
Abstract
Exosomes derived from mesenchymal stromal cells (MSCs) have emerged as novel drug and gene delivery tools. Current study aimed to elucidate the potential therapeutic role of human placental MSC (hPLMSC)-derived exosomes carrying AntagomiR-4450 (EXO-AntagomiR-4450) in intervertebral disc degeneration (IDD) progression. Initially, the differentially expressed miRNAs related to IDD were identified by microarray analysis, which provided data predicting the interaction between microRNA-4450 (miR-4450) and zinc finger protein-121 (ZNF121) in IDD. Next, miR-4450 and ZNF121 were elevated or silenced to determine their effects on the damage of nucleus pulposus cells (NPCs) treated with tumor necrosis factor α (TNF-α). The therapeutic effects of EXO-AntagomiR-4450 on NPCs were verified both in vitro and in vivo (15-week-old C57BL/6 male mice); especially gait analysis and fluorescent molecular tomography were used in live mice with IDD. Our results revealed that miR-4450 was highly expressed, while ZNF121 was poorly expressed in IDD patients and NPCs treated with TNF-α. Furthermore, miR-4450 was identified to specifically target ZNF121. In addition, the inhibition of miR-4450 exerted an alleviatory effect on the inflammation, apoptosis, and damage of the NPCs by upregulating ZNF121 (all P < 0.05). Moreover, EXO-AntagomiR-4450 retarded damage of NPCs in vitro, alleviated IDD damage, and ameliorated gait abnormality in vivo (all P < 0.05). hPLMSC-derived exosomes could be a feasible nanovehicle to deliver inhibitory oligonucleotides like AntagomiR-4450 in IDD.
Introduction
Intervertebral disc degeneration (IDD) is considered as the major cause of low back pain, placing a huge burden on global health care systems [1]. The intervertebral disc (IVD) consists of the inner nucleus pulposus (NP) and outer annulus fibrosus (AF), which functions as the load-bearing and buffering unit of the spine [2]. In the degeneration process, the inner NP changes the most, including cell death, extracellular matrix destruction, and accumulation of inflammatory factors [3]. Nucleus pulposus cells (NPCs) are the major cell types residing in NP and responsible for the synthesis and maintenance of the gelatinous extracellular matrix.
Therapeutic methods for IDD focus on the restoration of apoptotic, damaged, or aging NPCs. Mesenchymal stromal cells (MSCs) are considered as potential options for disc repair, which improve IDD progression by supplementing depleted NPCs or ameliorating excessive cell apoptosis during IDD [4]. MSCs are derived from various mesenchymal tissues such as bone marrow and placenta [5], which are the progenitor cells that differentiate into various other adult cells, such as chondrocytes and adipocytes. Compared to bone marrow derived MSCs, placenta derived MSCs (PLMSCs) have more unlimited differentiation potential, therapeutic effects, and lower immunogenicity [6]. Therefore, PLMSCs may be a promising therapeutic selection for IDD.
Recently, increasing evidence demonstrated that MSCs could release a type of specialized extracellular vesicles called exosomes to provide therapeutic benefits [7]. MSC-derived exosomes (MSC-exos) are bilayer vesicles with 50–200 nm in diameter, containing a variety of regulatory nucleic acids [7]. The bilayer membranous shell of exosome prevents the degradation of its contents [8]. Therefore, exosomes have emerged as carriers for the delivery of microRNAs and small interfering RNAs [9]. MSC-exos are likely to be safe and would not cause intrinsic adverse reactions or immune rejection when used as drug delivery vehicles [7]. A series of studies showed that MSC-exos could be potential alternative therapeutic methods in regenerative medicine [10 –12]. Similarly, a recent study showed that MSC-exos could promote NP cell proliferation, enhance extracellular matrix production, and inhibit NPC apoptosis in vitro [13]. Considering the advantages of PLMSCs and exosomes described above, PLMSC derived exosomes (PLMSC-exos) can also be used as natural drug delivery vehicles and may be a potential choice for IDD treatment.
There is growing evidence that strategies to downregulate or prevent the upregulation of specific miRNAs have the potential to treat and/or prevent a wide range of human diseases, including IDD [14]. A significant differential expression of some miRNAs between normal and degenerated NP tissues has been found and has been shown to be related to the occurrence and development of IDD, and many knockdown experiments have been conducted in vitro [14]. To find a new miRNA which is related to IDD progression, we have tried to conduct a miRNA microarray to identify a microRNA which was highly expressed in IDD patients. miR-targeting therapeutics such as miRNA inhibitor or AntagomiR oligonucleotide is an extensive field of study. These RNAs (miRNAs, shRNA, and modified miRNAs) fully function after incubation with recipient cells, leading to target gene knockdown [15]. Exosomes as nanocarriers could be used to deliver miRNAs and small interfering RNAs (siRNAs). Thus, a hypothesis was proposed that human placental MSC (hPLMSC)-derived exosomes carrying microRNA inhibitor of highly expressed microRNA may alleviate the progression of IDD.
Materials and Methods
Ethics approval and consent to participate
This study has been approved by the Human Ethics Committee of the First Affiliated Hospital of Xi'an Jiaotong University (XJTU1AF2017LSL-019). After giving written informed consent, all subjects participated in the study. The placentas were taken from pregnant women who gave birth normally in the First Affiliated Hospital of Xi'an Jiaotong University. The animal experiment protocols had been approved by the Institutional Animal Care and Use Committee of Xi'an Jiaotong University Health Science Center. All animal experiments were carried out in strict accordance with the recommendations of the National Institutes of Health's “Guidelines for the Care and Use of Laboratory Animals”. Institutional review board (IRB) approval of Xi'an Jiaotong University was obtained for this study.
Microarray analysis
The IDD-related gene expression dataset GSE63492 was retrieved from Gene Expression Omnibus database (GSE63492) [16], which included microRNA expression profiling of human NP derived from five patients with IDD in comparison with those derived from five healthy cadaveric discs as normal control. The limma 3.26.8 package was used for differential analyses [17]. Differentially expressed microRNAs were screened out based on the set P value <0.05 and Log FoldChange >2. Analyses were performed using R (v 3.6.1) with Rstudio (1.2.5019). Afterward, miRNA-target gene prediction databases, including miRTarBase (
Isolation and characterization of NPCs
Degenerative NP tissues from four males and six females, aged 32–69 years (average 53.9 years), were collected from patients undergoing surgery for IDD (Supplementary Table S1). Control NP tissues were collected from four non-IDD patients aged 16–22 years (average: 19 years), who underwent idiopathic scoliosis surgery or anterior decompression surgery due to fresh traumatic lumbar fractures with neurological deficits. These patients underwent conventional lumbar magnetic resonance imaging (MRI) scans before surgery. According to the Pfirrmann classification [18], the degree of disc degeneration was graded based on T2-weighted images.
Specimens were cut into small parts (∼1–2 mm3) immediately for various experiments. A portion was immediately immersed in RNA later (Invitrogen, Carlsbad, CA) and frozen in liquid nitrogen for RNA analysis. The other part was immediately immersed in 1 × phosphate buffered saline (PBS) for cell separation. The NP tissues were cut into small pieces of 1–2 mm3, washed three times with PBS supplemented with penicillin and streptomycin, and then digested with 0.5% type II collagenase (Sigma, St. Louis, MO) for 6 h. The tissue debris was then removed through a 75-μm filter. The resulting cells were centrifuged at 250g for 10 min and then resuspended in Dulbecco's modified Eagle's medium/F12 (DMEM/F-12) medium (Gibco, Grand Island, NY) containing 10% fetal bovine serum (FBS; Gibco) and 100 U/mL penicillin–streptomycin. Finally, the NPCs were incubated at 37°C in a humidified atmosphere of 5% CO2. After 1 week, the cells adhering to the well were considered as primary NPCs. The cells from the second passage were used for further experiments.
Characterization of NPCs was described in detail by Wang et al. [19]. Briefly, four-well chambers (Thomas Scientific) were used to culture NPCs. For toluidine staining, the chambers were washed with PBS and then fixed in 4% paraformaldehyde for 30 min, followed by immersing for 2 h in 1% toluidine blue solution (Fisher Scientific), and then they were rinsed in 95% ethanol. For type II collagen staining, endogenous peroxidase was blocked with methanol containing 3% H2O2 and then incubated with 1:100 diluted anti-human type II collagen antibody (Abcam, Cambridge, MA) for 30 min. The secondary antibody conjugated with avidin-biotin-peroxidase (Abcam) and diaminobenzidine substrate solutions were used to visualize the immunoreactivity, followed by counterstaining in hematoxylin, and the slides with cover slips were observed under a microscope.
Dual luciferase reporter gene assay
The target relationship between microRNA-4450 (miR-4450) and zinc finger protein-121 (ZNF121) was initially predicted by the biological prediction tool miRDB (
Twenty-four hours after transfection, the luciferase activity was detected using a dual luciferase reporter gene detection system (Dual-Luciferase® Reporter Assay System, E1910; Promega). Briefly, cells were washed with PBS and lysed with the recommended volume of 1 × Passive Lysis Buffer. The culture vessel was gently shaken/rocked at room temperature for 15 min. The lysate was then transferred to a test tube or vial, centrifuged at 3,200 g for 1 min, and the supernatant was collected. Next, 100 μL of LAR II (Luciferase Assay Substrate in Luciferase Assay Buffer II) equilibrated to room temperature was added to the photometer tube, and carefully draw 20 μL of cell lysate into the photometer tube. The firefly luciferase activity was then measured by shaking gently. Thereafter, 100 μL of Stop and Glo® reagent equilibrated to room temperature was added to measure Renilla luciferase activity.
The relative luciferase activity was expressed as the ratio of firefly luciferase activity to Renilla luciferase activity. Luciferase activity was detected using a GloMax® 20/20 luminometer with a programmed photometer to provide a pre-read delay of 2 s, followed by a measurement time of 10 s. The experiment was repeated three times independently.
NPCs in vitro inflammation induction and transfection
Tumor necrosis factor α (TNF-α) is a key pro-inflammatory cytokine that could induce NPC apoptosis [20]. Therefore, in vitro, TNF-α was usually used as an inducer of a model of IDD. TNF-α (10 ng/mL, 24 h) induced apoptosis of degenerative NPCs (DNPCs) and successfully established an apoptosis model suitable for subsequent experiments. TNF-α was used in subsequent experiments. The DNPCs were treated with negative control (NC) of miR-4450 mimic (NC-mimic), miR-4450 mimic, NC of miR-4450 inhibitor (NC-inhibitor), miR-4450 inhibitor, NC of Vector-ZNF121 (Vector-ZNF121 NC), and Vector-ZNF121. miR-4450 mimic and miR-4450 inhibitor were purchased from GenePharma (Shanghai, China). The respective sequence of miR-4450 mimic and miR-4450 inhibitor was 5′-UGGGGAUUUGGAGAAGUGGUGA-3′ and 5′-UCACCACUUCUCCAAAUCCCCA-3′.
The pCMV6-AC-GFP and pCMV6-AC-GFP-ZNF121 plasmids were purchased from Origene Company and were used as Vector-NC and Vector-ZNF121, respectively. The open reading frame of ZNF121 cloned into this vector will be expressed in mammalian cells as a C-terminal green fluorescent protein (GFP) tagged protein. GFP (maximum excitation/emission = 482/502 nm) was mainly used for applications where fast appearance of bright fluorescence is crucial. The transfection procedure of DNPCs was performed according to the manual of Lipofectamine 2000 (11668-019; Invitrogen, Inc., Carlsbad, CA). DNPCs were seeded in a 12-well culture plate at a density of 1.5 × 105 cells. After incubating overnight, cells were transiently transfected with Vector-ZNF121 NC (1.5 μg/well) or Vector-ZNF121 (1.5 μg/well). Cells were incubated in a CO2 incubator at 37°C for 6–8 h and then cultured for 24 h in fresh complete medium (DMEM/F-12 with 10% FBS and 100 U/mL penicillin–streptomycin) for further experiments.
