cells were allowed to recover. Seventy-two hours after transfection, cells were treated with or without nitrofen (0.1 mg/mL) in antibiotic-free medium, and, 24 h later, the cells were harvested and prepared for total cellular extracts as described above. The efficiency of target silencing was assessed by an immunoblotting assay with anti-LOX-1 antibody, and we further analyzed the expressions of LOX-1-associated pathway-related protein.
Cell treatment for the uptake of MSCEv by the clathrin-mediated pathway
To prepare fluorescently labeled exosomes, MSCEv were labeled with Exo-Glow Exosomes labeling kits (SBI System Biosciences, Palo Alto, CA), according to the manufacturer's protocol, with some modifications. Briefly, 50 μL 10 × Exo-Green was added to 500 μL of resuspended exosome pellets (1 × 1013/mL), and the solution was incubated at 37°C for 10 min. To stop the labeling reaction, 100 μL of the ExoQuick-TC reagent was added to the labeled exosome sample suspension and mixed by inverting. The labeled exosomes sample was then placed on ice for 30 min. Finally, the sample was centrifuged for 3 min at 14,000 rpm in a microfuge, the supernatant containing excess label removed, and the labeled exosome pellets resuspended in 500 μL phosphate-buffered saline (PBS). To measure MSCEv uptake, medium was replaced by MSCEv-containing medium (1 × 1010/mL) or medium without MSCEv (control) and incubated for 60 min. In the nitrofen treatment experiment, cells were incubated with nitrofen (0.1 mg/mL) for 24 h, and then the cells were washed and incubated with MSCEv. To evaluate the uptake of MSCEv by the clathrin-mediated pathway, cells were pretreated with 1 mM amantadine for 30 min before incubation with nitrofen, as described above. To evaluate the effects of amantadine on clathrin expression, cells were treated with nitrofen alone or pretreated with amantadine followed by nitrofen, proteins were extracted, and immunoblotting was performed using anti-clathrin heavy-chain antibody (1:1,000 dilution; Cell Signaling Technology, Inc.). β-Actin (Sigma-Aldrich) was used as a loading control. The uptake of fluorescent MSCEv was visualized by using a Leica SP5 confocal system with a Leica TCS SP5 II microscope. For quantitative analysis of fluorescence, cells were trypsinized, resuspended in ice-cold PBS, and analyzed by using a BD LSR II flow cytometer (BD Biosciences, Franklin Lakes, NJ) with a blue laser (488 nm). Automated compensation was performed by using FACSDiva Version 6.1.3 software (BD BioSciences, San Jose, CA).
Creation of CDH and experimental protocol
The animal experimental protocol was reviewed and approved by the Animal Welfare Committee (permit No. AWC 14–009), University of Texas McGovern Medical School (Houston, TX). To establish the CDH animal model, pregnant Sprague-Dawley rats (ENVIGO, Houston, TX) were fed 100 mg of nitrofen (Sigma-Aldrich) dissolved in 1 mL of olive oil on gestational day 9.5 (±6 h), while rats in the control group were fed the same dose of olive oil without nitrofen as previously described [3]. Power analyses were performed to determine the optimal number of animal subjects required to observe significant differences under the proposed conditions as well as to ensure reproducibility. To more closely approximate severe human disease, only pups with large (>50% of total surface area) diaphragmatic defects underwent treatment and tissue harvest.
Wire myography
The newborn rats were sacrificed, and PAs were isolated from both CDH (n = 29) and control groups (n = 20). Changes in force generated by PA segments were measured using wire myography, as previously described [12,13]. To evaluate the effects of MSCEv on PA vasoreactivity, newborn rodents with CDH were treated with intravascular MSCEv (1 × 1010/mL in PBS, CDH+MSCEv group, n = 9) or PBS (CDH group, n = 20). Briefly, vascular reactivity was compared utilizing a water-jacketed organ bath in which 1.5 mm cross-sections of PAs were allowed to equilibrate in oxygenated Krebs buffer (gassed with 5% CO2/21% O2/balanceN2) at 37°C then attached to a wire myograph for isometric tension measurements. After a vessel had equilibrated and optimal tension had been slowly attained from 0.1 to 0.6 mV, each segment was contracted four times (the first three with 40 mM hypertonic KCl and a final time with 60 mM hypertonic KCl) with washouts in between. Next, segments were adjusted to the resting tension of 0.6 mV before each experiment. To identify changes in ex vivo PA vasoreactivity, the basal tone and induced vascular tone were measured, creating a contraction-response curve as ET-1 (10−4, contraction) and fasudil (50 μM, dilation) were introduced into the system. These measurements were obtained in the presence of MSCEv (dosed at 1010/mL exosomes, CDH+MSCEv group) or PBS (control and CDH groups).
Immunofluorescence staining
To determine the uptake and distribution of MSCEv in PA tissue, rodents were treated with intravascular fluorescently labeled MSCEv (1 × 1010/mL in PBS, FL-MSCEv+CDH group, n = 3) or PBS (CDH group, n = 3), and PAs were isolated 5 min after the injection for immunofluorescence staining. Briefly, isolated PAs from both groups were fixed with 4% paraformaldehyde in PBS (pH 7.4) overnight at 4°C. The tissue was then rinsed in 15% sucrose for 24 h at 4°C, embedded in optimal cutting temperature compound (Tissue-Tek; Sakura Finetek, Torrance, CA), and made into slides (5–8 μm). Subsequently, the slides were washed with PBS and counterstained with 4′,6′-diamidino-2-phenylindole (DAPI) nucleic acid stain. For colocalization analysis, the slides were incubated in 1% bovine serum albumin (BSA)/10% normal goat serum in 0.1% PBS-Tween 20 for 1 h to permeabilize the tissue and block nonspecific protein–protein interactions. The slides were then incubated with rabbit monoclonal anti-LOX-1 (1:50 dilution; Abcam) at 4°C overnight, washed three times with 1 × PBS, and incubated with anti-rabbit Alexa Fluor 546 conjugated secondary antibody (1:2,000 dilution; Invitrogen) at room temperature for 60 min. Images were observed on a Leica SP5 confocal system with a Leica TCS SP5 II microscope (Leica Microsystems, Mannheim, Germany), DAPI and fluorescein isothiocyanate filters, and LAS AF image acquisition software (Leica Microsystems). For immunofluorescence staining, isolated PAs from both CDH (n = 3) and control (n = 3) groups were fixed and made into slides as described above. The slides were incubated in 1% BSA/10% normal goat serum in 0.1% PBS-Tween 20 for 1 h to permeabilize the tissue and block nonspecific protein–protein interactions. The slides were then incubated with rabbit monoclonal anti-LOX-1 (1:50 dilution; Abcam) and mouse monoclonal anti-ET-1 (1:50 dilution; Abcam) at 4°C overnight, washed three times with 1 × PBS, and incubated with anti-rabbit Cy5 conjugated secondary antibody (1:2,000 dilution; Abcam) and anti-mouse Alexa Fluor 488 conjugated secondary antibody (1:2,000 dilution; Abcam) at room temperature for 60 min. Images were analyzed on a Leica SP5 confocal system with a Leica TCS SP5 II microscope as described above.
