
Abstract
Forced coexpression of the transcription factors Oct3/4, Klf4, Sox2, and c-Myc reprograms somatic cells into pluripotent stem cells (PSCs). Such induced PSCs (iPSCs) can generate any cell type of the adult body or indefinitely proliferate without losing their potential. Accordingly, iPSCs can serve as an unlimited cell source for the development of various disease models and regenerative therapies for animals and humans. Although canine peripheral blood mononuclear cells (PBMCs) can be easily obtained, they have a very low iPSC reprogramming efficiency. In this study, we determined the reprogramming efficiency of canine PBMCs under several conditions involving three types of media supplemented with small-molecule compounds. We found that canine iPSCs (ciPSCs) could be efficiently generated from PBMCs using N2B27 medium supplemented with leukemia inhibitory factor (LIF), basic fibroblast growth factor (bFGF), and a small-molecule cocktail (Y-27632, PD0325901, CHIR99021, A-83-01, Forskolin, and
Introduction
Embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) can differentiate into most cell types and potentially be used to repair damaged tissues [1 –3]. However, there are ethical concerns regarding the use of ESCs because these cells are derived from the inner cell mass of an early embryo, which is destroyed during the process.
Instead, iPSCs can be generated from differentiated cells by upregulating pluripotency factors. Thus, iPSCs are expected to provide abundant cell resources, which are otherwise rather difficult to obtain, for regenerative medicine. Accordingly, the development of regenerative medicine research involving iPSCs has been remarkable, and the resulting therapeutic approaches have already begun to undergo clinical trials [4]. Furthermore, patient-derived iPSCs have the potential to produce disease-associated specialized cells and related organoids, helping researchers to understand the underlying pathophysiology [5].
Canines were the first species to be domesticated by humans and have been companion animals since the early days of human civilization. Consequently, canine life expectancy has increased along with the incidence of degenerative diseases. Novel advanced interventions, such as regenerative approaches may be effective in treating such diseases.
Accordingly, canine iPSCs (ciPSCs) may enable such approaches by providing the required cell resource, whose transplantation into the patient can hypothetically replenish the lost functional cells in the body. Furthermore, there are many types of genetic disorders in canines, such as ceroid lipofuscinosis [6], rheumatoid arthritis [7], and narcolepsy [8], as in humans. Patient-specific ciPSCs generated from dogs with such disorders can also be useful to elucidate the pathogenesis of canine genetic disorders. To use ciPSCs for such applications in the veterinary field, it is instrumental to develop a simple and efficient method of ciPSC generation [9].
To date, several studies have assessed the generation of cPSCs from canine fetal or skin fibroblasts by using lentiviral or retroviral vectors expressing pluripotency factors [10 –17]. However, because these viral vectors are inserted into the host genome, re-expression of the transgenic pluripotency factors in reprogrammed cells can lead to tumorigenesis after these cells are transplanted into patients [18].
Instead, our group has successfully generated footprint-free ciPSCs from canine embryonic fibroblasts (CEFs) by using the Sendai viral vector SeVdp(KOSM)302L [19]. This vector can generate clinically useful iPSCs because it expresses the transgenic pluripotency factors without genomic insertion and can be automatically erased by microRNA-302 (miR-302) expressed in pluripotent stem cells (PSCs) [20]. Furthermore, SeVdp(KOSM)302L expresses all the four transcription factors (Oct3/4, Klf4, Sox2, and c-Myc) required for reprogramming into iPSCs [21]. Accordingly, the usage of this vector ensures stable expression of all the four factors, thereby enabling efficient generation of high-quality iPSCs.
In addition to the viral vector, the cell source used for reprogramming is very important. Human iPSCs have been generated from various cell types, including skin fibroblasts [3] and blood cells, such as peripheral blood mononuclear cells (PBMCs) [22], CD34(+) cells [23], and T cells [24]. Relative to a skin biopsy, blood collection is less invasive, and the constituent cells can be isolated directly at the site of clinical care. In addition, PBMCs can be reprogrammed without in vitro expansion. Therefore, PBMCs are attractive cell sources to generate iPSCs for disease modeling or regenerative medicine [25]. Recently, our group successfully generated ciPSC-like cells from canine PBMCs using SeVdp(KOSM)302L [26]. However, the efficiency of the formation of primary iPSC colonies was very low, and the ciPSCs did not differentiate into the three germ layers in vivo.
Small-molecule compounds that modulate intracellular signal transductions increase the iPSC reprogramming efficiency of human fibroblasts [27] and mouse blood cells, such as granulocyte–monocyte precursor cells and B cells [28]. In this study, we show that the efficiency of primary-colony formation of PBMCs undergoing iPSC reprogramming can be increased by a combination of small-molecule compounds, and footprint-free ciPSCs with teratoma-forming ability can be reproducibly generated from PBMCs.
Materials and Methods
This study was approved by the Institutional Animal Experiment Committee of the Osaka Prefecture University (Permission number: 19–83, 19–85, and 19–86) and carried out according to the Animal Experimentation Regulations of the Osaka Prefecture University.
Preparation of feeder cells
Mouse embryonic fibroblasts (MEFs) were isolated from embryonic day 13.5 ICR mouse fetuses (Japan SLC, Shizuoka, Japan). The fetuses were washed with Dulbecco's (D-) phosphate-buffered saline without Mg2+ and Ca2+ [PBS(–); Nacalai Tesque, Kyoto, Japan] and minced in a sterile dish. It was then cultured in the “feeder” medium composed of Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO), 2 mM
Preparation of canine PBMCs
Canine PBMCs were obtained from three beagles aged 7 years. The previously reported method was modified to collect mainly canine monocytes [29]. In brief, 10 mL of whole blood was collected in heparinized syringes from each donor and diluted with the same volume of D-PBS(–). The diluted blood was carefully poured onto the lymphocyte separation solution (Nacalai Tesque) and centrifuged for 35 min at 1,000 g. The PBMC layer was collected and washed twice with D-PBS(–). The PBMCs were cultured for 2 days in RPMI medium (RPMI 1640; Nacalai Tesque) containing 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin in 60 mm tissue culture dishes (Iwaki, Tokyo, Japan). Afterward, nonadherent cells were removed, and adherent cells were infected with SeVdp(KOSM)302L to generate ciPSCs.