Isolation and culture of PLMSCs
After signing the written consent, the placentas were collected from 11 female donors immediately after selective cesarean section without artificial labor, premature rupture of membranes, chromosomal abnormalities, or chorioamnionitis. The average maternal age was 29 years (between 26 and 33 years), and the average gestational age is 37.5 weeks. The average placenta weight is 501.23 g. The amnion was dissected from the deflected part of the fetal membranes to minimize the presence of maternal cells. Then the amniotic membranes (3 × 3 × 1 cm size) were dissected under sterile conditions. The tissues were washed thoroughly with 1 × PBS pH 7.4 and cut into small pieces (about 1–2 mm3). Subsequently, the tissue was digested with 1.6 mg/mL collagenase (Sigma-Aldrich) and 200 mg/mL deoxyribonuclease I (Sigma-Aldrich) with shaking at 37°C for 4 h.
After washing twice with PBS, the cells and all pellets were resuspended in MSC growth medium (DMEM; Gibco) supplemented with 10% human serum or 10% FBS (Gibco®) and plated in a 25 cm2 tissue culture flask (Costa, Corning). The flask was placed in a humidified tissue incubator with 5% carbon dioxide and kept at 37°C. The medium was replaced every 3–4 days. The cells were continuously observed until the developmental colonies of fibroblast-like cells were formed. Adherent cells (∼80%–90% confluence) were subcultured with 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA; Gibco) and replated at a density of 1 × 104 cells/cm2 for further expansion. Some batches of continuous subcultured cells were cryopreserved in freezing medium (90% FBS and 10% dimethylsulfoxide) and stored in liquid nitrogen for future use.
Characterization of hPLMSCs
PLMSCs at passage 3–5 cultured in DMEM supplemented with FBS or human serum were detached with 0.25% trypsin-EDTA and washed twice with PBS. In each sample, 4 × 105 cells were resuspended in 50 μL PBS and incubated with 10 μL fluorescein isothiocyanate (FITC) or phycoerythrin (PE)-conjugated antibodies against CD34 (Bio Legend), CD44 (Bio Legend), CD45 (Bio Legend), CD73 (Bio Legend), CD90 (Bio Legend), or CD105 (BD Bioscience), placed in a dark environment at 4°C for 30 min. After washing with PBS, the cells were fixed with PBS containing 1% paraformaldehyde. Positive cells were identified by comparison with isotype matching controls [PE-conjugated mouse immunoglobulin G1 (IgG1) and FITC-conjugated mouse immunoglobulin G2a (IgG2a)]. At least 20,000 labeled cells were collected and analyzed using a flow cytometer (FACS caliburTM; Becton Dickinson), and then the results were analyzed using FlowJo V10 software.
Lentivirus infection in PLMSCs
By the miRNA lentiviral vector system (OriGene, Inc.), recombinant lentivirus containing the AntagomiR-4450 sequence and negative recombinant lentivirus without the AntagomiR-4450 (AntagomiR-NC) were prepared. The synthesized AntagomiR-4450 sequence was cloned into the pLenti-C-mGFP-P2A-Puro miR plasmid of the retroviral expression vector system (OriGene, Inc.) and transformed into DH5α competent cells. Then the pLenti-C-mGFP-P2A-Puro AntagomiR-4450 plasmid DNA was prepared and purified. Using liposomes, the recombinant plasmid and the packaging plasmid containing the target gene were cotransfected into HEK293T cells to construct an experimental lentivirus. Similarly, the plasmid without the target gene and the packaging plasmid were cotransfected into HEK293T cells to construct NC lentivirus. After incubating in 5% CO2 at 37°C for 4–6 h, the cells were further cultured in DMEM supplemented with 10% FBS for 72 h. The virus-containing supernatant was collected and then filtered with a 0.45 μm cellulose acetate filter (Merck Millipore, Burlington, MA) and stored at −80°C.
Subsequently, PLMSCs in the logarithmic growth phase were inoculated into a six-well plate at a seeding density of 1 × 106 cells/mL and incubated for 24 h at 37°C with 5% CO2. A lentivirus solution was prepared by dilution with complete medium supplemented with 10% FBS, and Coagel was added at 5 μg/mL to enhance infection ability. After setting the NC group, 24 h after infection, the medium was replaced with fresh complete medium. After 72 h of further cultivation, exosomes were extracted for subsequent experiments.
Exosome purification and tracking analysis
The cells were cultured in medium supplemented with 10% exosome-depleted FBS (ultracentrifugation at 100,000g overnight) for 48–72 h. The supernatant was collected by differential centrifugation methods. The supernatant was collected and centrifuged at 300g for 10 min to remove the cell pellet. The supernatant was then centrifuged at 2,000g for 10 min to remove dead cells and at 10,000g for 30 min to remove cell debris. The exosomes were then harvested by centrifugation at 100,000g for 70 min. The exosome pellet was washed twice by resuspending in 20 mL PBS and ultracentrifuging at 100,000g for 70 min (Sorvall SureSpin 630 rotor). A nanoparticle characterization system (NanoSight NS-300 Instrument, Malvern, United Kingdom) equipped with a blue laser (405 nm) was used to characterize vesicles in real time.
Electron microscopy
The exosomes purified as described above were fixed in 200 mM phosphate buffer (pH 7.4) containing 2% paraformaldehyde (w/v). The fixed exosomes were dropped onto the grid of the carbon coating of Forval and dried at room temperature for 20 min. After washing with PBS, the exosomes were fixed in 1% glutaraldehyde for 5 min, washed in water, and stained with saturated aqueous uranyl oxalate solution for 5 min. The samples were then embedded in 0.4% (w/v) uranyl acetate and 1.8% (w/v) methyl cellulose and incubated on ice for 10 min. Then the excess liquid was removed. The grid was dried at room temperature for 10 min and observed using an electron microscope (Model 910; Carl Zeiss) at 20,000 and 50,000 magnifications.
Uptake assay of exosomes by NPCs
The purified exosomes were directly labeled with 1 μM Vybrant Cell Tracers DiL (Life Technologies) by incubating at 37°C for 30 min. The labeled exosomes were washed twice in 20 mL PBS, collected by ultracentrifugation as described above, and resuspended in PBS. We verified that no dye contamination occurred in the DiL-labeled exosome preparation by ultracentrifugation as described above. NPCs were stained with 1 μM DiL (Life Technologies) and seeded into the eight-well chamber and incubated with DiL-labeled exosomes (1 × 1010 particles/mL) at 37°C for 24 h under 5% CO2.
Exosome treatment in vitro
The DNPCs were seeded into 6-well or 12-well plates the day before treatment. When the cell concentration reached 70%, 1 × 1010 particles/mL exosomes (Exo-AntagomiR-NC or Exo-AntagomiR-4450) were introduced into the DNPC medium. DNPCs treated with PBS were regarded as blank controls. After 48 h of treatment, cells were collected for later use.
5-bromo-2-deoxyuridine cell proliferation assay
Cell proliferation rate was evaluated using a commercially available 5-bromo-2-deoxyuridine (BrdU) immunohistochemistry kit (Abcam). This method was based on the incorporation of BrdU into the newly synthesized DNA chain of proliferating cells. All steps were performed according to the manufacturer's manual. BrdU analysis was used to study the role of miR-4450, ZNF121, and related exosomes in DNPC proliferation. In short, the cultured cells (1 × 105/well NPCs) were seeded into a four-well chamber (Thomas Scientific) and incubated with 10 ng/mL TNF-α for 24 h and then treated with relevant reagents.
The vector or lentivirus was transfected and incubated for 24 h or exosomes treated and incubated for 48 h, then the medium was removed, the cells were washed twice with 1 × PBS, and labeled with 10 μM BrdU at 37°C for 6 h. Cells were fixed with 4% paraformaldehyde (Sigma-Aldrich; Merck-Millipore) for 30 min and incubated with biotinylated anti-BrdU antibody for 90 min at room temperature. Subsequently, streptavidin–horseradish peroxidase conjugate and diaminobenzidine substrate were added, and the slides with coverslips were observed under a microscope. The BrdU incorporation was quantified by ImageJ 1.52a software (National Institutes of Health). The cell proliferation rate was expressed as the number of positively stained cells divided by the total number of cells (set at 100%).
MTT assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazole bromide (MTT, MTT Cell Proliferation Assay Kit; Abcam) was used to measure cell viability. Cells (1 × 105/well) were seeded into 96-well microplates and exposed to the indicated reagents at 37°C for 48 h before adding MTT. After treatment, the medium was carefully aspirated, and then 50 μL of serum-free medium and 50 μL of MTT reagents were added into each well and incubated at 37°C for 3 h until a purple precipitate was visible. After incubation, 150 μL of MTT solvent was added to each well. The absorbance at 590 nm was measured by a microplate reader (ELx800 Absorbance Microplate Reader). All measurements were repeated three times.
Cell migration assay
The cell migration rate was measured using a transwell chamber (BD Biosciences, Franklin Lakes, NJ). Trypsin-digested DNPCs were diluted to a final concentration of 2 × 106 cells/mL in serum-free medium, and 100 μL of cell suspension was added to the upper chamber, while 0.6 mL of DMEM containing 10% FBS was added to the lower chamber. DNPCs were transfected with vector or lentivirus and incubated for 24 h, or treated with exosomes and incubated for 48 h, then the medium was removed, and the cells were washed twice with 1 × PBS. After removal of the medium, chambers were incubated with 4% paraformaldehyde (Sigma-Aldrich; Merck-Millipore) for 15 min to fix the migrated cells on the lower side of the insert filter membrane, and the cells on the upper side of the filter membrane were removed with cotton swabs.
The cells on the underside of the filter membrane were stained with 0.1% crystal violet (Sigma-Aldrich; Merck-Millipore) for 10 min. Then, after washing with 1 × PBS (Gibco; Thermo Fisher Scientific, Inc.), the cells on the lower side of the filter membrane were clearly visible and counted under a microscope (IX-70; Olympus Corporation, Tokyo, Japan). The cells that have migrated through the membrane were stained and counted, and NPC migration was expressed as the total number of cells that have migrated.
Annexin-V propidium iodide staining
Apoptosis Detection Kit (Abcam) of Annexin V-FITC was used for the detection of cell apoptosis. After 48 h of treatment, the cells were detached with trypsin (YB15050057; Yubo Biotechnology Co., Ltd., Shanghai) free of EDTA. DNPCs (1–5 × 105) were collected by centrifugation, washed once with cold serum-containing medium, and then resuspended in 500 μL 1 × Binding Buffer. Five microliters of Annexin V-FITC and 5 μL of propidium iodide (PI, 50 μg/mL) were added to the suspended cells and incubated for 5 min at room temperature in the dark. Then, FITC signal detector (usually FL1) was used to perform annexin V-FITC binding analysis on the prepared sample by flow cytometry (Ex = 488 nm; Em = 530 nm) within 1 h. The PE emission signal detector (usually FL2) performed PI staining analysis.
To compensate and do gate settings, a NC (binding buffer alone, unstained), a positive control, and single staining of annexin V-FITC (no PI) and PI (no annexin V-FITC) were used. Usually, FITC Annexin V and PI double negative (feasible or no measurable apoptosis) were used to track cells, FITC Annexin V positive and PI negative (early apoptosis, membrane integrity) were used to trace early apoptotic cells, and finally, FITC Annexin V and PI positive (final stage apoptosis and death) were applied to trace late apoptosis or dead cells.