Isolation and characterization of rodent PA endothelial cells
Newborn rats were euthanized, and autopsies were performed. The thorax was disinfected with 70% ethanol, and a thoracotomy was performed using dissection scissors and forceps to expose the heart and lungs. One milliliter of cold PBS containing 1,000 U/mL of heparin was injected into the right ventricle and flushed into the pulmonary and systemic circulation. The main pulmonary arteries were dissected microsurgically from the thoracic cavity as described in the Supplementary Materials and Methods section in Supplementary Data. Endothelial cell phenotype was confirmed with positive fluorescence for platelet/endothelial cell adhesion molecule-1 (PECAM-1 or CD31) by flow cytometry. The expression of endothelial dysfunction-associated proteins in the isolated primary cells was measured by immunoblotting assay (as described in the Supplementary Materials and Methods section in Supplementary Data).
Statistical analyses
For all analyses, comparisons among groups were made using one-way analysis of variance followed by post hoc comparison (Newman-Keuls multiple) tests. Differences between the means were considered significant when two tailed. The significance level was set at P < 0.05. Results were shown as the mean + standard error of the mean. Statistical analysis was performed using GraphPad Prism (v7.0 software).
Results
Nitrofen induced cytotoxicity in HPAECs
HPAECs were exposed to different concentrations of nitrofen for 24 h and cell viability was determined by MTT assays. The results (Fig. 1A) show that increasing the nitrofen concentration (range: 0.05–1 mg/mL) resulted in dose-dependent decreases in HPAEC viability (cell viabilities were 60.9%, 50.7%, 19.5%, and 3.6%, respectively). Given these results, we selected a 0.1 mg/mL nitrofen concentration and a 24-h incubation time point for further studies. MSCEv alleviated the cytotoxic effects of nitrofen (Fig. 1B, C). More specifically, the morphological analysis showed that nitrofen induced dramatic morphological changes and significant cell death in HPAECs after 24-h treatment. MSCEv treatment reversed the morphological changes (Fig. 1B). However, these morphological changes were not visualized in the MSCEv+control treatment group (Fig. 1B). In Fig. 1C, quantification analysis showed that cultured HPAECs exposed to 0.1 mg/mL of nitrofen decreased the cell viability by 56% (P < 0.01) in comparison with untreated controls. Importantly, the treatment with MSCEv recovered the cell viability by 51% (P < 0.05) compared with nitrofen-treated HPAECs. We found no significant difference in the cell viability between the untreated control and MSCEv+control groups (Fig. 1C). Given these results, we selected untreated controls, nitrofen, and MSCEv+nitrofen as our groups for further study.
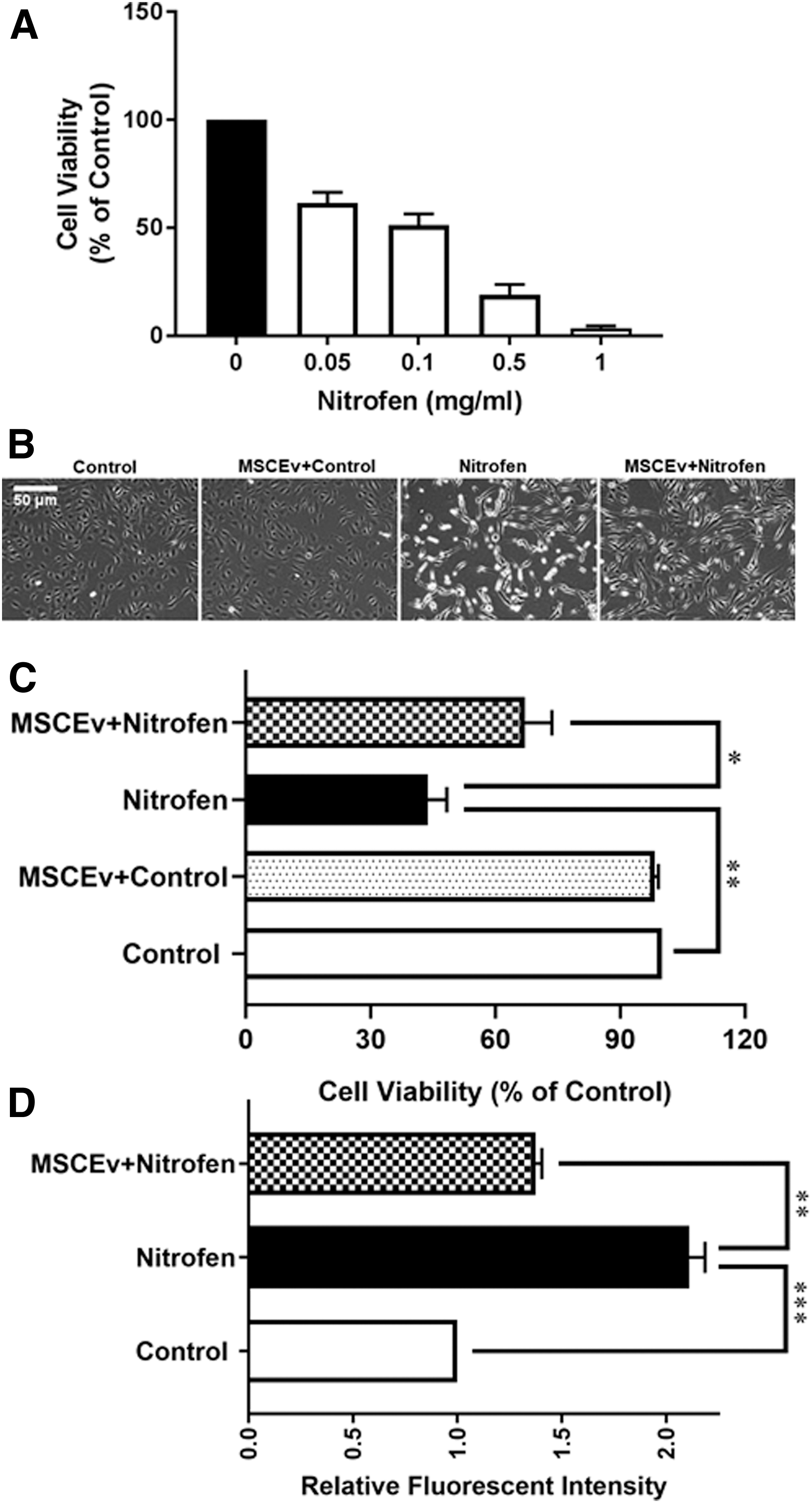