Sendai virus infection
The SeVdp(KOSM)302L vector was produced as previously reported [20]. Canine PBMCs were infected with SeVdp(KOSM)302L encoding human Kruppel-like factor (KLF) 4, octamer-binding transcription factor (OCT) 3/4, sex-determining region Y-box (SOX) 2, and C-MYC, at a multiplicity of infection (MOI) of 2. The MOI was determined according to our previous report [26]. Infected canine PBMCs were incubated at 37°C in 5% CO2 for 2 h. The infected cells were resuspended in the PBMC culture medium indicated previously and seeded at the density of 3 × 105 cells/well in an MEF-seeded 12-well tissue culture plate (Iwaki).
Generation of ciPSCs
On day 1 postinfection, the medium was replaced with either of three different reprogramming media—N2B27, knockout serum replacement (KSR), or StemFit® Basic02 (Ajinomoto, Tokyo, Japan)—supplemented with 10 ng/mL human leukemia inhibitory factor (hLIF; Peprotech, Rocky Hill, NJ) and 10 ng/mL human basic fibroblast growth factor (bFGF) (Peprotech).
N2B27 medium consisted of 50% DMEM/Nutrient Mixture F-12 Ham (Nacalai Tesque) and 50% Neuro Basal Medium (Thermo Fisher Scientific, Waltham, MA) supplemented with N-2 supplement (1 × ; Thermo Fisher Scientific), B-27 supplement (1 × ; Thermo Fisher Scientific), GlutaMAX (1 × ; Thermo Fisher Scientific), 100 U/mL penicillin, 100 μg/mL streptomycin, 0.1 mM minimal essential medium nonessential amino acids (MEM NEAA; Thermo Fisher Scientific), and 0.1 mM 2-mercaptoethanol (Sigma-Aldrich).
KSR medium consisted of DMEM/Nutrient Mixture F-12 Ham (Nacalai Tesque) supplemented with 20% knockout serum replacement (Thermo Fisher Scientific), 2 mM
In addition, a small-molecule cocktail was added into each reprogramming medium. The cocktail consisted of 10 μM Y-27632 (Nacalai Tesque), 0.5 μM PD0325901 (Reprocell, Kanagawa, Japan), 3 μM CHIR99021 (Fujifilm Wako Pure Chemical Corp., Osaka, Japan), 0.5 μM A-83-01(Stemgent, San Diego, CA), 10 μM Forskolin (Tocris, Bristol, Avon, United Kingdom), and 50 μg/mL
On day 8 postinfection, the media were replaced with StemFit® AK02N (StemFit; Ajinomoto) containing 10 ng/mL hLIF.
On day 22 postinfection, primary ciPSC-like colonies were counted to calculate the reprogramming efficiency and manually transferred onto fresh feeder cells by using a Pasteur pipette. ciPSCs were cultured with StemFit supplemented with hLIF, and the medium was changed daily. ciPSCs were mechanically passaged every 5–7 days until passage 5–8. Thereafter, they were passaged by using the dissociation solution for human ES/iPS Cells (CTK solution; Reprocell). In brief, ciPSCs were incubated with CTK solution for 8 min at 37°C. They were then seeded in a dish containing a fresh layer of MEFs. The split ratio was 1:3 or 1:5.
Expression analysis of the pluripotency transgenes in SeVdp(KOSM)302L
Removal of SeVdp(KOSM)302L was assessed using quantitative reverse transcription (RT)–polymerase chain reaction (PCR). In brief, total RNA was extracted using the RNeasy Micro Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. RT was performed using random primers and ReverTra Ace kit (Toyobo, Osaka, Japan). PCR was carried out using Blend Taq Plus (Toyobo) with the following parameters: 94°C for 3 min; 40 cycles of 95°C for 15 s, primer-specific annealing temperature for 20 s, and 72°C for 15 s; and 72°C for 5 min. PCR products were resolved on a 2% agarose gel, followed by staining in gel with ethidium bromide, and visualization under an ultraviolet transilluminator (AE-9020; ATTO, Tokyo, Japan). The primers used are listed in Supplementary Table S1.
Expression levels of pluripotency markers in generated ciPSCs
The pluripotency of generated ciPSCs was assessed using alkaline phosphatase (AP) staining, quantitative RT-PCR, and immunocytochemical analysis.
AP staining was performed using the Stemgent™ Alkaline Phosphatase Staining Kit II (Reprocell) according to the manufacturer's instructions.
Quantitative RT-PCR was performed three times with three different clones of the same ciPSC lines by using the Plus one system (Thermo Fisher Scientific) with PowerUp™ SYBR™ Green Master Mix (Thermo Fisher Scientific) according to the manufacturer's instructions. The average value of the three runs was used. The mRNA expression levels of pluripotency markers were compared with those of a CEF-derived iPSC line that were previously reported [19] and with those of three different passages of the same line. The primers used are listed in Supplementary Table S2.
Immunocytochemical analyses were performed for the pluripotency markers OCT3/4, NANOG, SSEA-1, SSEA-4, TRA-1-60, and TRA-1-81. In brief, ciPSCs cultured in an eight-well glass chamber slide (Iwaki) were immunolabeled as described hereunder. The cells were first washed with D-PBS(–) and fixed in 4% paraformaldehyde (PFA) for 5 min and then permeabilized with 0.1% Tween-20 in D-PBS(–) for 5 min at 25°C. Afterward, they were incubated in D-PBS(–) containing 10% bovine serum albumin (F-V) (Nacalai Tesque, code: 08163-77) for 30 min at 25°C, followed by overnight incubation at 4°C with the primary antibodies against OCT3/4 (5 μg/mL, H-134; Santa Cruz Biotechnology, Dallas), NANOG (5 μg/mL, ab77095; Abcam, Cambridge, MA, United Kingdom), SSEA-1 (10 μg/mL, MAB4301; Merck, Darmstadt, Germany), SSEA-4 (5 μg/mL, 09-0006; Reprocell), TRA-1-60 (5 μg/mL, 09-0010; Reprocell), or TRA-1-81 (5 μg/mL, 09-0011; Reprocell). Negative-control cells were incubated in D-PBS(–) without the primary antibodies.