Establishment of a mouse model of IDD
Forty 15-week-old C57BL/6 male mice were obtained from the Experimental Animal Center of the Health Science Center of Xi'an Jiaotong University, and the study protocol passed the review and approval of the Ethics Committee of the Health Science Center of Xi'an Jiaotong University. The mice were randomly divided into sham operation, IDD + PBS, IDD + Exo-AntagomiR-NC, and IDD + Exo-AntagomiR-4450 groups, 10 mice each group. The mice were placed in a room under standard laboratory conditions (temperature controlled at 21°C ± 1°C) and reared with a normal 12-h light/12-h dark cycle. The research protocol involving animal procedures was consistent with the guidelines of the Rush Institutional Animal Care and Use Committee (IACUC) and the IACUC Health Science Center of Xi'an Jiaotong University.
All surgical operations were performed according to the protocol by Shi et al. [21]. The mice (15 weeks old) were placed in a supine position and anesthetized with oxygen containing 1.5% isoflurane (Abbott Laboratories, North Chicago) through the mask at a rate of 2 L/min. After shaving and disinfecting the skin, use a #15 blade scalpel to make a longitudinal incision on the outside of the left abdomen with a length of about 1.5 cm. The peritoneum was separated from the subcutaneous fat and pulled to the right to expose the space between the back of the peritoneum and the left psoas muscle. Through this space, the posterior peritoneum was gently pulled to the right to make the spine visible and easily accessible. Use the pelvic edge as an anatomical landmark to identify L5/6 and L6/S1 IVDs.
To induce IDD, first, under the guidance of a polyethylene stopper sleeve (BD Intramedic™ polyethylene tube; BD Biosciences, Franklin Lakes, NJ), a sharp micro-scalpel was used to puncture the IVD at a depth of 0.7 mm. Through the same incision, another micro-scalpel with a bending point was used for a second puncture to destroy and remove the NP tissue. Except for the absence of IVD puncture, the mice in the sham operation group received all the operations before IDD surgical puncture. Then the IDD + PBS, IDD + Exo-AntagomiR-NC, and IDD + Exo-AntagomiR-4450 were injected with 2 μL PBS, 2 μL 1 × 1010 particles/mL Exo-AntagomiR-NC, and Exo-AntagomiR-4450, respectively. Then, the skin incision was sutured with 4-0 vein suture. After the operation, penicillin (Beijing Bayerdi Biotechnology Co. Ltd.) was injected into the thigh muscle of the mice, 40,000 U/kg per day according to the instructions for three consecutive days.
On the 42nd day after surgery and injection, the mice were sacrificed with carbon dioxide. After removing the fur and tendon tissue, under a dissecting microscope (VistaVision; VWR International, Radnor, PA), the IVD tissue was separated from the adjacent cartilage endplate and bone with a scalpel.
Gait analysis
The gait was recorded using the DigiGait 9.0 analysis system (Mouse Specifics, Inc., Quincy, MA) [22]. In short, the mouse ran at a specific speed on a transparent flat treadmill, and the camera captured ventral images. For each measurement, the animal ran for a maximum of 30 s, of which 5 s (equivalent to 10 consecutive steps) were used for analysis. Each mouse underwent a habituation test before the experimental test, and the animal was required to walk at a slightly faster speed (10 cm/s) for 10 s. DigiGait image analysis software automatically defined each paw area, generated a waveform describing the forward/backward movement of each limb in a continuous stride, and determined the time period during which each paw contacted on the treadmill was the standing phase and the middle period was the swing phase.
We could also calculate posture and movement gait measurements, including stride time, stride length, and paw area. Brake and propel times were defined as the time before and after the maximum paw area during the stance phase, and the paw angle was expressed as the angle of the paw relative to the long axis [22]. The symmetry index is defined as the absolute value of the difference between the contralateral hindlimbs divided by its average value. Before conducting the experimental tests, all settings (ie, camera, lighting, belt speed) were optimized.
After studying a series of speeds (Study 1) and reproducibility (Study 2), the treadmill speed used for the subsequent research (Study 3) was set to 15 cm/s. It also recorded that the mouse treadmill task was not in compliance (ie, refuses to perform or complete the treadmill running task, which was considered to be the mouse's inability or reluctance to perform more than two consecutive steps). Data were recorded at the time points of day 7 and 42 after surgery and injection. Considering that IDD mainly affected two hind legs, and the interference of any one of the four legs would affect the remaining legs, the right hind leg which was researched in multiple studies was selected for analysis.
Fluorescence molecular tomography
Inflammation-related factors and matrix metalloproteinases (MMPs) were monitored by fluorescence molecular tomography (FMT) in vivo at day 21 and 42 after surgery and injection, respectively. FMT could perform real-time three-dimensional quantitative analysis of the distribution of fluorescent dyes in living animal tissues. Twenty-four hours before imaging, mice were injected with single dose of ProSense 680 or MMPSense 680 fluorescent imaging agents (PerkinElmer, Waltham, MA). ProSense detected changes in the activity of lysosomal cathepsin (a protease), which could indicate many processes related to disease, such as cancer, inflammation, arthritis, and so on. MMPs were calcium-dependent zinc-containing endopeptidases, responsible for the degradation of most extracellular matrix proteins, and were associated with a variety of diseases, including inflammation and arthritis.
MMPSense fluorescent agent was designed to detect the activity of MMPs (MMP-2, MMP-3, MMP-9, and MMP-13) and to evaluate many disease-related processes, including cancer progression, rheumatoid arthritis, lung disease, and so on. Moreover, MMPSense also was used to evaluate the therapeutic efficacy of MMPs and potential drug candidates. The mice were anesthetized by intraperitoneal injection of ketamine (75 mg/kg) and medetomidine (1 mg/kg), placed in the imaging cassette, and then imaged using the VisEn FMT optical imaging system (PerkinElmer). A near-infrared laser diode emitting continuous wave radiation at wavelengths of 670 nm transilluminated the lower body of each animal from posterior to anterior, and both excitation and emission signals were detected by a charge-coupled device camera and appropriate band-pass filters.
The fluorescence intensity was quantified by evaluating the total radiation efficiency of the signal in the region of interest (ROI) ([photon/s]/[mW/cm2]). The ROI was a 12.3 mm2 rectangle. It was selected from the anatomy of the disc segment on the grayscale photo of the mouse. Therefore, the ROI selection included the entire disc and was not affected by the fluorescent signal. A threshold of 10% of the maximum fluorescence in each reconstructed volume was used. The peak concentration (nmol/L) and total amount (pmol) of the fluorescent dye were automatically calculated relative to the internal standard generated by the appropriate dye of known concentration.
Histological evaluation
On the 42nd day after the surgery and injection, mice were sacrificed. The lumbar disc specimens L5/6 were taken, fixed by 4% paraformaldehyde, decalcified by EDTA, paraffin-embedded, sectioned, and histologically evaluated. The IVD was sectioned in a coronal plane using a microtome. Three to four representative 5-μm-thick mid-coronal sections were selected and stained with Safranin O (SO)/Fast Green to evaluate the pathological changes of IVD. The histological score was evaluated using the modified grading scale reported by Masuda et al. [23] (Supplementary Table S2).
Enzyme linked immunosorbent assay
The lumbar disc specimens L6/S1 were taken for enzyme linked immunosorbent assay (ELISA). The expression of TNF-α and MMP13 was determined according to the operational manual provided by ELISA Kit (Neobioscience Biotechnology Co., Ltd., Shenzhen, China). The optical density value of each well was determined at a wavelength of 450 nm within 20 min.
Reverse transcription–quantitative polymerase chain reaction
According to the manufacturer's instructions, total RNA was extracted from cells or tissues using TRIzol reagent (Invitrogen) and quantified spectrophotometrically at 260 nm. The acceptable optical density 260/280 ratio was between 1.8 and 2.0. RNA quality was also determined by 1% agarose gel electrophoresis and stained with 1 mg/mL ethidium bromide. RNA was incubated with RNase-free DNase (Promega) to remove residual genomic DNA. The expression level of miR-4450 was quantified by TaqMan microRNA analysis (Life Technologies) specific for mature miR-4450. Briefly, 10 ng RNA was transcribed by TaqMan microRNA RT Kit (Life Technologies), and real-time polymerase chain reaction (PCR) was performed using TaqMan MicroRNA Assays.
The ubiquitously expressed miRNA snRNA U6 was used as an endogenous control. Levels of mRNA were quantified by real-time PCR using SYBR Green PCR Master Mix (Qiagen). Real-time PCR was performed on the ABI Prism 7900HT fast system (Applied Biosystems). Primers of ZNF121 were designed by Primer 5 software according to the related sequences provided by GeneBank, and the primers of miR-4450 were designed and synthesized by Shanghai Sanyuan Bioengineering Technology and Service Co., Ltd. (Shanghai, China). BLAST software was used to design primers for downstream target genes by homology analysis. The primer sequences are shown in Supplementary Table S3. Amplifications were performed in a total volume of 20 μL with 1 μL cDNA.
Reverse transcription quantitative polymerase chain reaction (RT-qPCR) was conducted with the following condition: 1 cycle of predenaturation at 95°C for 15 min, and 45 cycles of denaturation at 94°C for 15 s, annealing at 55°C for 30 s, and extension at 70°C for 30 s. The relative mRNA expression was quantified by a comparison of the cycle threshold (Ct) values. The relative transcript level was calculated using the 2−ΔΔCt method and normalized to the internal reference gene. Each experiment was repeated three times.
Western blot analysis
Exosomes and cells were lysed using RIPA lysis buffer (Thermo Scientific), then total protein was collected, and protein concentration was determined using BCA Kit (20201ES76; Yasen Company, Shanghai, China). Next, 20 μg/well protein was loaded and separated by 8% sodium dodecyl sulfate–polyacrylamide gel electrophoresis for 1.5 h. The protein was then transferred to a polyvinylidene fluoride membrane and then sealed with 5% skim milk. Thereafter, rabbit polyclonal antibodies against β-actin (ab8227; 1:1,000), CD9 (ab223052; 1:500), CD63 (ab134045; 1:400), ZNF121 (PA5-63043; 1:200), MMP13 (ab39012; 1:500), cleaved-Caspase3 (c-Caspase3, ab2302; 1:400), and collagen II (COL2, ab34712; 1:500) were incubated with the membrane at 4°C overnight.
The membrane was then incubated with the secondary antibody goat anti-rabbit IgG H & L (488) (ab150077; 1:1,000) at room temperature for 1 h. All the above antibodies were purchased from Abcam, Inc. (Cambridge, United Kingdom) except for ZNF121, which was from Invitrogen Company. Next, the membrane was imaged using an Odyssey fluorescence scanner (LI-COR Biosciences, Lincoln, NE). Finally, ImageJ was used to analyze the relative protein expression by scanning the protein strips in gray. Each reaction was repeated three times.
Statistical analysis
All data analyses were performed using SPSS 21.0 software (IBM Corp., Armonk, NY). The measurement data are expressed as mean ± standard deviation. t-Test was applied to the comparison of two groups. For data comparison between multiple groups, one-way analysis of variance was used, and Tukey test was used for post hoc test. P < 0.05 was considered a statistically significant indicator.
Results
miR-4450 is significantly upregulated in IDD
Monolayer cultured NPCs changed from rounded or polygonal primary NPCs (Passage 0) (Fig. 1A[a]) to a spindle shape after subculture (≥Passage 3) (Fig. 1A[b]). NPCs were chondrocyte-like cells, the cytoplasm of the NPCs was stained blue by toluidine blue staining (Fig. 1A[c]) and brown by type II collagen immunochemistry staining (Fig. 1A[d]), respectively, which verified the expression of glycosaminoglycan and type II collagen. IDD-related microRNA expression dataset GSE63492 was analyzed by R language. Both heatmap and volcano plot identified that miR-4450 (miRBase: MI0016795) was highly expressed in IDD relative to normal controls (Fig. 1B, C).