Effect of nitrofen on cell viability in HPAECs. Cell viability of HPAECs treated with different concentrations of nitrofen (0, 0.05, 0.1, 0.5, and 1 mg/mL) obtained from MTT assays after 24-h exposure (
MSCEv attenuated nitrofen-induced elevation of total ROS levels in HPAECs
It has been reported that nitrofen induced a dose-dependent increase in ROS and cell death in teratocarcinoma cells [14]. In this study, we showed that nitrofen induced a dose-dependent decrease in HPAEC viability. Increases in ROS are often associated with cell death. Thus, we detected the intracellular ROS levels in nitrofen-exposed HPAECs, and the effect of MSCEv was further investigated. As shown in Fig. 1D, nitrofen resulted in a significant increase of intracellular ROS levels. Importantly, MSCEv treatment significantly attenuated the nitrofen-induced increases of ROS levels in HPAECs. Quantification analysis showed that there was a 2.1-fold increase in ROS levels in nitrofen-exposed HPAECs versus control (P < 0.001), and MSCEv treatment decreased ROS levels by 1.5-fold versus nitrofen-exposed HPAECs (P < 0.01), respectively (Fig. 1D). The value for the untreated control was set to be 1, and the nitrofen only and MSCEv+nitrofen-treated group value were normalized to the control. Given previous data suggesting that ROS cause cell injury and dysfunction through directly oxidizing and damaging DNA, proteins, and lipids, as well as by activating several intracellular signaling pathways [15,16], we investigated the alteration of proteins associated with endothelial cell dysfunction and the effect of MSCEv on those proteins.
MSCEv reduced expression of nitrofen-induced endothelial dysfunction-associated proteins in HPAECs
Nitric oxide (NO) and ET produced in endothelial cells are important molecules that regulate vascular function. Failure of the physiological balance between these two molecules is usually referred to as endothelial dysfunction [17,18]. We recently elucidated changes in endothelial dysfunction-associated pathways in nitrofen-induced CDH in rodents. In particular, we found that ET-1 and endothelial NO synthase (eNOS), two major vasoactive mediators, are imbalanced in CDH PA tissues [3]. Thus, this study examined nitrofen-exposed HPAECs to determine how MSCEv influences the expressions of endothelial dysfunction-associated proteins. The immunoblotting analysis shows that the expression of ET-1 was significantly increased in nitrofen-treated HPAECs (46%, P < 0.05). However, the expression of p-eNOS was significantly decreased (62%, P < 0.01) in nitrofen-treated HPAECs compared with controls. Importantly, MSCEv treatment significantly reversed the alterations of the major vasoactive mediators in nitrofen-treated HPAECs. The treatment with MSCEv reduced the expression of ET-1 by 42% (P < 0.05) and increased the expression of eNOS by 72% (P < 0.01) compared to the nitrofen-treated group (Fig. 2A, B).