The next day, the cells were washed with 0.1% saponin (Sigma-Aldrich) in D-PBS(–) and incubated for 30 min at 25°C with the appropriate secondary antibodies from the following: Alexa Fluor 488 goat anti-mouse immunoglobulin G (IgG; 1 μg/mL; Thermo Fisher Scientific), Cy3 goat anti-mouse IgM (1 μg/mL; Merck), Alexa Fluor 488 rabbit anti-goat IgG (1 μg/mL; Thermo Fisher Scientific), and Alexa Fluor 546 goat anti-rabbit IgG (2 μg/mL; Thermo Fisher Scientific).
The cells were subsequently washed with 0.1% saponin and mounted with ProLong®Gold antifade reagent with DAPI (Thermo Fisher Scientific) to label DNA. The stained samples were examined by a confocal laser microscope (FV3000; Olympus, Tokyo, Japan).
In vitro differentiation
To assess the differentiation potentials of generated ciPSCs, they were induced to form embryoid bodies (EBs), and the expression levels of differentiation markers were examined using RT-PCR and immunocytochemical analyses. In brief, ∼20–30 ciPSC colonies were collected using CTK solution and transferred to Costar® six-well Clear Flat Bottom Ultra-Low Attachment Multiple Well Plates (Corning, New York) containing the FBS medium with/without 10 ng/mL human bone morphogenetic protein (BMP) 4 (Miltenyi Biotec, Bergisch Gladbach, Germany) and 10 ng/mL human activin A (Peprotech) to induce the formation of EBs. The FBS medium consisted of DMEM/Nutrient Mixture F-12 Ham supplemented with 20% FBS (Sigma-Aldrich), 2 mM
After 7 days, some of the EBs were transferred onto a gelatin-coated eight-well glass chamber slide containing the same medium, and the remaining EBs were continued to be cultured in suspension. The medium was replenished every 2 days.
At days 21–28 after seeding, total RNA was extracted from these remaining EBs and used for marker-gene expression analysis using RT-PCR. The EBs in the chamber slide were fixed and immunolabeled.
Tubb3 (ectoderm), Desmin and Nkx2.5 (mesoderm), Foxa2, and Gata4 (endoderm) were used as the differentiation marker genes for analysis by RT-PCR. The primers used are listed in Supplementary Table S3.
For immunocytochemistry, antibodies against TUBB3 (1:400, MAB1637; Merck), DESMIN (1 μg/mL, ab82506; Abcam), and SOX17 (1 μg/mL, ab84990; Abcam) were used as the primary antibodies. Alexa Fluor 546 goat anti-rabbit IgG (2 μg/mL) and Alexa Fluor 488 goat anti-mouse IgG (1 μg/mL) were used as the secondary antibodies.
In vivo differentiation
The in vivo differentiation potential of generated ciPSCs was assessed by the teratoma assay. In brief, ∼1–2 × 106 ciPSCs (OPUiD01-A, passage 13; or OPUiD01-B, passage 14) suspended in Matrigel (Corning) diluted 1:1 in DMEM/Nutrient Mixture F-12 Ham were injected into the testicular capsule of NOD/SCID mice (OPUiD01-A: n = 3, OPUiD01-B: n = 2). The mice were killed 2–3 months later; the tumors were fixed in 4% PFA and paraffin embedded. The paraffin-embedded samples were sliced and then stained with hematoxylin and eosin.
Karyotyping
ciPSCs were incubated in a medium containing 0.04 μg/mL colcemid (Thermo Fisher Scientific) for 2 h, followed by trypsinization and incubation in 0.075 M KCl at 37°C for 20 min. The cells were fixed in acetic acid:methanol (1:3) mixture, stained with Giemsa (Merck), and analyzed under a light microscope.
Generation and characterization of ciPSCs from the PBMCs of multiple donors
To prove that our new method could generate ciPSCs from a larger number of donor samples, canine PBMCs were collected from two different donors with ciPSC lines OPUiD01-A and -B, and were reprogrammed by the method described previously. From day 1 to 8 after SeVdp(KOSM)302L infection, N2B27 medium supplemented with a small-molecule cocktail was used. Each of the two ciPSC lines from both donors was analyzed for SeVdp(KOSM)302L removal, and each iPSC line of both donors was characterized regarding pluripotency markers expression, in vitro differentiation, and karyotyping using the method described previously. The mRNA expression levels of pluripotency markers were compared with those of ciPSC line OPUiD01-B.
Statistical analysis
Data were expressed as mean ± standard deviation. Statistical significance was determined using Turkey–Kramer multiple comparison using the Statcel software (OMS Ltd, Tokorozawa, Japan).
Results
A small-molecule cocktail improves the efficiency of the formation of primary ciPSC colonies from PBMCs
To determine the optimum culture conditions to reprogram canine PBMCs, we tested three media and a small-molecule cocktail modified from a previous report [27] and calculated the primary-colony formation efficiency under each condition. In the group cultured with the cocktail and N2B27 medium, the primary colony formation efficiency of PBMCs isolated from three dogs was 0.0096% ± 0.0033% and significantly higher than that in any other group (Fig. 1A). Moreover, only under this condition was it possible to obtain primary colonies from the PBMCs of all the three dogs.