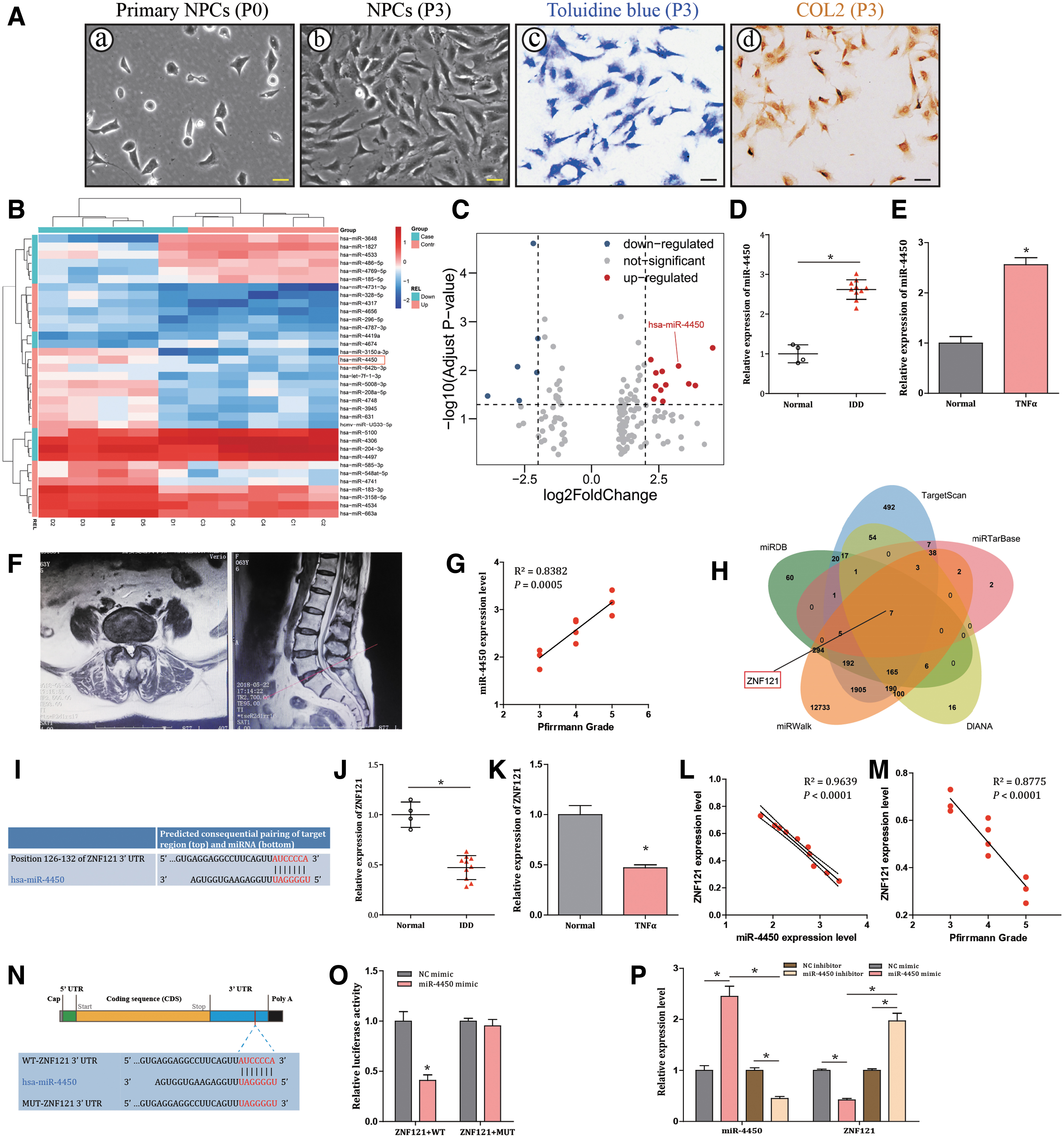
miR-4450 is assumed as a putative microRNA that might target ZNF121 to influence IDD progression.
Furthermore, our experimental results revealed that miR-4450 expression level was upregulated in both IDD tissues and NPCs treated with TNF-α compared with normal control (both P < 0.001; Fig. 1D, E). Figure 1F showed a representative of a MRI sagittal and axial view of an IDD patient with Pfirrmann-grade-IV L5/S1 IVD herniation. Meanwhile, a positive correlation was observed between miR-4450 and Pfirrmann grade in IDD patients (R 2 = 0.9639, P < 0.0001; Fig. 1G). Thus, miR-4450 was predicted to be implicated in IDD progression.
ZNF121 is a putative target of miR-4450
A Venn map based on target-predicting database (miRTarBase, DIANA, miRDB, miRWalk, TargetScan, and StarBase) showed that ZNF121 (MIM: 194628) was likely regulated by miR-4450 (Fig. 1H). Moreover, miR-4450 was predicted to bind to 3′ UTR of ZNF121 with conserved target sequence (AUCCCCA) (Fig. 1I). Furthermore, ZNF121 expression level was downregulated in both IDD tissues and NPCs treated with TNF-α compared with normal control (both P < 0.001; Fig. 1J, K). Besides, ZNF121 was found to be negatively correlated with miR-4450 (Fig. 1L), as well as Pfirrmann grade (Fig. 1M), in IDD patients. Moreover, dual luciferase reporter gene assay was designed (Fig. 1N) and further verified that miR-4450 could specifically bind to ZNF121 (P < 0.001; Fig. 1O). Furthermore, the results revealed that miR-4450 mimic triggered a decrease in the expression of ZNF121 in NPCs (P < 0.001), while miR-4450 inhibitor induced reverse effects (P < 0.001; Fig. 1P). Taken together, the results indicated that ZNF121 was poorly expressed in IDD and could specifically be bound by miR-4450.
Overexpression of miR-4450 aggravates the damage of NPCs
Afterward, to investigate the effect of miR-4450 in NPCs, the NPCs were transfected with miR-4450 mimic or miR-4450 inhibitor, followed by the TNF-α treatment to induce the inflammation in the cells. The findings revealed that miR-4450 mimic triggered an increase in the expression of miR-4450 in NPCs (P < 0.001), while miR-4450 inhibitor induced reverse effects (P < 0.001; Fig. 2A). Moreover, the results also revealed that there was a decrease in proliferation (P < 0.001) and migration (P < 0.001), as well as an increase in apoptosis (P < 0.001), following the treatment of miR-4450 mimic (Fig. 2B–H). These trends could be reversed following treatment with miR-4450 inhibitor (all P < 0.001; Fig. 2B–H).
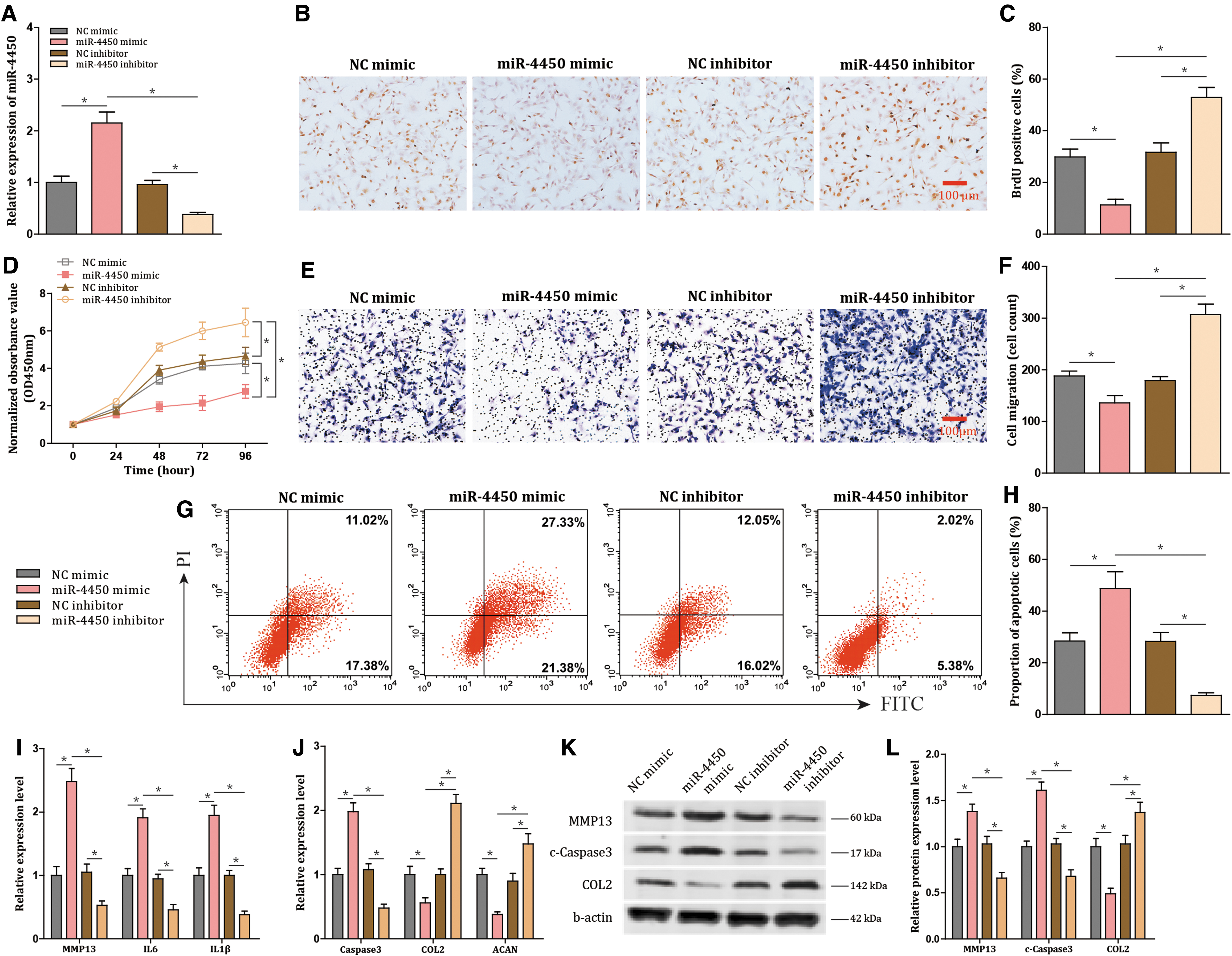
miR-4450 elevation aggravates the damage of NPCs. NPCs treated with TNF-α are transfected with miR-4450 mimic or miR-4450 inhibitor.
Moreover, as shown in Fig. 2I and J, the treatment of miR-4450 mimic led to an elevation in the expression of MMP13 (P < 0.001), interleukin (IL)-6 (P < 0.001), IL-1β (P < 0.001), and Caspase-3 (P < 0.001), which was accompanied by a decline in expression of COL2 (P < 0.001) and aggrecan (ACAN) (P < 0.001), while treatment with miR-4450 inhibitor resulted in an opposite tendency (all P < 0.001). Furthermore, the protein expression levels of MMP13, c-Caspase3, and COL2 showed the same tendencies as their mRNA expression levels (all P < 0.001; Fig. 2K, L). Therefore, overexpressed miR-4450 increased apoptosis and inflammatory response of NPCs and decreased protective extracellular matrix, proliferation, and migration; vice versa the damage of NPCs was alleviated by downregulating miR-4450 expression.
miR-4450 exacerbates NPC damage potentially by targeting ZNF121
Next, NPCs treated with TNF-α were transfected with miR-4450 mimic and Vector-ZNF121. Vector-ZNF121 increased expression of ZNF121, cell proliferation, and cell migration and declined cell apoptosis (all P < 0.001; Fig. 3A, H). Moreover, Vector-ZNF121 decreased expression of Caspase-3, MMP13, IL-6, and IL-1β and increased the expression of COL2 and ACAN (all P < 0.001; Fig. 3I, J). However, miR-4450 mimic reversed the aforementioned results with the above indicators (all P < 0.001; Fig. 3A–J). Furthermore, the protein expression levels of ZNF121, MMP13, c-Caspase3, and COL2 demonstrated the same tendencies as their mRNA expression levels (all P < 0.001; Fig. 3K, L). Thereby, miR-4450 exacerbated NPC damages mediated by downregulating its target ZNF121.