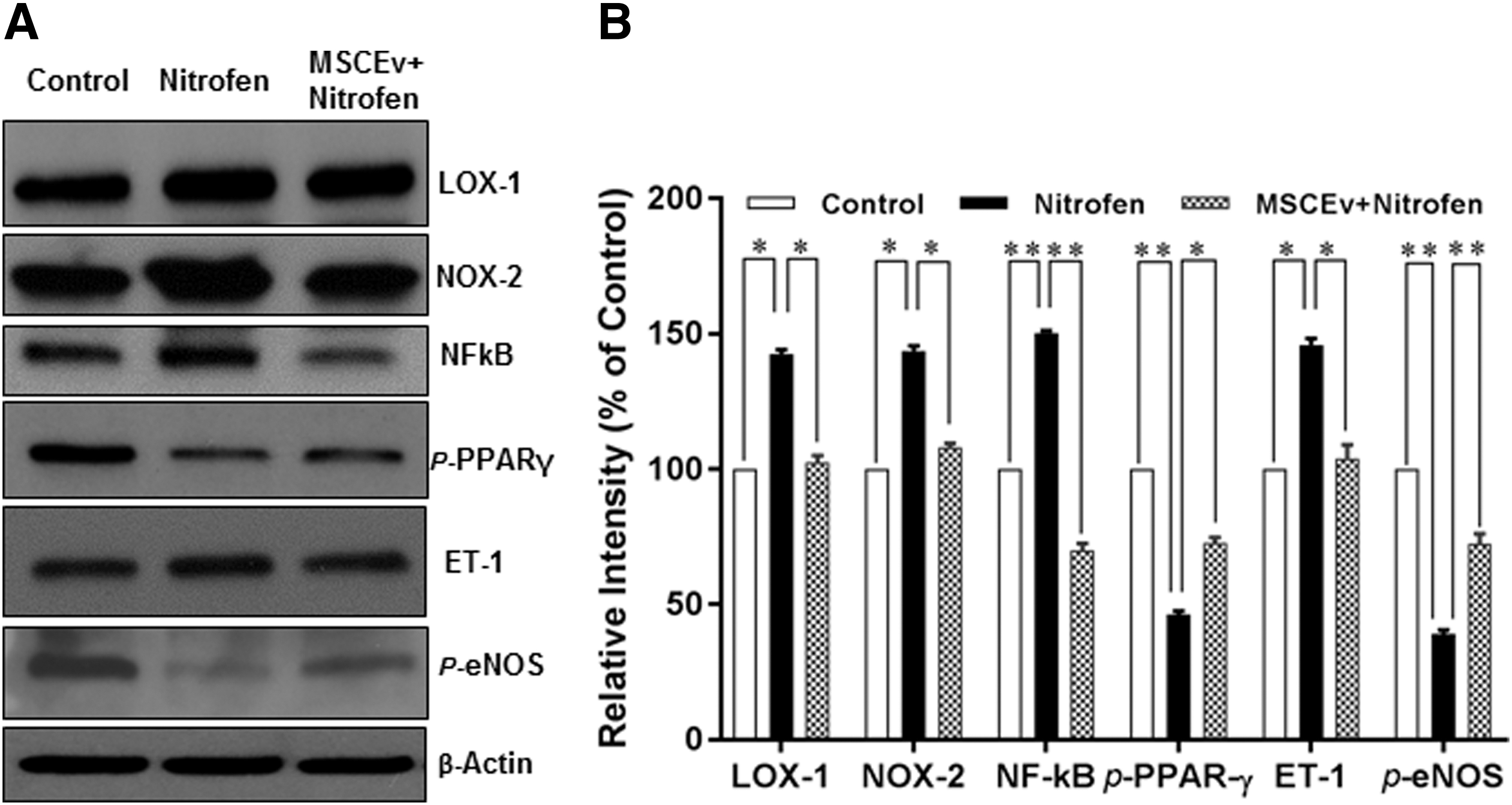
MSCEv reversed the expression of nitrofen-induced endothelial dysfunction-associated proteins in HPACs. Cultured HPAECs were treated with nitrofen (0.1 mg/mL), either with or without MSCEv (1 × 1010/mL), for 24 h. Total soluble proteins were prepared, and endothelial dysfunction-associated protein levels were determined by immunoblot analysis as described in Materials and Methods section. Antibodies against LOX-1, NOX-2, NF-κB, p-PPAR γ, ET-1, and p-eNOS were used for detection of the respective proteins. B-Actin was used as a loading control. Representatives of protein expression are shown (
LOX-1, an OxLDL receptor expressed on endothelial cells, has been implicated in PH through its activation of NOX-2, which generates ROS, and its activation of NF-κB, which upregulates ET-1 and inactivates eNOS. LOX-1 might be a key molecule in the generation of endothelial dysfunction [17,18]. Thus, in this study, we hypothesized that nitrofen treatment would activate this pathway, and that MSCEv exposure would lead to recovery from the resulting endothelial dysfunction. Confirming our hypothesis, the immunoblotting analysis showed that cultured HPAECs exposed to nitrofen significantly increased the expressions of endothelial NF-κB, NOX-2, and LOX-1 (50%, P < 0.01; and 44% and 46%, P < 0.05, respectively), compared with controls (Fig. 2A, B). As expected, MSCEv treatment significantly reversed the alterations of the endothelial dysfunction-associated proteins in nitrofen-treated HPAECs. The treatment with MSCEv reduced the expressions of NF-κB, NOX-2, and LOX-1 by 80% (P < 0.01), and 36% and 40% (P < 0.05), respectively (Fig. 2A, B).
PPARγ dysregulation is also implicated in PH, as it normally downregulates ET-1, NOX-2, and NF-κB, and maintains the activity of eNOS, thus opposing endothelial dysfunction and vascular remodeling [19 –21]. Therefore, we examined the expression of PPARγ in nitrofen-treated HPAECs and investigated the effects of MSCEv on this expression. As shown in Fig. 2A, p-PPARγ, a marker for PPARγ catalytic activity, was significantly decreased in nitrofen-treated HPAECs. Quantitative analysis showed that nitrofen treatment decreased p-PPARγ by 54% (P < 0.01) in comparison with untreated controls (Fig. 2B). Importantly, the treatment with MSCEv increased the expression of p-PPARγ by 27% (P < 0.01) compared with nitrofen-treated HPAECs (Fig. 2).
Taken together, our data showed that nitrofen induced endothelial dysfunction in HPAECs and exposure to MSCEv restores a more physiologic expression of the endothelial dysfunction-associated proteins.