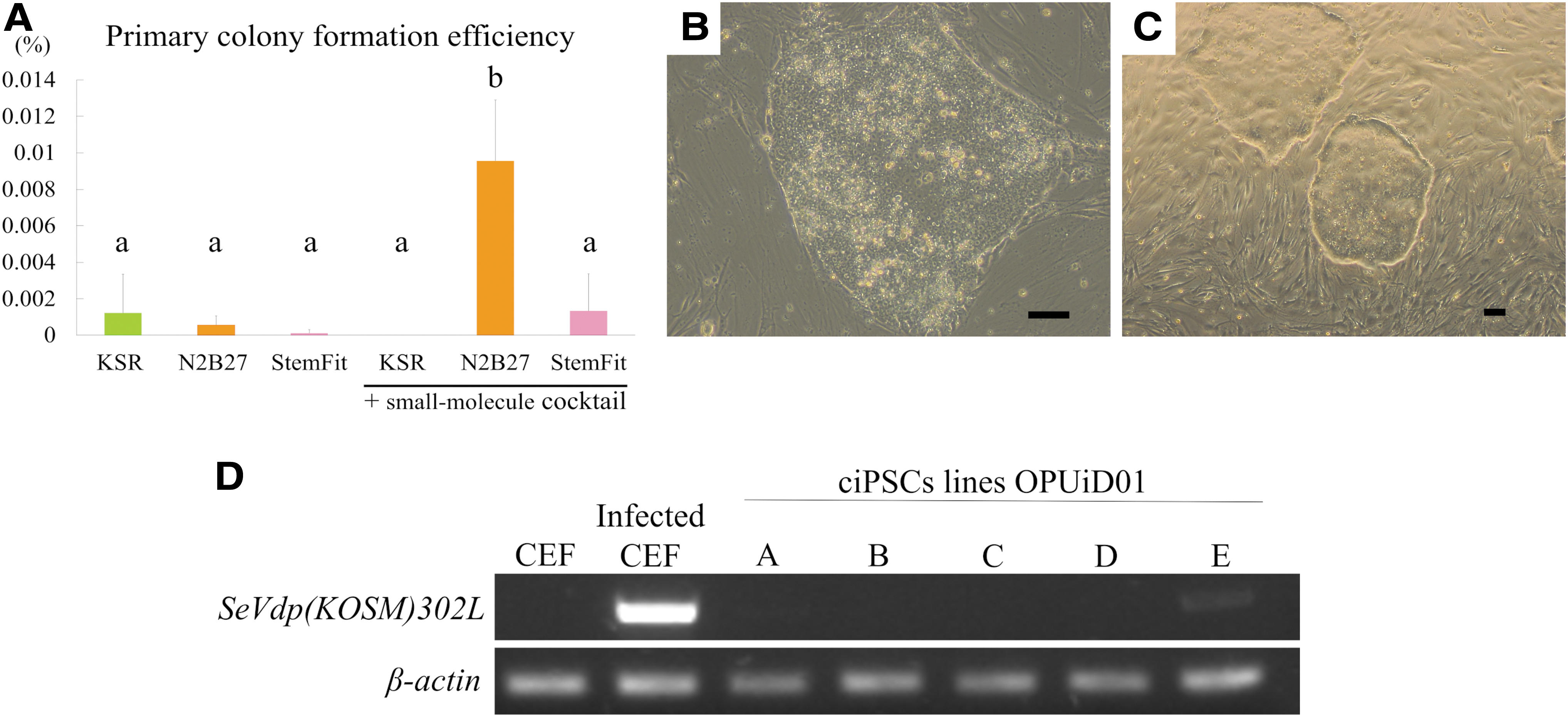
Generation of PBMC-derived ciPSC lines and confirmation of SeVdp(KOSM)302L removal
Multiple primary colonies emerged ∼10 days after infecting the PBMCs with SeVdp(KOSM)302L. Approximately 22 days postinfection, the primary colonies grew to a size large enough to pick up. Their morphologies were similar to that of canine ES cells, with clear borders, a high nuclear–-cytoplasmic ratio, clear nuclei, and a flat and tightly packed morphology (Fig. 1B, C). By further passaging, we generated five ciPSC lines (OPUiD01-A, -B, -C, -D, and -E) from one of the PBMC samples. RT-PCR was used to determine whether SeVdp(KOSM)302L was removed from the reprogrammed cells. OPUiD01-A, -B, -C, and -D were negative for SeVdp(KOSM)302L (Fig. 1D). However, OPUiD01-E still retained the virus (Fig. 1D). All the lines survived long-term passage while maintaining a flat colony morphology. However, only OPUiD01-A and -B could be passaged more than 40 times while maintaining their proliferative rate. Therefore, OPUiD01-A and -B were selected for further analyses.
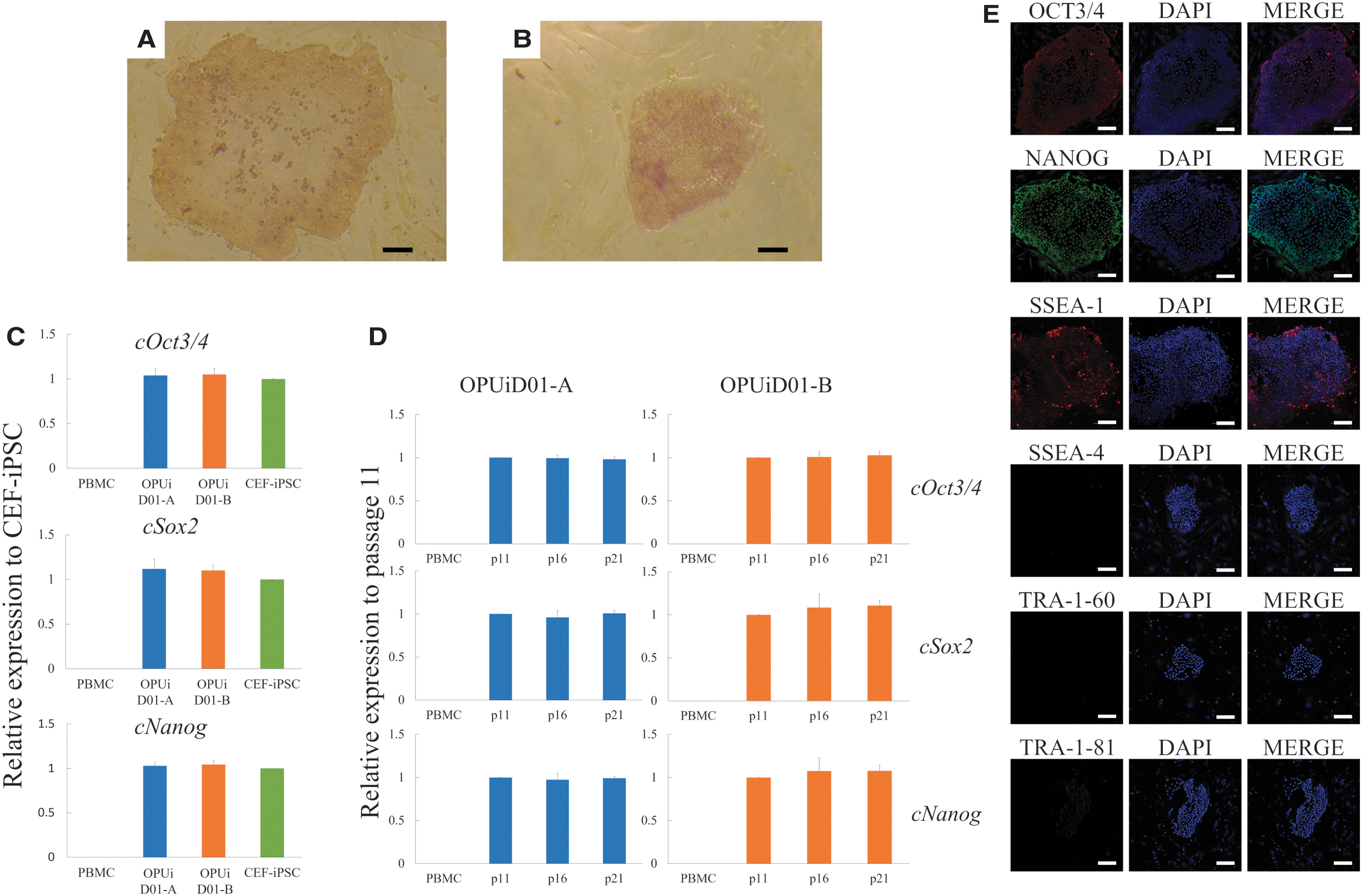
Expression of endogenous pluripotency markers