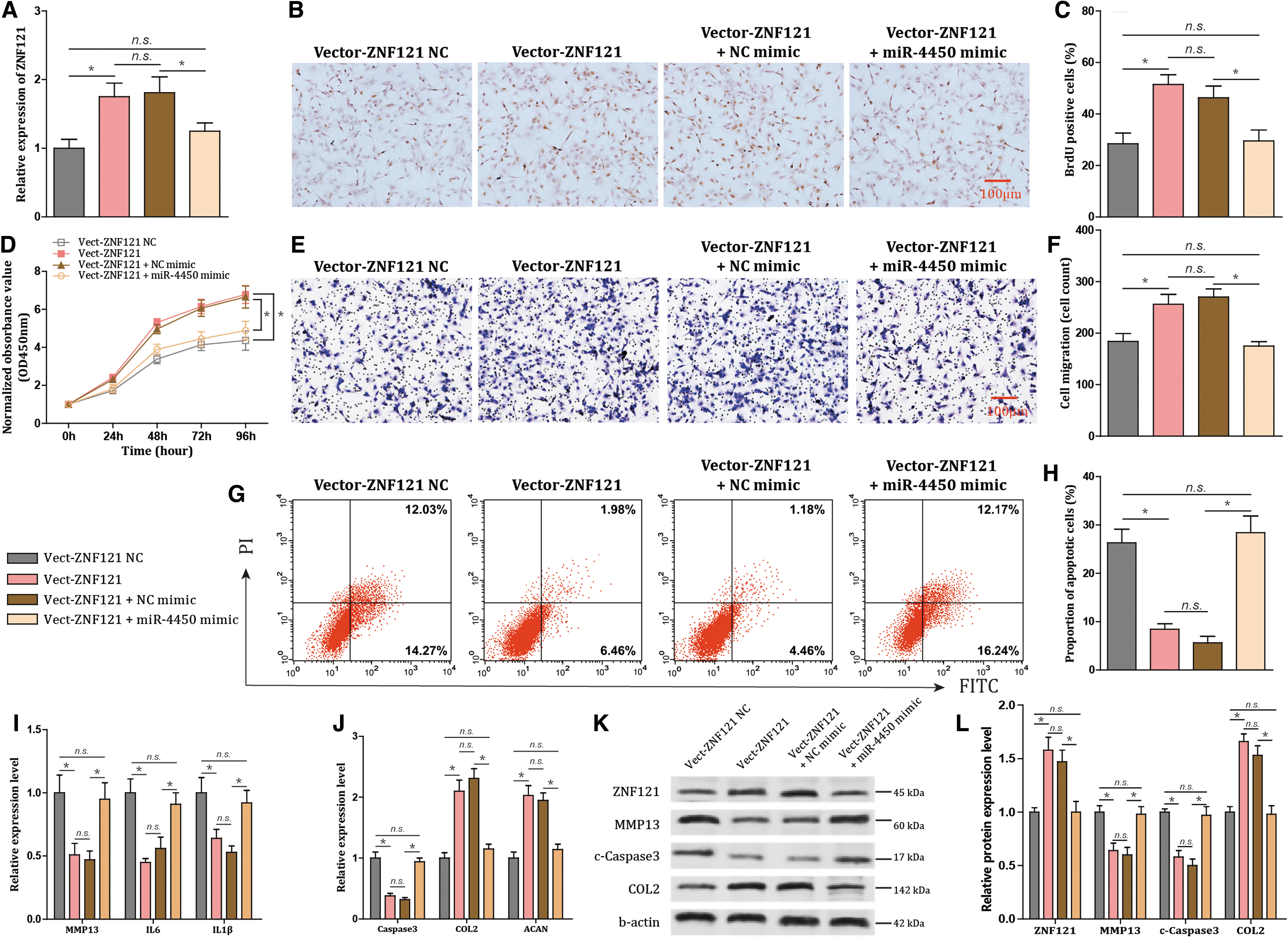
miR-4450 contributes to exacerbated damage of NPCs by downregulating ZNF121.
hPLMSC-derived exosomes transfer AntagomiR-4450 into NPCs
Next, further investigations were performed to assess the vector function of hPLMSC-derived exosomes transporting AntagomiR-4450 to NPCs. Initially, hPLMSCs were isolated from amniotic membranes of placentas, adherent to the bottom of culture dish and had the morphology of fibroblasts. The appearance of hPLMSCs was spindle shaped, refractive, and spiral shaped (Fig. 4A). Next, both three-lineage differentiation assay and flow cytometry revealed that hPLMSCs had preserved the stem cell characteristics (Fig. 4B, C). Moreover, hPLMSC-derived exosomes were isolated by differential centrifuge (Fig. 5A) and characterized with classical cup-shaped bubbles with diameters ranging from 30 to 150 nm (Fig. 5B–E) and positive CD9 and CD63 markers (Fig. 5F). In addition, exosomes labeled with DiL (red) were cocultured with NPCs labeled with DiO (green), and the exosomes were uptaken by the NPCs (Fig. 5G). These results demonstrated the successful delivery of AntagomiR-4450 into NPCs by hPLMSC-derived exosomes.
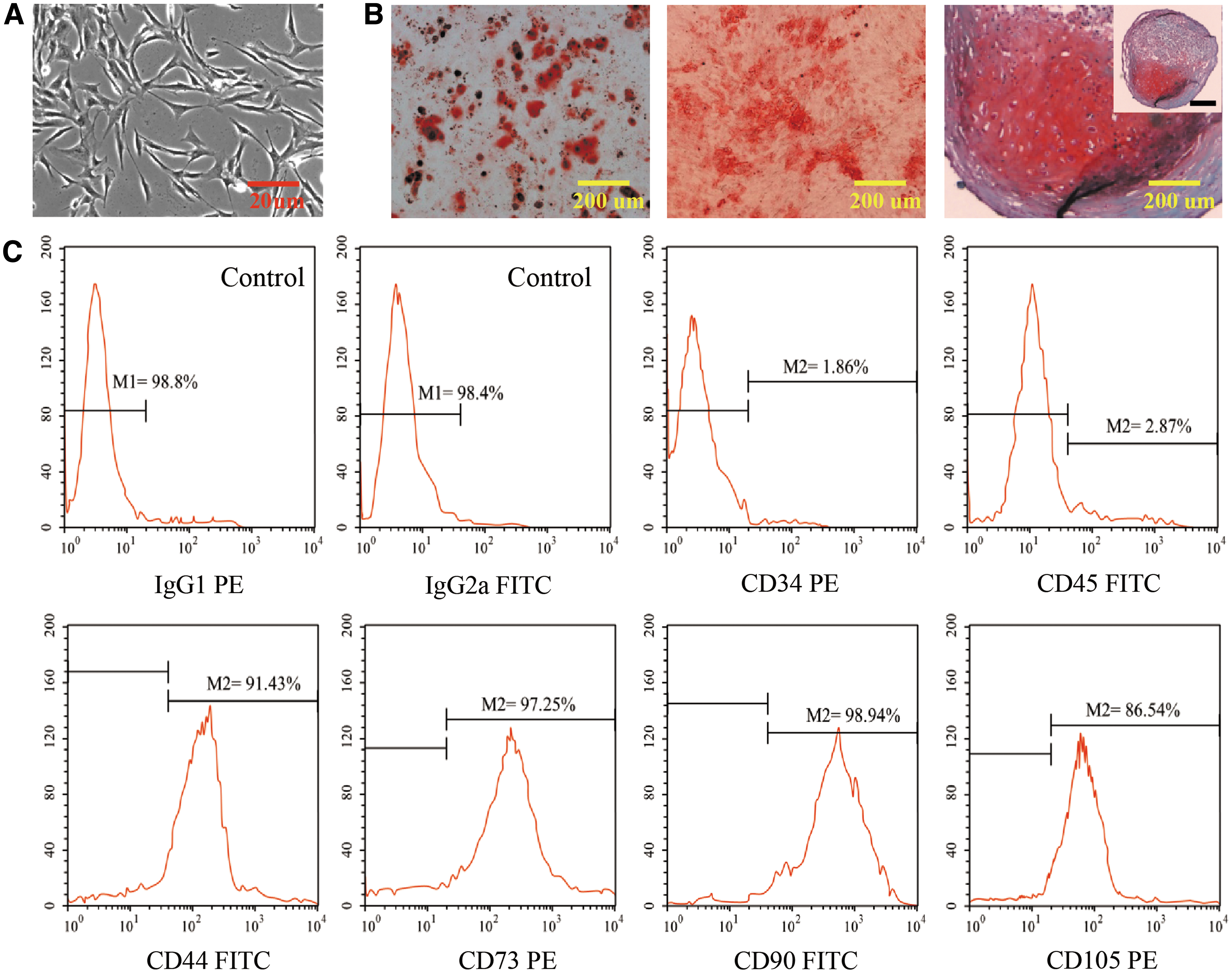
Characterization and differentiation assay of hPLMSCs.