Nitrofen induces endothelial dysfunction, in part, through NF-κB generation, which can be blunted by siLOX-1 treatment
We showed an overexpression of LOX-1 in nitrofen-treated HPAECs, and we hypothesized that endothelial LOX-1 plays a critical role in nitrofen-induced endothelial dysfunction. To test this hypothesis, we examined the impact of LOX-1 silencing on the expressions of NF-κB and ET-1. As shown in Fig. 3A, immunoblot analysis demonstrated that siLOX-1 greatly reduced the levels of LOX-1 expression in both nitrofen-treated and control cells. Importantly, nitrofen-induced upregulation of NF-κB was significantly reduced by siLOX-1 treatment. Quantification analysis showed that the treatment with siLOX-1 decreased the expressions of LOX-1 by 55% (P < 0.01) compared with nontreated HPAECs and by 74.3% (P < 0.01) compared with nitrofen-treated HPAECs (Fig. 3C). In addition, the treatment with siLOX-1 decreased the expressions of NF-κB by 54.1% (P < 0.01) compared with nontreated HPAECs, and by 79.3% (P < 0.01) compared with nitrofen-treated HPAECs (Fig. 3C). Interestingly, treatment with siLOX-1 did not significantly decrease the expressions of ET-1 (5%, P > 0.05) compared with nitrofen-treated HPAECs (Fig. 3B, D), implying that nitrofen-induced overexpression of ET-1 in the endothelial cells is not primarily mediated by an LOX-1-associated pathway. Taken together, the LOX-1-associated pathway is at least partially responsible for nitrofen-induced endothelial dysfunction.

Immunoblot analysis showing siLOX-1 greatly reduced the levels of LOX-1 expression in both nitrofen-treated or untreated cells (
Clathrin-mediated endocytosis in MSCEv uptake by HPAECs
Confocal microscopy was used to evaluate the degree of MSCEv uptake by both control and nitrofen-treated HPAECs. Moreover, to characterize the role of clathrin-mediated endocytosis in the uptake of MSCEv by HPAECs, we studied the effects of amantadine, an independent inhibitor of clathrin-mediated endocytosis, on MSCEv uptake in both control and nitrofen-treated HPAECs [22,23]. We first examined the cytotoxicity of amantadine and found that a concentration of 1 mM amantadine was not toxic to HPAECs (<5% of cell death; data not shown); thus, this concentration was used in subsequent experiments. Confocal microscopy analysis showed that the uptake of MSCEv was greater in HPAECs treated with nitrofen than in untreated HPAECs (MSCEv only) (Fig. 4A). Importantly, the pretreatment of HPAECs with amantadine reduced the uptake of MSCEv in both nitrofen-treated and untreated HPAECs (Fig. 4A). The quantification of fluorescently labeled liposomes by flow cytometry (expressed as intensity of fluorescently labeled MSCEv) showed that the uptake of MSCEv by HPAECs was 1.8-fold higher in nitrofen-treated HPAECs than in untreated HPAECs (Fig. 4B). Furthermore, in untreated HPAECs or nitrofen-treated HPAECs, pretreatment with amantadine significantly reduced MSCEv uptake by 30% and 46%, respectively (Fig. 4B). In addition, we examined whether the nitrofen-induced increase in the uptake of MSCEv was mediated by an increase in clathrin expression. Immunoblot analysis showed that the basal expression of endogenous clathrin in PAECs was low (Fig. 4C). However, in cells treated with nitrofen, clathrin expression was significantly increased (Fig. 4C). Importantly, amantadine greatly inhibited clathrin expression in nitrofen-treated cells (Fig. 4C). Our data thus indicate that the nitrofen-induced increase in MSCEv uptake was mediated by clathrin-dependent endocytosis.
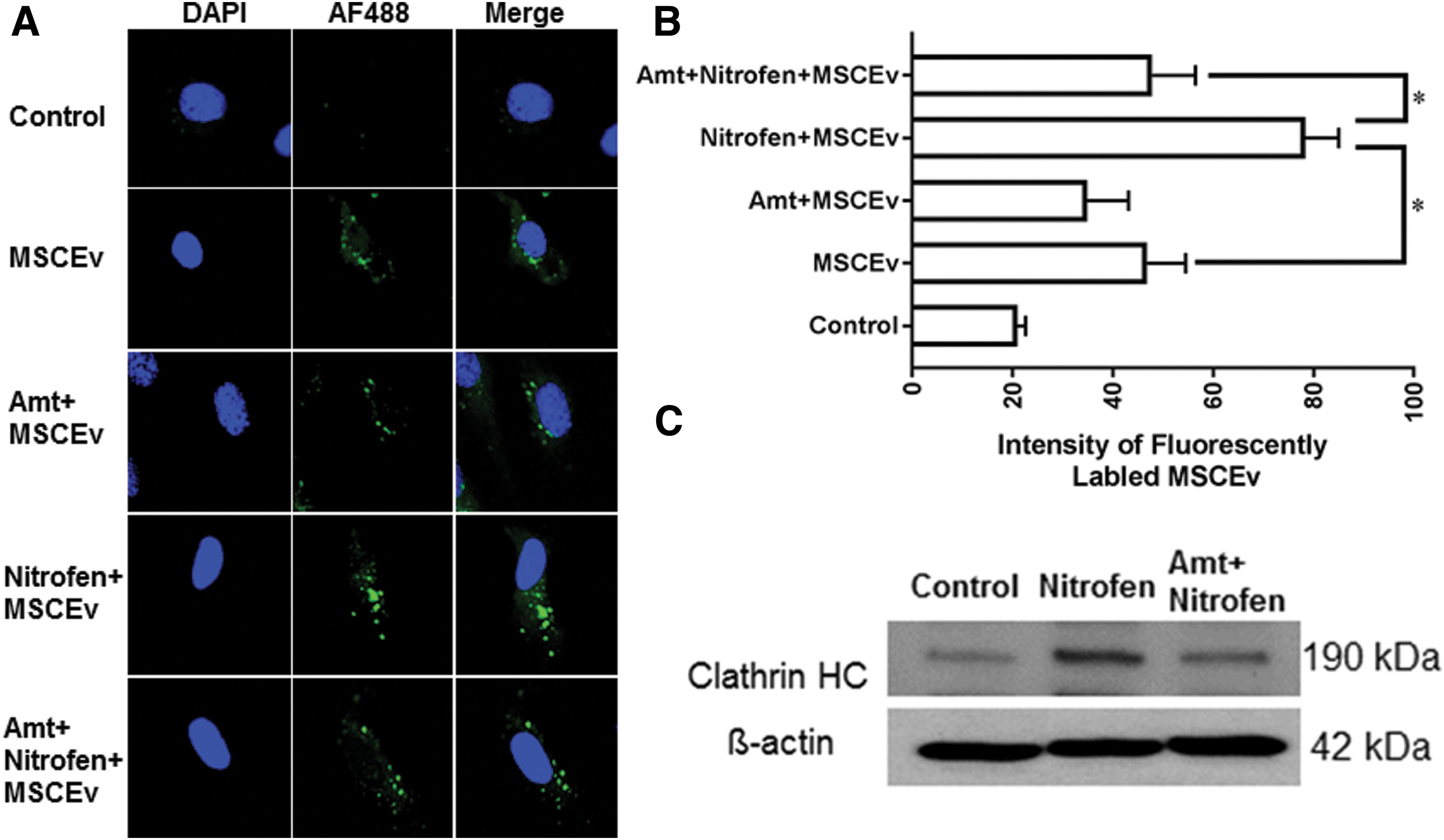
Effect of amantadine on the uptake of fluorescently labeled MSCEv into HPAECs.