Even after the removal of SeVdp (KOSM)302L, OPUiD01-A and -B were positive for AP activity (Fig. 2A, B). The mRNA levels of Oct3/4, Sox2, and Nanog in both lines were comparable with those in CEF-derived iPSCs, which we previously reported, as revealed by quantitative RT-PCR (Fig. 2C). Furthermore, after multiple passages, both lines maintained the mRNA levels of Oct3/4, Sox2, and Nanog (Fig. 2D). Immunocytochemical staining showed that both lines were positive for the pluripotency markers, such as NANOG, OCT3/4, and SSEA-1, but negative for SSEA-4, TRA-1-60, and TRA-1-81 (Fig. 2E).
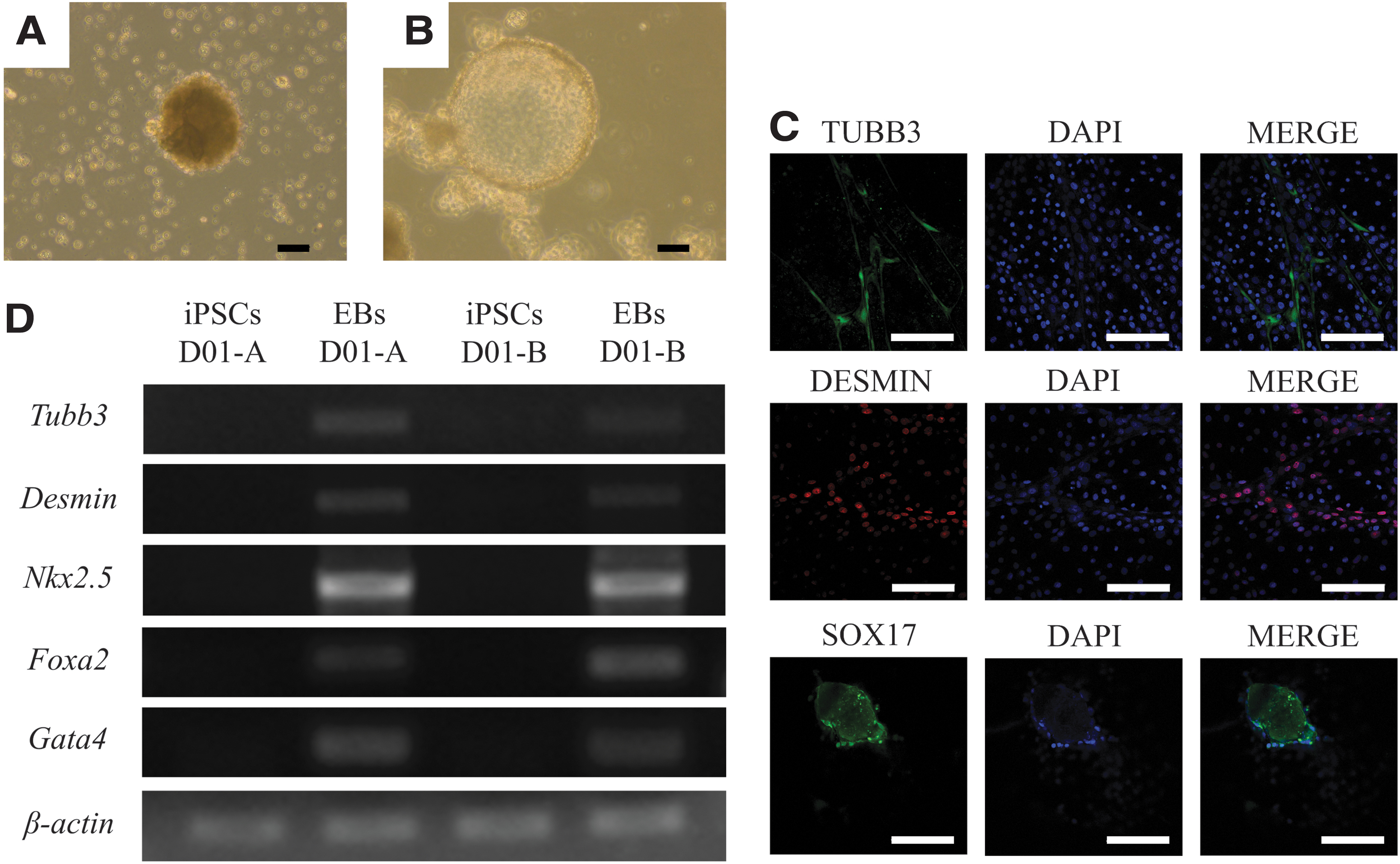
In vitro differentiation into all the three germ layers
OPUiD01-A formed EBs when cultured with the FBS medium containing activin A and BMP4 (Fig. 3A). Similarly, OPUiD01-B also formed EBs with the FBS medium; however, unlike OPUiD01-A, OPUiD01-B did not require activin A or BMP4 for EB formation. In addition, the EBs derived from OPUiD01-B exhibited a cystic morphology (Fig. 3B). After transferring onto gelatin-coated dishes, these EBs differentiated and expressed the germ layer-specific markers (TUBB3, DESMIN, and SOX17 for ectoderm, mesoderm, and endoderm, respectively) (Fig. 3C). The remaining EBs also expressed the differentiation markers, such as Tubb3 (ectoderm), Desmin and Nkx2.5 (mesoderm), and Foxa2 and Gata4 (endoderm), as determined by RT-PCR (Fig. 3D).