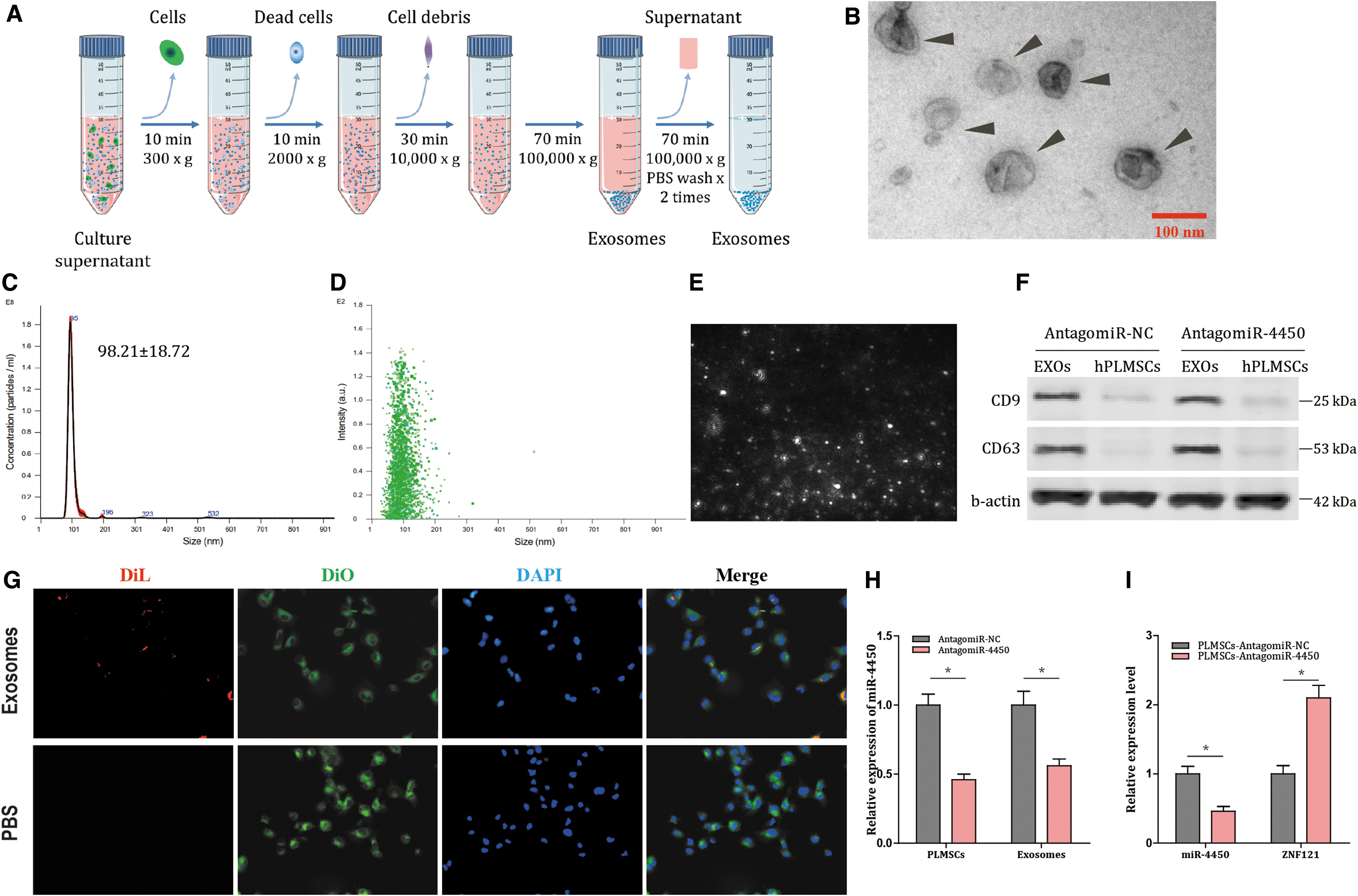
Isolation and characterization of hPLMSC-derived exosomes, and AntagomiR-4450 is transferred into NPCs by hPLMSC-derived exosomes. miR-4450 is transferred into NPCs by hPLMSC-derived exosomes.
hPLMSC-derived exosomes expressing AntagomiR-4450 alleviate damage of NPCs
To investigate the effects associated with the exosomes derived from hPLMSCs with AntagomiR-4450 expression (Exo-AntagomiR-4450) or not (Exo-AntagomiR-NC) in NPCs, exosomes were extracted from hPLMSCs and added into NPCs treated with TNF-α (Fig. 6A). The NPCs treated with exosomes exhibited decreased expression of miR-4450 but increased ZNF121 expression (all P < 0.001; Fig. 6B). Meanwhile, after the NPCs were cocultured with Exo-AntagomiR-4450, cell proliferation (P < 0.001; Fig. 6C–E) and migration were enhanced (P < 0.001; Fig. 6G, H), and cell apoptosis was decreased (P < 0.001; Fig. 6J, K). In addition, Exo-AntagomiR-4450 treatment declined Caspase-3, MMP13, IL-6, and IL-1β expression (all P < 0.001; Fig. 6F, I). Furthermore, the protein expression levels of ZNF121, MMP13, c-Caspase3, and COL2 demonstrated the same tendencies as their mRNA expression levels (all P < 0.001; Fig. 6L, M). These results demonstrated that damage of NPCs was retarded by transfer of AntagomiR-4450 by hPLMSC-derived exosomes.
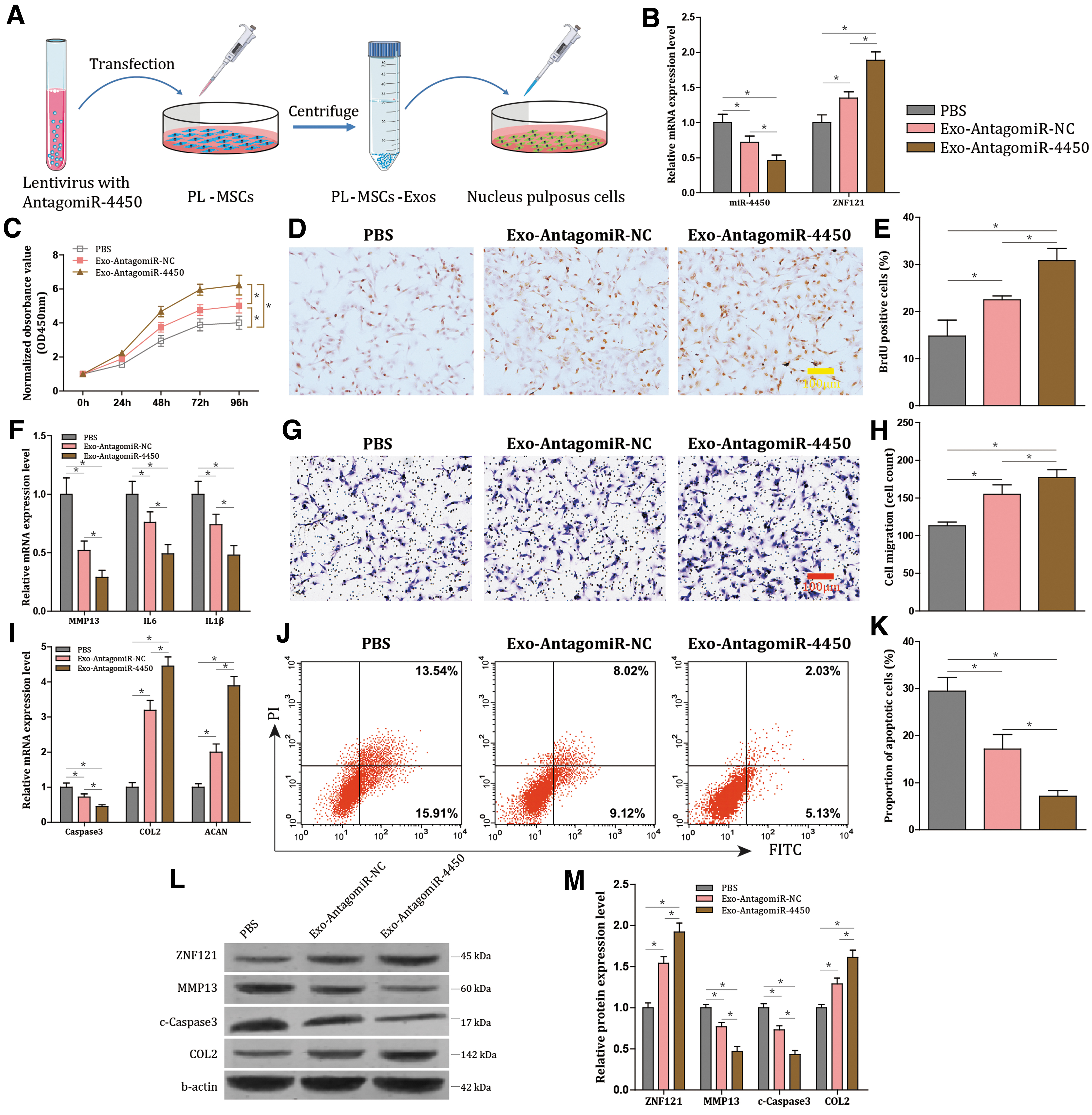
hPLMSC-derived exosomes harboring AntagomiR-4450 ameliorate damage of NPCs.
PLMSC-derived exosomes harboring AntagomiR-4450 retard IDD damage in vivo
Finally, the effect of hPLMSC-derived exosomes on mice with IDD was evaluated in vivo. Gait analysis evaluated the gait of mice at day 7 and 42 after injection (Fig. 7A). A representative gait analysis result was showed (Fig. 7B). At day 7, the results revealed that compared with the sham group, the remaining three groups with IDD model showed decreased swing time, brake time, and paw area and increased stride frequency (all P < 0.001; Fig. 7C), and no differences were observed among the groups with IDD model regardless of what reagents were injected (all P > 0.500; Fig. 7C). At day 42, compared with the IDD + PBS group, IDD + EXO-AntagomiR-4450 or EXO-AntagomiR-NC exhibited decreased swing time, brake time, and paw area and increased stride frequency (all P < 0.001; Fig. 7D). However, compared with sham group, both EXO-AntagomiR-4450 and EXO-AntagomiR-NC showed no difference for the aforementioned indices (all P > 0.380; Fig. 7D), which indicated that both these exosome groups could restore the gait abnormality. In brief, these results suggested that EXO-AntagomiR-4450 could partially improve the gait patterns of IDD mice.
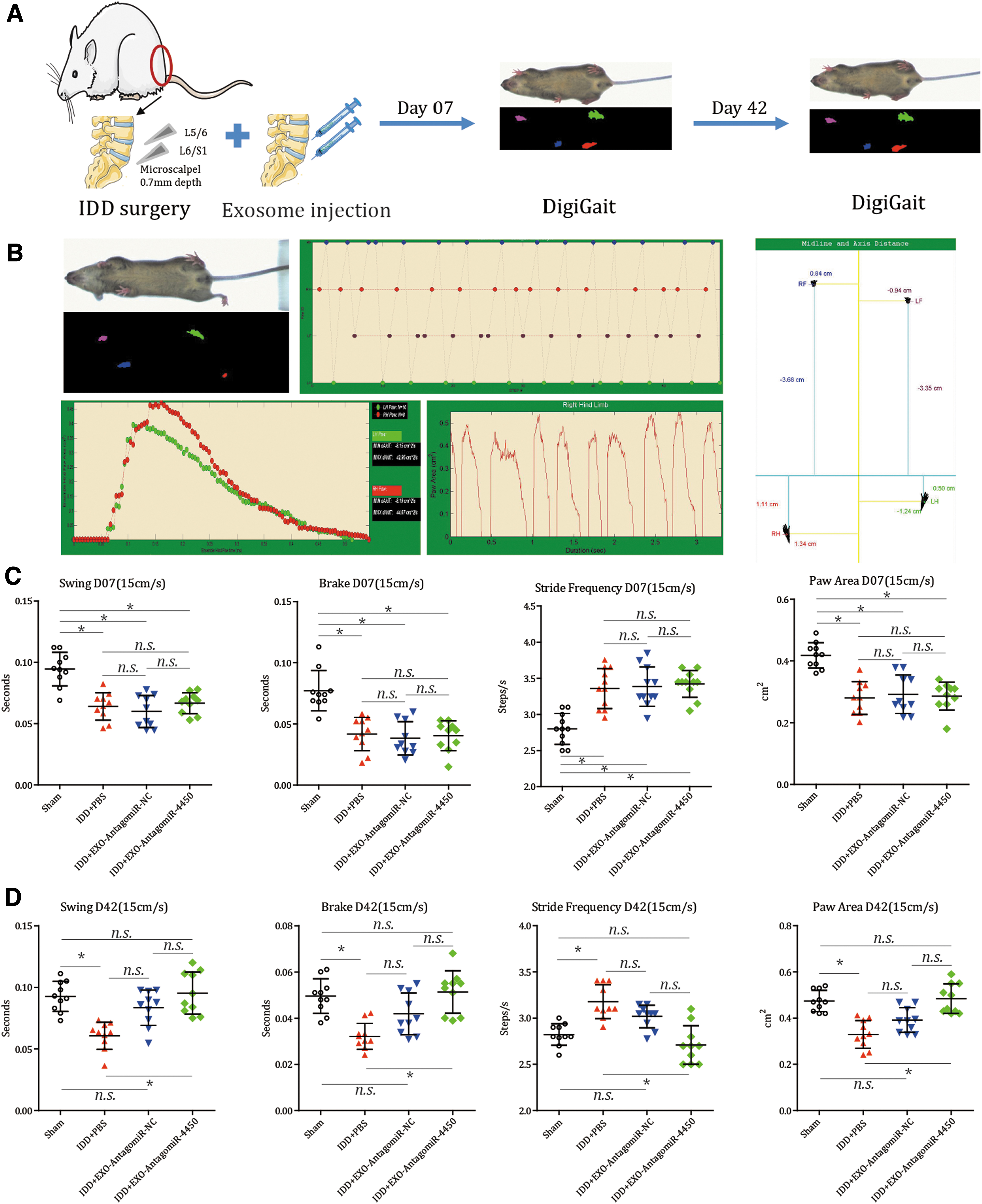
hPLMSC-derived exosomes carrying AntagomiR-4450 could relieve the abnormality of gait patterns in IDD mice. IDD mice are injected with hPLMSC-derived exosomes harboring AntagomiR-4450 and AntagomiR-NC immediately after surgery. Gait analysis was conducted by DigiGait systromal to observe the behavior changes of gait of the mice at day 7 and 42 after surgery and injection.