MSCEv treatment improves CDH PA contraction
As shown in Fig. 5, CDH PA contraction was significantly impaired with ET-1 (118.8% ± 1.8% vs. 131.2% ± 1.8%, P < 0.0001) (Fig. 5A). CDH PA relaxation was significantly impaired with fasudil (30.5% ± 1.3% vs. 42.1% ± 1.6%, P < 0.001) (Fig. 5B). After MSCEv treatment, CDH PA contraction was shifted closer toward controls (125.6% ± 3.4% vs. 118.8 ± 1.8, P = 0.06) and relaxation unchanged (30.5% ± 1.3% vs. 32.4% ± 2.5%, P = 0.496) (Fig. 5A, B).

Effect of MSCEv on CDH PA contractility. Newborn rodents with or without CDH were treated with intravascular MSCEv (or vehicle control); their PAs were isolated, and PA contractility was assessed by wire myography. The contractile (ET-1) and relaxation (fasudil) responses were evaluated. Bars represent the mean ± standard error (control n = 20; CDH n = 20; MSCEv+CDH n = 9). MSCEv treatment improves CDH PA contraction (
Uptake of fluorescently labeled MSCEv into CDH PA tissue and colocalized with LOX-1 in PA tissue
In the in vitro study, we showed that treatment with nitrofen significantly increased MSCEv uptake by HPAECs (Fig. 4). To determine whether intravascular treatment of MSCEv resulted in MSCEv uptake by the endothelium in CDH PA tissue, we performed immunofluorescence staining and confocal fluorescence imaging of CDH PA tissue after the injection of PBS (Fig. 6A) or fluorescently labeled MSCEv (Fig. 6B). As shown in Fig. 6B, uptake of fluorescently labeled MSCEv was significant in PA tissue, and most of them were located in the endothelial cell cytoplasm. There is increasing evidence that LOX-1 might be a key molecule in the generation of endothelial dysfunction [17,18,24]. Our data showed that nitrofen induced endothelial dysfunction in HPAECs through elevating the expression of LOX-1, and treatment with MSCEv significantly reduced the expression of LOX-1 (Fig. 2). The finding implies that LOX-1 may be a target for MSCEv treatment. Thus, we performed colocalization analysis to determine whether MSCEV colocalized with local LOX-1 in the CDH PA tissue after the injection. As expected, fluorescently labeled MSCEv (Fig. 6C) were colocalized with local LOX-1 (Fig. 6D) in the CDH rodent PA tissue (Fig. 6E). Quantitative analysis showed that ∼64% of the expression of LOX-1 was colocalized with in CDH PA tissue.

Uptake of fluorescently labeled MSCEv into CDH PA tissue (
Colocalization of LOX-1 and ET-1 in PA tissue
Accumulating evidence has shown the interaction of LOX-1 and ET-1 plays a critical role in endothelial dysfunction process [17,18,25 –27]. To determine whether endothelial LOX-1 colocalized with ET-1 in the CDH PA tissue, we performed colocalization analysis. As expected, the expressions of LOX-1 and ET-1 (Fig. 6I, J) were significantly increased in the CDH rodent PA tissue compared with controls (Fig. 6F, G). Quantitative analysis showed that ∼75% of expression of ET-1 was colocalized within CDH PA tissue (Fig. 6K).
Discussion
In this study, we demonstrated that MSCEv administration attenuated the expression of various endothelial dysfunction-associated proteins linked to LOX-1 signaling in an experimental in vitro model of nitrofen-induced CDH. Furthermore, we showed that MSCEv were preferentially taken up by cells treated with nitrofen by clathrin-dependent endocytosis. Finally, we showed that PAs from nitrofen-treated rodents with CDH had impaired contractility and relaxation, and that MSCEv treatment was able to improve contractility in vivo. Taken together, these results reveal a potential mechanism of endothelial cellular interaction and signaling pathway alterations that characterize MSCEv-mediated attenuation of pulmonary vascular dysfunction in CDH-associated PH (Supplementary Fig. S3).
Nitrofen is a compound that has long been used to create experimental CDH models in rodents [4], owing to its ability to induce diaphragmatic defects, hypoplastic lungs, and pulmonary arterial abnormalities, secondary to alteration of the retinoic acid pathway [28], in the offspring of exposed dams [29]. The nitrofen model is a cornerstone for early, preclinical investigation in CDH and PH due to its diaphragmatic and pulmonary developmental and anatomic insults, which are highly analogous to the human condition [30]. HPAEC and PAEC from nitrofen-CDH pups display similar markers of endothelial dysfunction as described in the Supplementary Materials and Methods section in Supplementary Data. Therefore, evaluating nitrofen's influence in vitro on cultured HPAECs is critical for evaluating pathways involved in the pathogenesis of endothelial dysfunction in CDH, and, thus, potential targets for therapeutic intervention. However, there is currently no published standard for an in vitro nitrofen CDH model of HPAECs. We calculated the approximate endothelial cellular exposure dose to nitrofen in the rodent model, using 100 mg dose in a 250 g dam. Subsequently, we mapped out a dose–response relationship between nitrofen treatment and HPAEC viability and refined the appropriate nitrofen treatment concentration and incubation time for our studies.