In vivo differentiation
To assess the in vivo differentiation potential of ciPSCs, the two lines were injected into the testes of NOD/SCID mice. OPUiD01-A and -B formed tumors (OPUiD01-A: 1/3, OPUiD01-B: 2/2) (Fig. 4A, B). However, in OPUiD01-A tumor, only undifferentiated cells, and not any differentiated tissues or cells, were observed. In contrast, OPUiD01-B formed teratomas that comprised various types of cells of the three germ layers (2/2) (Fig. 4C–H).
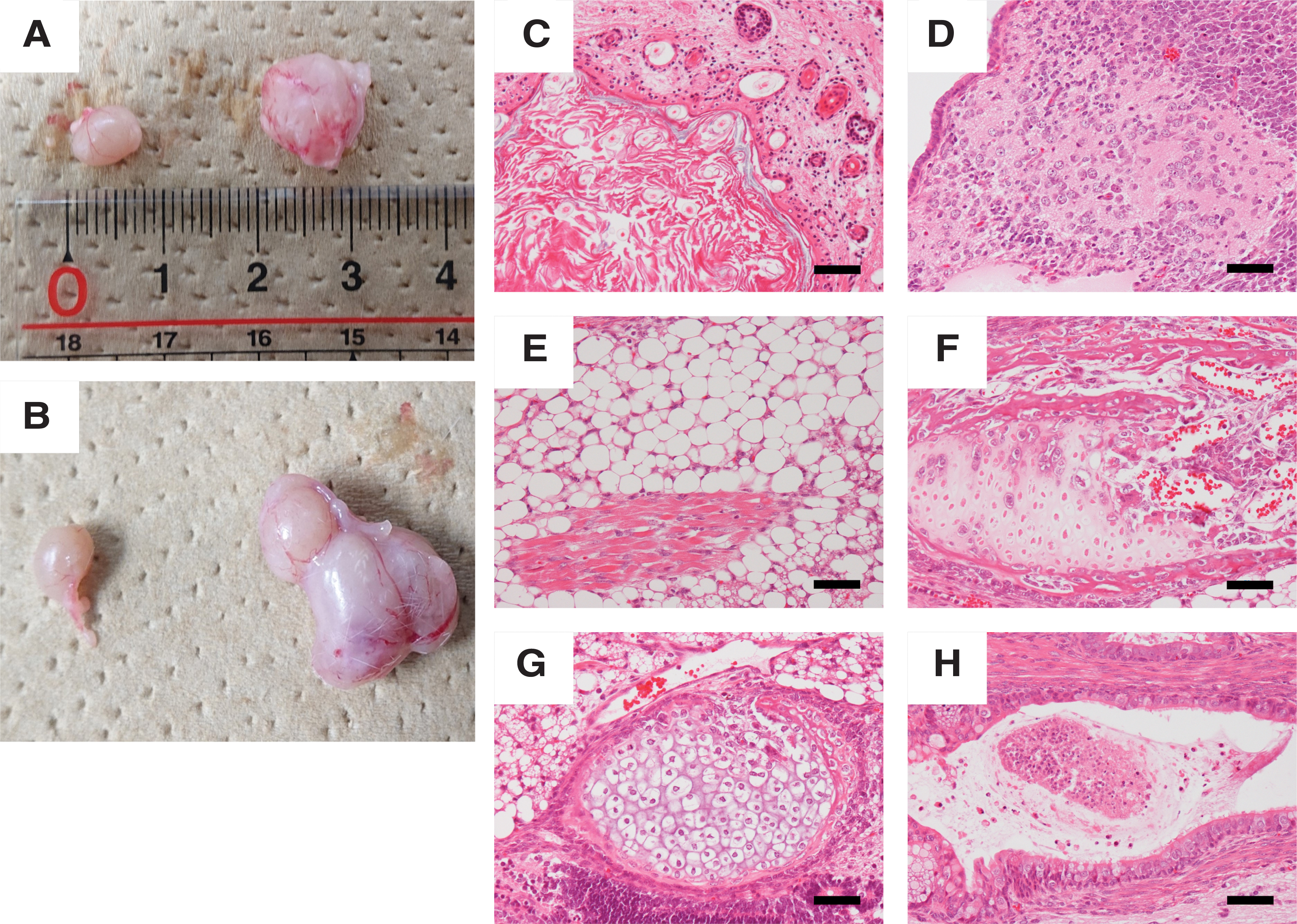
Teratoma formation of ciPSCs.
Karyotyping
The two ciPSC lines had normal karyotypes, with 38 matched pairs of autosomes and XX gonosomes at passage 28 (OPUiD01-A) or 26 (OPUiD01-B) (Fig. 5).