Inflammation-associated factors and MMPs were monitored by FMT in vivo at day 21 and 42 after injection, respectively (Fig. 8A). Compared to sham group, PBS + IDD group had more signaling detected by both ProSense and MMPSense, indicating that IDD surgery elevated both pro-inflammatory factors and MMPs in the IVD (all P < 0.001; Fig. 8B–D). Moreover, these elevated signaling detected by both ProSense and MMPSense were weakened by both EXO-AntagomiR-NC and EXO-AntagomiR-4450 (all P < 0.001; Fig. 8B–D), particularly EXO-AntagomiR-4450 group which showed even lower signaling intensity compared to EXO-AntagomiR-NC group (P < 0.001). Therefore, the EXO-AntagomiR-4450 could partially decline the pro-inflammatory factors and MMPs in IDD mice.
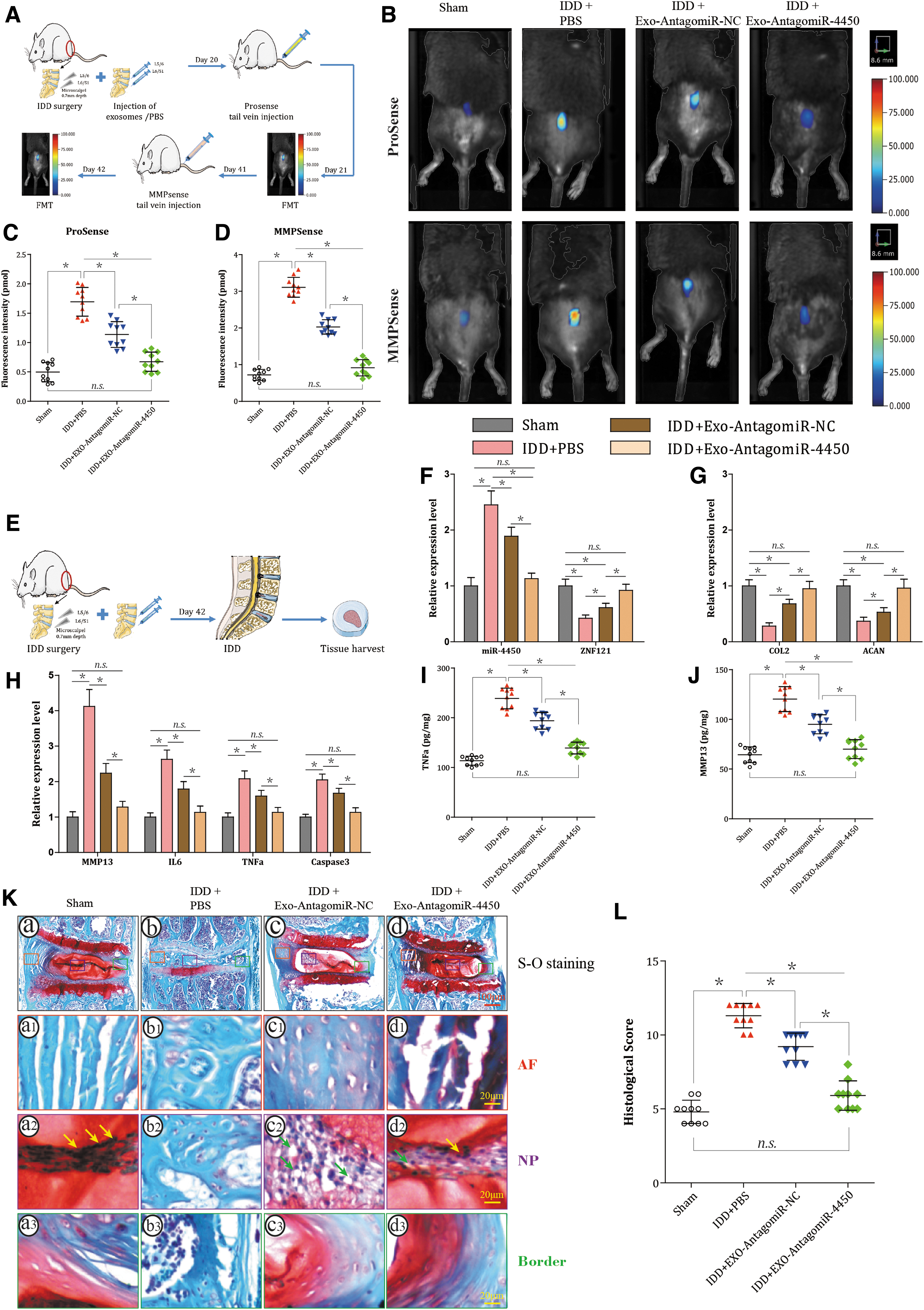
hPLMSC-derived exosomes harboring AntagomiR-4450 attenuate damage of IDD in vivo by repressing miR-4450 and thus increasing ZNF121.
At day 42 after injection, all the mice were sacrificed, then IVD tissues were separated and analyzed (Fig. 8E). The lumbar disc specimens L6/S1 were taken for RT-qPCR and ELISA. Compared to sham group, PBS + IDD group showed an increased expression of miR-4450, Caspase-3, MMP13, IL-6, and IL-1β and decreased that of ZNF121, COL2, and ACAN (all P < 0.001). Compared with IDD + PBS, IDD + EXO-AntagomiR-NC or IDD + EXO-AntagomiR-4450 declined the expression of miR-4450, Caspase-3, MMP13, IL-6, and IL-1β and elevated that of ZNF121, COL2, and ACAN (all P < 0.001), especially IDD + EXO-AntagomiR-4450 (P < 0.001; Fig. 8F–H). In addition, ELISA showed the protein level of TNF-α and MMP13 was the same as their mRNA expression level (all P < 0.001; Fig. 8I, J).
The lumbar disc specimens L5/6 were taken for histological evaluation. Disc morphology was evaluated using SO staining (Fig. 8K[a–d]). The sham discs displayed a rounded NP structure that contains stellar-shaped NPCs clustered at the center with proteoglycan matrix located at the periphery (yellow arrows), an intact AF with a normal pattern of fibrocartilage lamellae, a well-defined border between the AF and NP, and preserved proteoglycan content (red staining) (Fig. 8K[a, a1, a2]). After surgery, the discs in the IDD + PBS group revealed progressive degeneration in which the NP area was prominently decreased and collapsed, the NPCs were lost and the proteoglycan content (even no proteoglycan content preserved) was reduced, the AF lamellae were disorganized and even lost, and the border between the NP and AF became less distinct (Fig. 8K[b, b1, b2]). Interestingly, EXO-AntagomiR-NC restored some of the NP area, increased proteoglycan content, reorganized AF lamellae, and preserved some border between the NP and AF (Fig. 8K[c, c1, c2]). Particularly, EXO-AntagomiR-4450 even showed stronger recovery effect and has more NPCs (yellow arrows) and less cell vacuoles (green arrows) (Fig. 8K[d, d1, d2]). A modified Masuda's grading score was used to quantify IDD score (Fig. 8L). Compared to sham group, PBS + IDD group had higher score, indicating that IDD surgery elevated histological score in the IVD (P < 0.001). Moreover, the score was declined by both EXO-AntagomiR-NC and EXO-AntagomiR-4450 (both P < 0.001), particularly EXO-AntagomiR-4450 group which showed even lower score compared to EXO-AntagomiR-NC group (P < 0.001). Therefore, as demonstrated by the in vivo experiments, the hPLMSC-derived exosomes harboring AntagomiR-4450 could hinder IDD damage.
Discussion
There is increasing evidence that MSC transplantation can improve the progress of IDD through paracrine [24]. Exosomes secreted by MSCs have been shown to have anti-inflammatory, antiapoptotic, and discoprotective effects in vitro and can delay or reverse the progress of IDD in vivo [13,25]. Exosomes are key bioactive components secreted by MSCs and can replace MSC-based therapies. In the treatment of IDD, the growing diagnostic and therapeutic potential of miRNA has been emphasized [26]. Exosomes can be used as nanocarriers to deliver specific molecules, such as siRNA or AntagomiR [27], and deliver their contents to recipient cells through endocytosis and membrane fusion [28].
In this study, we found that miR-4450 was significantly upregulated while ZNF121 was downregulated in IDD, and miR-4450 exacerbated NPC damage by targeting ZNF121 while knockdown of miR-4450 showed a protective effect on NPCs by AntagomiR-4450, and hPLMSC-derived exosomes could transfer AntagomiR-4450 to NPCs and exhibited the same effects both in vitro and in vivo. Collectively, the key findings of our study provided evidence suggesting that hPLMSC-derived exosomes carrying AntagomiR-4450 alleviated damage of NPCs in vitro and retarded IDD damage in vivo.
miRNAs are a class of endogenous small noncoding single-stranded RNA molecules that can regulate gene expression and affect a variety of cellular processes, including proliferation, development, differentiation, apoptosis, and angiogenesis. Gain or loss of specific miRNA function can lead to the onset of IDD [26]. Since many upregulated miRNAs can function as IDD promoters, miR treatments (such as miRNA inhibitors or AntagomiR oligonucleotides) have become areas of extensive research. Compared with traditional gene therapy vectors, exosomes as natural membrane nanocarriers can protect and transfer biomolecules [29]. In the clinical field, MSCs have become more prominent because they are more functional than other cells. MSCs have a greater ability to expand in vitro and have a strong ability to differentiate into multiple cell types [30]. MSCs also have the potential to produce a large number of exosomes, so these cells can be used to produce exosomes clinically. Because MSCs do not cause inherent adverse reactions or immune rejection, MSC-derived exosomes have low immunogenic properties [7]. In short, in addition to their own therapeutic effects, these exosomes can also be used as natural drug delivery vehicles.
Our results found that miR-4450 was significantly upregulated in IDD, which highlights its possible role in the pathogenesis of IDD, and the inhibition of miR-4450 may help protect NP. Two other studies further confirmed this trend [31,32]. Therefore, the overexpression of miR-4450 in NP of IDD patients highlights its possible role in the pathogenesis of IDD, and the inhibition of miR-4450 may help protect NP. Our results showed that overexpressed miR-4450 increased the apoptosis and inflammatory response of NPCs and reduced the protective extracellular matrix (COL2 and ACAN), and vice versa reduction of miR-4450 expression could reduce NPC damage. A study showed that miR-222 was upregulated in human IDD tissues, and miR-222 inhibitors attenuated apoptosis, reduced TNF-α, IL-1β, and IL-6, and enhanced COL2 and ACAN expressions by regulating tissue inhibitor of metalloproteinase 3 [33]. Besides, Zhang et al. found that miR-3150a-3p was significantly upregulated in IDD; inhibition of miR-3150a-3p increased ACAN expression [31]. In addition, another study showed that miR-138-5p was significantly upregulated in degenerative NP tissue [34]; miR-138-5p silencing could ameliorate TNF-α-induced apoptosis and expression of cleaved caspase-3. These findings were consistent with our results.