The contribution of endothelial dysfunction to pulmonary vascular remodeling, leading to PH, has been extensively investigated. Endothelial cells play important roles in regulating vascular function, particularly in maintaining the balance of vasoconstrictor and vasodilator production, modulating pulmonary arterial smooth muscle cell (PASMC) proliferation, and mediating inflammatory responses [31]. Our previous work, using the in vivo nitrofen model, demonstrated that the upregulation of the potent vasoconstrictor ET-1 on PAECs and its receptor, ETA, on PASMCs is especially significant in the increased PA contractility and PASMC hypertrophy seen in CDH-associated PH [3]. In addition, eNOS activity in HPAECs was suppressed in this model, which explains the decreased bioavailability of NO seen in PH. NO, a strong vasodilator, acts through the protein kinase G pathway in PASMCs to promote muscle relaxation [32]. NO also has antiproliferative effects on PASMCs [33]. Nevertheless, the dysregulation of ET-1, eNOS, and other associated molecular pathways by the upstream components of endothelial dysfunction in CDH-associated PH remains ill defined. We utilized an in vitro nitrofen model to elucidate the pathways associated with this pathological process, uncovering changes in PPARγ and LOX-1 signaling.
LOX-1 is the major scavenger receptor for OxLDL in endothelial cells [34] and is implicated in hypoxic PH. Although LOX-1 is normally expressed in low levels by HPAECs, it can be upregulated by proinflammatory factors, hypoxia, and mechanical stimulation [35]. It has been shown that activation of LOX-1 by OxLDL causes endothelial changes that are characterized by the generation of ROS, facilitated by the induction of NOX-2 in endothelial cells [36]. This, in turn, induces NF-κB, which decreases the activity of eNOS [37] and generation of NO [38], while upregulating ET-1 expression [39]. In addition, through LOX-1, OxLDL also induces the expression of chemokines and various adhesion molecules on HPAECs, which stimulate inflammatory responses [40,41]. Together, these greatly contribute to endothelial dysfunction. In this study, we hypothesized that nitrofen treatment would result in endothelial dysfunction by the involvement of the LOX-1 pathway.
PPARγ, a member of the PPAR family of highly conserved nuclear hormone receptors, is widely known for its important role in regulating various genes involved in lipid metabolism, glucose homeostasis, cell differentiation, and immune inflammation [42 –44]. Mounting evidence has shown that activation of PPARγ attenuates PASMC proliferation and suppresses the development of PH in several animal models [45]. Moreover, PPARγ expression is reduced in the lungs and pulmonary vasculature of patients with PH and in several experimental models of PH [46]. These data are certainly consistent with the finding that PPARγ is downregulated in our model. Numerous studies have demonstrated that PPARγ plays an important role in the development of endothelial dysfunction in PH through its regulation of the expression of target genes that mediate PASMC proliferation, contraction, and damage, including ET-1 [20], eNOS [47], NOX-2 [19], NF-κB [48] and more. Specifically, PPARγ is able to repress NF-κB [49,50] and, thus, indirectly downregulate ET-1 and prevent the deactivation of eNOS. PPARγ is also able to downregulate NOX-2 and, therefore, decrease ROS production [48,49]. It has been shown that the expression of PPARγ is markedly decreased in nitrofen-induced hypoplastic lungs in the CDH model, supporting the role of PPARγ dysregulation in the pathogenesis of CDH-PH [51].
MSCs, found in bone marrow, subcutaneous adipose tissue, lungs, and other organs [52,53], have been found to confer therapeutic effects in various cardiopulmonary diseases, including PH [54,55]. Previous investigation suggests that they exert various immunosuppressive and antifibrotic actions and restore damaged vascular cells to their physiologic state [56,57]. Although it was presumed that MSCs exert their effects by differentiating into pulmonary and vascular cell types, subsequent studies have shown that MSCs may carry out their stabilizing actions through a paracrine mechanism, possibly with secreted EVs as an intermediary. There have been a cadre of studies demonstrating the therapeutic effects of MSCEv in many experimental disease models, including acute myocardial infarction [58,59], stroke [60], renal injuries [61], and PH [8,62].
In a model of hypoxia-induced PH, Lee et al. found that MSCEv were able to attenuate pulmonary vascular wall thickening and remodeling by preventing the activation of pulmonary inflammation and vascular cell hyperproliferation pathways [8]. We evaluated the extent to which MSCEv treatment could attenuate endothelial dysfunction in the in vitro nitrofen CDH model, identifying significant attenuation of pathophysiologic alterations among key endothelial dysfunction-associated proteins. We also show that while nitrofen and CDH alter PA vasoreactivity in nitrofen-treated dam offspring, MSCEv change these contractile responses. Furthermore, we elucidated an important part of the mechanism by which MSCEv are internalized by HPAECs, clathrin-mediated cellular uptake.
To qualitatively assess the ability of MSCEv to restore viability and proliferation in injured endothelial cells, we first performed viability analysis on nitrofen-treated HPAECs. As expected, nitrofen treatment resulted in prominent morphological changes and cell death among HPAECs. A simultaneous treatment of nitrofen and MSCEv, however, led to recovered cell viability compared to nitrofen treatment alone. As higher levels of ROS are associated with cell death, we explored how nitrofen and MSCEv affected ROS levels in HPAECs. We found that nitrofen increased intracellular ROS levels, and MSCEv treatment diminished this increase by 1.5-fold. Our immunoblots demonstrated that nitrofen treatment in cultured HPAECs increased the expression of LOX-1 and its downstream targets NOX-2 and NF-κB. Furthermore, nitrofen treatment decreased the activity of PPARγ. These results support our theory that the activation of LOX-1 and the dysregulation of PPARγ are important in the pathogenesis of CDH-associated PH. In a previous study, we showed that ET-1 is elevated, and eNOS activity is decreased in the nitrofen CDH model [3]. These results expound on the involved upstream pathways, confirming that, in CDH, LOX-1 leads to ROS production by NOX-2, which activates NF-κB and results in ET-1 upregulation and eNOS deactivation. In addition, we confirm that the normal actions of PPARγ in suppressing NOX-2, NF-κB, and ET-1, and maintaining eNOS activity, are impaired. However, following simultaneous treatment with nitrofen and MSCEv, the expression of NF-κB, NOX-2, and LOX-1 are decreased, while the activity of PPARγ is increased, indicating that MSCEv are able to reduce the degree of endothelial dysfunction in nitrofen-induced CDH.