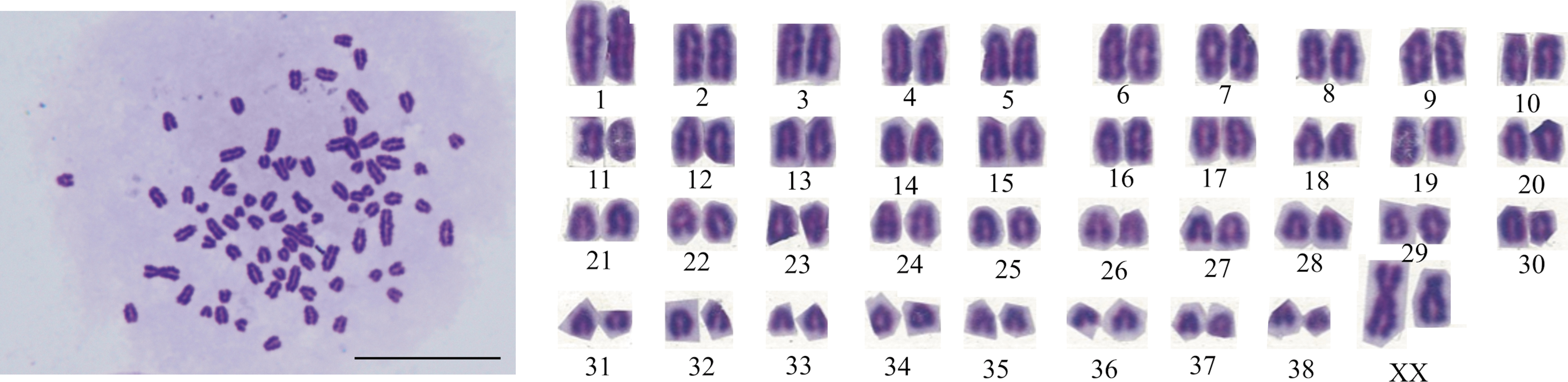
Karyotype analysis of ciPSCs. Normal: 78, XX with 38 matched pairs of autosomes in cells derived from OPUiD01-B at passage 26. Magnification, 1,000 × . Scale bar, 100 μm.
Generation and characterization of ciPSCs from PBMCs of multiple donors
About 20 days after infection, we were able to pick up multiple primary colonies in both donor samples. Two ciPSC lines were generated from each donor's PBMCs (Fig. 6A, B) and all four ciPSC lines were negative for SeVdp(KOSM)302L (Fig. 6C). All lines were positive for the pluripotency markers as assessed using immunocytochemistry and AP activity (Fig. 6D, E). There were no significant differences in the mRNA levels of pluripotency markers between these ciPSC lines and OPUiD01-B as shown by RT-qPCR and statistical analysis (Fig. 6F). These ciPSC lines also formed EBs, which differentiated and expressed germ layer-specific markers (Fig. 6G). Furthermore, these lines had normal karyotypes.

Discussion
We have previously generated ciPSCs from canine PBMCs collected in the same way as in this study, by reprogramming using KSR medium; however, the primary-colony formation efficiency was very low (0.00037%) [26]. To improve the efficiency, we explored a more effective culture condition in this study. When N2B27 medium supplemented with a cocktail of small-molecule compounds was used, the efficiency was maximal (0.0096% ± 0.0033%), which was 25.8 times higher than that observed in our previous report [26]. Furthermore, every PBMC sample, isolated from a different dog, formed primary colonies in this culture condition, whereas some of the samples did not form colonies in other culture conditions. This observation suggests that the optimized method is amenable to generating ciPSCs from the PBMCs of healthy beagles regardless of individual background differences, although with different efficiencies.
We demonstrated that N2B27, along with a small-molecule cocktail composed of Y-27632, PD0325901, CHIR99021, A-83-01, Forskolin, and
The TGFβ inhibitor A-83-01 and the MEK inhibitor PD0325901 promote reprogramming by inducing the mesenchymal-to-epithelial transition during the reprogramming of human cells to iPSCs [31], whereas the GSK3β inhibitor CHIR99021 and the adenylate cyclase activator Forskolin do so by modulating intracellular metabolism [32
–34]. Y-27632, a Rho-associated coiled-coil forming kinase (ROCK) inhibitor, and
However, it is currently unclear whether these factors synergistically improve the reprogramming efficiency. Therefore, further research is required to determine the contribution of each factor individually and the underlying mechanism to improve reprogramming further.
In previous studies on canine ESCs or ciPSCs, the FBS or KSR medium has been used for reprogramming and maintenance [12 –15,17,19,38 –41]. We have also used the KSR medium in our previous study for both the reprogramming of canine PBMCs and ciPSC maintenance [26]. However, in our preliminary experiments, the primary colonies obtained with N2B27 medium supplemented with the small-molecule cocktail did not grow large enough to pick up and establish ciPSC lines, even when cultured under the same conditions or changed to KSR or FBS medium during the late reprogramming phase (data not shown).
In the late reprogramming phase, unlike in the early phase, the activation of the core pluripotency circuitry, silencing of transgenes, and complete epigenetic resetting are necessary [30]. Thus, we concluded that FBS, KSR, or N2B27 medium with the cocktail was not suitable for the late reprogramming phase of canine PBMCs or expansion of ciPSCs.
In contrast, StemFit is effective for both the iPSC reprogramming of human cells, including PBMCs and the maintenance of the derived iPSCs in feeder-free conditions and has great potential for the maintenance of iPSC survival and proliferation [42]. Therefore, we attempted to use StemFit for the late reprogramming stage of canine PBMCs and for ciPSC maintenance although StemFit had not previously been used in on-feeder conditions, even for human cells. Our results suggest that StemFit is advantageous for the late reprogramming stage of canine PBMCs and for the maintenance of ciPSCs in on-feeder condition, whereas N2B27 medium with the small-molecule cocktail is effective for early reprogramming.
Furthermore, The StemFit Basic02 used for early phase of reprogramming does not contain bFGF, whereas StemFit AK02N contains high concentration of bFGF [42]. Therefore, both LIF and bFGF were included in the medium of early phase of reprogramming and maintenance of these ciPSCs. In this study, it is not clear which of the LIF, high concentrations of bFGF or/and StemFit medium itself produced good results. There were no previous reports that the StemFit or high concentrations of bFGF were used for ciPSC maintenance. In addition, some studies reported that LIF was used for reprogramming and maintenance of ciPSCs [10,12 –16,19,26], whereas there is a report that LIF is not used for the same [17]. In the future, it is necessary to investigate which factor is required for reprogramming and ciPSC maintenance.