Our findings identified that ZNF121 was a target of miR-4450. Moreover, a negative correlation between miR-4450 and ZNF121 was demonstrated in IDD NP samples and NPCs treated with TNF-α. As one of the largest families of regulatory proteins in human cells, ZNFs play vital roles in development, differentiation, and human diseases [35]. ZNF121 had been shown to regulate cell proliferation, apoptosis, and possible breast cancer development [36]. Accumulating studies showed that many ZNFs were regulated by multiple microRNAs, some of which were even direct targets of specific microRNAs. One study found that miRNA-1247 inhibited cell proliferation by directly targeting ZNF346 in childhood neuroblastoma [37]. Besides, miRNA-204-5p negatively regulated ZNF521, thereby inhibiting the proliferation, movement, and invasion of gastric cancer cells [38].
hPLMSC-derived exosomes could deliver AntagomiR-4450 into NPCs and thus retard IDD damage in vitro and in vivo. Increasing studies confirmed that MSC-exos had therapeutic potential in IVD regeneration. A recent study showed that MSC-exos could promote the proliferation of NP cells and enhance the production of extracellular matrix in vitro [13]. The use of oligonucleotide-based drugs to regulate gene expression has opened up a novel avenue for drug discovery. In particular, siRNAs have been quickly recognized as promising therapeutic tools, but their low bioavailability limits the full realization of its clinical potential [39]. One way to incorporate RNA into exosomes is through overexpressing cargo RNAs in the exosome-producing cells. These exosomal RNAs (miRNAs, modified miRNAs, and shRNAs) are fully functional when incubated with recipient cells and lead to target gene knockdown [15]. For example, Naseri et al. demonstrated that MSC-derived exosomes could be used as viable nanocarriers to deliver drug molecules, such as locked nucleic acid modified anti-miR-142-3p oligonucleotides to inhibit miR-142-3p expression levels in vitro and in vivo in breast cancer [40]. Using a quantitative RT-PCR approach, we demonstrated that mature lentivirus-encoded AntagomiR-4450 was secreted by lentivirus-infected hPLMSCs through exosomes. In this study we demonstrated that exosomal AntagomiR-4450 secreted by lentivirus-infected hPLMSCs was successfully transferred into and act in NPCs.
hPLMSC-exos with AntagomiR-4450 enhanced the capacity to attenuate apoptosis and inflammation and elevated cell migration and proliferation partially through elevating ZNF121. Extracellular vesicles, including exosomes, are filled with miRNAs that can regulate the ZNF genes. For example, Lu et al. [41] found that 118 and 116 miRNAs from B and T acute lymphoblastic leukemia microvesicles regulated ZNF gene expressions, respectively. Besides, gene ontology and signaling pathway analysis indicated that the altered microvesicle miRNAs targeted a subset of ZNF genes and participated in cell biology processes, including differentiation, proliferation, apoptosis, and cell cycle regulation [41]. We further studied the function of EXO-AntagomiR-4450 in the IDD mouse model. Consistent with the results of in vitro studies, the findings of in vivo experiments demonstrated the inhibitory effect of hPLMSC-derived EXO-AntagomiR-4450 on NPC apoptosis and inflammation.
FMT and gait analysis enabled a real-time monitor of the changes of gait and inflammatory factors in IVD. In our present study, we found that the hPLMSC-derived exosomes carrying AntagomiR-4450 could partially decline the pro-inflammatory factors and MMPs in IDD mice. These findings obtained using the ProSense and MMPSense fluorescent imaging agents were consistent with our cellular and tissue results. Therefore, FMT provided a new means by which to assess the effects of IDD treatments on NP inflammation in the murine model in vivo. This noninvasive technique has been used in inflammation, oncology, lung, cardiovascular, and skeletal diseases [42]. The advantage of this technology is that it can longitudinally monitor and quantify biological targets in vivo at multiple time points and can better understand the mechanism and progress of the disease [43].
The present study explored the use of the noninvasive video-capture gait-analysis instrumentation in the evaluation of a mouse model of IDD. Gait analysis has been used as a powerful technique for evaluating locomotion humans and laboratory animals with osteoarthritis or rheumatoid arthritis [44]. Our study demonstrated that EXO-AntagomiR-4450 could partially alleviate gait abnormality of IDD mice. To the best of our knowledge, this was the first work seeking to apply comprehensive gait analysis in rodent IDD models. There is increasing evidence that anti-TNF-α or anti-IL-1β drugs could reduce IDD-related low back pain and leg pain in IDD patients [45]. Similarly, because EXO-AntagomiR-4450 could downregulate the expression of TNF-α and IL-1β, we speculated that EXO-AntagomiR-4450 could reduce the pain and thus improve the abnormal gait. In addition, ELISA enhanced the reliability of FMT and gait analysis. Finally, the histological analysis showed that the surgery successfully induced IDD in mice and further verified the therapeutic effects of both Exo-AntagomiR-4450 and Exo-AntagomiR-NC, especially the Exo-AntagomiR-4450. The tendency of the histological results was showed to be the same as FMT, gait analysis, and ELISA, which further enhanced the reliability of our experimental findings that hPLMSC-derived exosomes harboring AntagomiR-4450 could hinder IDD damage both in vitro and in vivo.
It is worth noting that PLMSC-EXO alone (Exo-AntagomiR-NC) without AntagomiR-4450 also showed improvement in IDD. In vitro, EXO-AntagomiR-NC inhibited the apoptosis and inflammation of NPCs and promoted the NPC proliferation. In vivo, EXO-AntagomiR-NC also inhibited the apoptosis and inflammation of NPCs, inhibited the degradation of NP matrix, improved the gait of mice, and partially improved and reversed the IDD process. These findings suggest that PLMSC-exo itself has a therapeutic effect on IDD. Lu et al. found that MSC-exos alone promoted NPC proliferation and healthier extracellular matrix production in the degenerated NPCs [13]. Moreover, Cheng et al. observed that MSC-exos alone could inhibit NPC apoptosis and reduce IVD degeneration [46]. Furthermore, Liao et al. found that MSC-exos alone could modulate endoplasmic reticulum stress to protect against NPC death and ameliorate IDD in vivo [25]. All these findings further verified the therapeutic effects of PLMSC-exos alone for IDD and also confirmed the natural advantages of PLMSC-exos as drug or oligonucleotide carriers for IDD treatment.
Conclusions
In summary, we demonstrated that hPLMSC-derived exosomes could be used as a feasible nanovehicle to deliver drug molecules like AntagomiR-4450 in both in vitro and in vivo studies. On the basis of our findings, we had drawn a brief diagram to present the mechanisms clearly (Fig. 9). hPLMSC-derived exosomes carrying AntagomiR-4450 alleviated IDD and ameliorated gait disturbances through upregulation of ZNF121. These findings offer new therapeutic opportunities for extracellular vesicles or exosomes in the treatment of IDD.
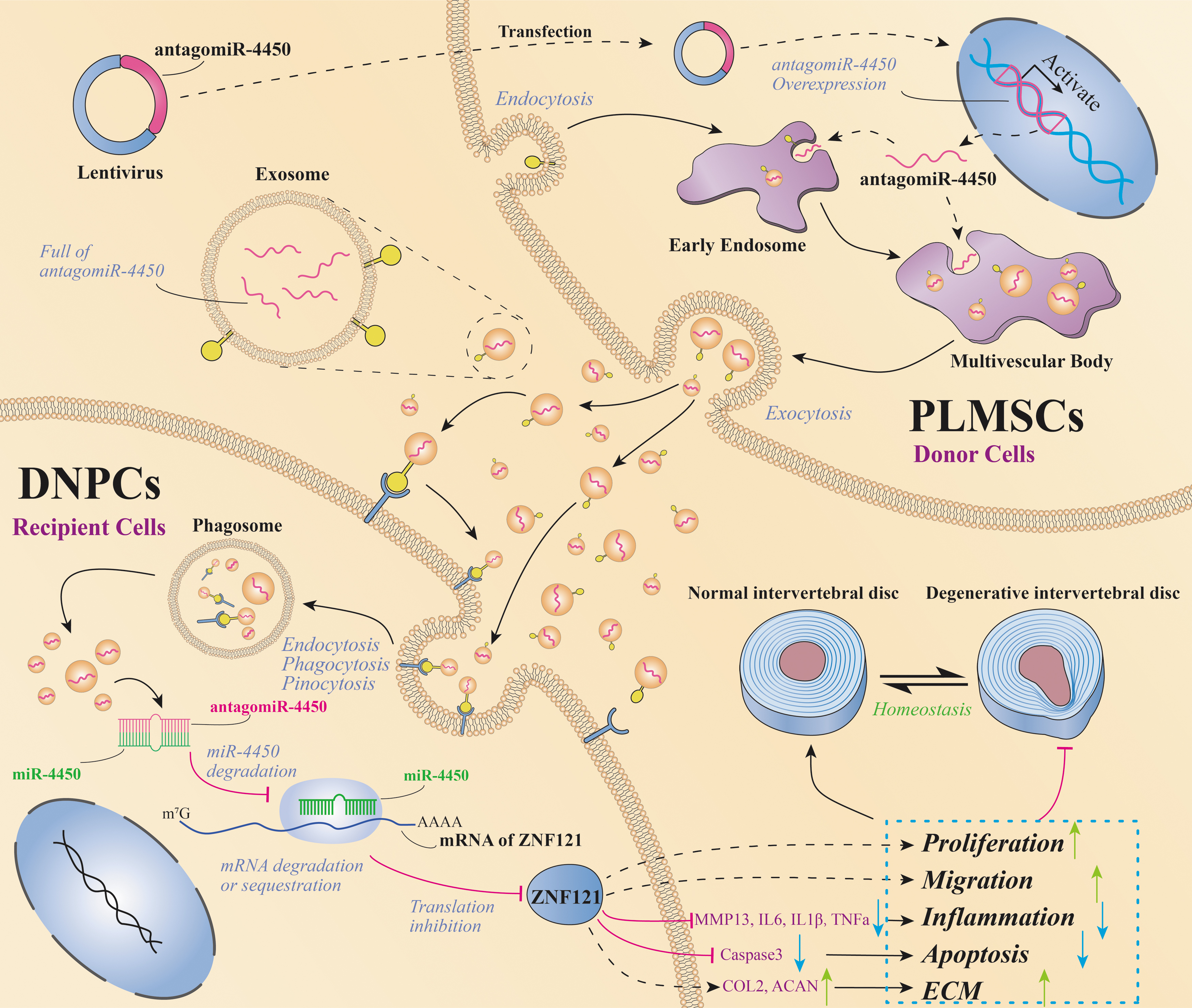
hPLMSC-derived exosomes carrying AntagomiR-4450 alleviate IDD damage by repressing miR-4450 and thus increasing ZNF121. Lentiviruses with AntagomiR-4450 are transfected into hPLMSCs and expressed in hPLMSCs, then hPLMSC-derived exosomes carry AntagomiR-4450 into DNPCs to repress miR-4450 and thus upregulate ZNF121 expression through the pathways such as endocytosis and membrane fusion, subsequently facilitate cell proliferation and migration, suppress regional inflammation by decreasing expression of IL-6, IL-8, IL-1β, TNF-α, MMP-13, and inhibiting cell apoptosis by reducing expression of caspase 3. DNPCs, degenerative NPCs; ECM, extracellular matrix.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by National Natural Science Foundation of China (grant number 81371987).
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.