In addition to the in vitro findings, MSCEv also showed a capacity to improve PA function in nitrofen-treated rats. PAs harvested from rats exposed to nitrofen exhibited impaired contraction and relaxation. Following MSCEv treatment, PA contraction was improved, although relaxation did not change to a significant degree. These results demonstrate the physiologic manifestations of the alterations in molecular pathways seen in the protein assays of CDH PAs, as there is an imbalance of the vasoconstrictor ET-1 and vasodilator NO.
Confocal microscopy was used to further characterize the uptake of MSCEv by HPAECs. Both untreated and nitrofen-treated HPAECs were treated with fluorescently labeled MSCEv, and an increased uptake of MSCEv by nitrofen-treated HPAECs was observed. There have been many proposed mechanisms for MSCEv uptake, including clathrin-mediated endocytosis, caveolin-mediated endocytosis, phagocytosis, and micropinocytosis [63]. Clathrin-mediated endocytosis of MSCEv has been commonly described in various disease models, including acute lung injury and PH [64]. To assess the extent to which clathrin-mediated endocytosis is involved in MSCEv internalization in the nitrofen CDH model, amandatine, an inhibitor of clathrin-mediated endocytosis, was administered to both untreated and nitrofen-treated HPAECs before incubation with MSCEv. We found that amantadine significantly reduced, but did not completely eliminate, MSCEv uptake by both untreated and nitrofen-treated HPAECs, with uptake by the latter decreased by a higher proportion. This result suggests that clathrin-mediated endocytosis is partially responsible for MSCEv uptake by HPAECs. Next, we attempted to explore the preferential uptake of MSCEv by nitrofen-treated HPAECs and found that nitrofen treatment greatly increased clathrin expression on the surface of HPAECs. Conversely, amantadine reduced clathrin expression. Taken together, our results validate the important role of clathrin-mediated endocytosis in MSCEv uptake by HPAECs in the CDH model, and we demonstrate that nitrofen treatment causes increased MSCEv uptake, in part, due to the upregulation of clathrin expression on HPAEC surfaces.
These results have important and novel implications for CDH. Due to the complex and incompletely elucidated pathogenesis of CDH, treatment options have been limited. Our study used the nitrofen rodent model to determine the key roles of the LOX-1 and PPARγ pathways and the specific components involved in the disease. These findings suggest potential therapeutic targets to explore in future studies. Our study furthermore demonstrates the effectiveness of MSCEv in alleviating the endothelial dysfunction in CDH by mediating the aforementioned pathways and restoring the vasoreactivity of PAs from rodents with CDH. MSCEv are able to affect multiple dysregulated pathways due to their diverse cargo, ranging from proteins to RNA to microRNA (miRNA) [65]. Therefore, MSCEv carry the wide-ranging therapeutic effects of MSCs without the difficulty of dose control and target accuracy. They also avoid the risks of emboli or uncontrolled growth that come with intravenous administration of MSCs. The fact that MSCEv are naturally secreted by various cell types in the body suggests their therapeutic safety and the extensive potential for cargo customization [7,66]. This study demonstrated the potent cytoprotective and reparative effects of MSCEv in vitro and ex vivo, and future studies will need to explore methods for delivering this therapy in vivo and to determine feasibility in translating this to human disease models.
There are important limitations to this study. First, our in vitro model may not be indicative of pathologic cellular responses to nitrofen in vivo. As opposed to direct effects on the endothelial (or endothelial progenitor) cells, maternal nitrofen exposure may affect endothelial cells (or other components of vasculogenesis) through the retinoic acid pathway or other mechanisms. Second, we have not examined the numerous other pathway alterations that may be critical. Third, despite colocalization, MSCEv may exert effects through interactions outside of LOX-1 and/or ET-1. This study utilized a single “normal” MSCEv product, and future optimization studies are needed to understand and exploit the therapeutic diversity between MSC donors, tissue sources, culture conditions, and pharmacokinetics of MSCEv treatments. Finally, other pathways may be involved in the PA contraction and relaxation responses. For example, our work focuses on specifically endothelial cell dysfunction and does not elucidate the direct effects of MSCEv on nitrofen-induced changes in other important cell types, including vascular smooth muscle cells.
Conclusions
In conclusion, HPAEC exposure to nitrofen led to pathophysiologic cellular changes consistent with known and previously published vasculopathy in CDH. MSCEv treatment led to a more physiologic cellular response, including expression of critical endothelial dysfunction-associated proteins in this experimental in vitro model of nitrofen-induced CDH. Furthermore, we showed that MSCEv were preferentially taken up by cells treated with nitrofen by clathrin-dependent endocytosis. In vivo, MSCEv exposure improved PA contractile response. These data reveal a potential mechanism of cellular interaction and signaling pathway alterations that characterize MSCEv-mediated attenuation of pulmonary vascular dysfunction in CDH-associated PH.
Footnotes
Author Disclosure Statement
There are no disclosures or potential conflicts of interest. All authors have read and approved the submission of the article; the article has not been published and is not being considered for publication elsewhere, in whole or in part, in any language, except as an abstract for presentation at a meeting.
Funding Information
Center for Clinical and Translational Sciences Training Award, University of Texas McGovern Medical School (5KL2-TR000370–10; M.T.H.); Ladybug Foundation (C.S.C. and M.T.H.); and Men of Distinction Foundation (M.T.H.).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.