Our ciPSC lines generated from canine PBMCs using SeVdp(KOSM)302L exhibited the typical morphology of canine ESCs. After several passages, reprogrammed canine PBMCs were free of SeVdp(KOSM)302L. This feature is also observed in primed human ESCs and iPSCs as well as CEF- or PBMC-derived ciPSC lines generated using the same vector [19,26]. Therefore, it is suggested that ciPSCs generated using SeVdp(KOSM)302L are morphologically similar to human ESCs and iPSCs.
In the two ciPSC lines in which the removal of SeVdp(KOSM)302L was confirmed, the pluripotency markers, such as AP activity, NANOG, OCT3/4, and SSEA-1, were still detected. Furthermore, these ciPSC lines maintained Oct3/4, Sox2, and Nanog transcription after several passages. These results suggest that our ciPSC lines were maintained in the undifferentiated state independent of the transgene expression. These results are similar to those observed with CEF- or PBMC-derived ciPSCs established using the same vector [19,26].
Our iPSC line OPUiD01-B differentiated into all the three germ layers in vitro. Furthermore, this line formed teratomas composed of various cell types, including mature cells. In humans and mice, teratoma formation is the gold standard for pluripotency [43]. Our results accordingly indicated that OPUiD01-B was pluripotent.
However, we did not confirm that any tissue that was formed was derived from the injected iPSCs, and not from the host. Labeling the teratoma-forming iPSC line with eGFP or another label is a valid way of confirming cell lineage identity and should be performed in further studies. In contrast, teratoma formation has not been assessed for the most reported ciPSCs [10,12,15,16]. Although some of these ciPSC lines form teratomas and differentiate into three germ layers, they still express the transgenic pluripotency factors, or the transgene-silencing has not been assessed at all in these lines [11,13,14], hampering their clinical use. Our footprint-free ciPSC line OPUiD01-B is thus superior to the available ciPSC lines.
We have previously generated PBMC-derived ciPSCs that form incomplete teratomas containing only the neuronal tissue [26]. The difference between the two studies is the cell culture method used for reprogramming and ciPSC maintenance. Epigenetic differences induced by either incomplete reprogramming or culture conditions cause functional variability in human iPSCs [44]. Therefore, the optimized conditions in this study likely enable complete reprogramming and promote teratoma formation in addition to colony formation efficiency.
Of interest, although OPUiD01-A differentiated into all the three germ layers only when activin A and BMP4 were added, OPUiD01-B differentiated and exhibited a cystic morphology, which likely indicates the differentiation into several cell types including epithelial and endoderm cells [38]. In addition, OPUiD01-A did not form teratomas unlike OPUiD01-B. Human iPSC lines derived from the same individual under the same conditions also show similar variability [45]. This variability is likely to be reprogramming based and may be because of the low levels of core pluripotency factors and high levels of differentiation markers [45]. In the future, to decrease the variability between ciPSC lines, it might be necessary to further improve the methods of reprogramming, such as by using canine, instead of human, pluripotency transgenes.
To show that our method described above can generate ciPSCs from the PBMCs of multiple donors, we reprogrammed PBMCs of two donors, which were different from the donor of OPUiD01-A and -B. Our results revealed that our new method can consistently generate iPSCs from a greater number of donors. Although we were not able to assess the in vivo differentiation potential of these ciPSC lines, we proved that our new method enabled a sufficiently robust generation of footprint-free PBMC-derived ciPSCs.
PBMCs are composed of nucleated red blood cells, monocytes, granulocytes, and lymphocytes (T and B cells). Human iPSCs have been generated from several types of blood cells, such as CD34(+) cells [23], T cells [24], and monocyte-derived dendritic cells [46]. In this study, we collected canine PBMCs, which mainly adhered to the cell culture dishes. Such adherent cells are mostly (≥95%) CD14(+) monocytes, whereas the nonadherent cells are mostly (∼68%) lymphocytes [29]. Therefore, the canine PBMCs used in this study are expected to be mostly monocytes. SeVdp(KOSM)302L can presumably infect human monocytes and CD4(–)/CD8(–) T cells but not B or CD4(+) T cells [47]. Our results suggested that SeVdp(KOSM)302L likely infects canine monocytes. iPSCs derived from monocytes, which have the exact genomic background of the donor, are expected to be useful in disease modeling because monocytes, unlike T cells, do not undergo major genetic rearrangements in T cell receptor and immunoglobulin regions.
This study provided an effective method for generating ciPSCs from canine PBMCs, which can be easily collected with minimum invasiveness. In addition, human peripheral blood samples remain viable at 25°C for ∼48 h and can be transported to long distances after cryopreservation [48]. Thus, it may be possible to generate ciPSCs from stored canine blood samples and freshly isolated samples from donors at the site of clinical care. Therefore, our method of generating footprint-free ciPSCs from PBMCs may allow easy generation of disease-specific ciPSCs from patients and large-scale ciPSCs for clinical use. For a more robust translation of ciPSCs to clinics, on-feeder culture systems should be replaced with a feeder-free system that allows easy amplification of derived iPSCs.
Moreover, to use ciPSC lines for clinical purposes, the appropriate quality standards are required in the veterinary stem cell field. The standards, policies, and practices to ensure the highest quality and uniformity of stem cell lines and their derivatives for human use are established [49]. In the same way, more kinds of tests for assessing the quality of ciPSC lines, such as genomic sequencing, STP analysis, and karyotyping with G-banding, should also be performed in the future.
In conclusion, relative to the available methods, we developed a more effective strategy for the iPSC reprogramming of canine PBMCs by using SeVdp(KOSM)302L, N2B27 medium supplemented with a small-molecule cocktail, and StemFit, resulting in the generation of footprint-free ciPSCs. We believe that our method can facilitate the research involving disease modeling, and regenerative therapies in the veterinary field.
Footnotes
Author Disclosure Statement
None of the authors have any conflicts of interest to declare.
The authors thank Editage for English language editing.
Funding Information
This work was supported by JSPS KAKENHI Grants JP18K19273, JP18H02349, and JP19J22851 and by Sasakawa Scientific Research Grants 2020-4079 from The Japan Science Society.
Supplementary Material
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.