
Abstract
The cell-type-specific response of neural cells to oxidative stress, a crucial mechanism for accelerating aging and cognitive dysfunction in Alzheimer's disease (AD), is still far from understood. Here, we employed human-induced pluripotent stem cells (hiPSCs)-derived neural stem cells (hiPSC-NSCs), neurons (hiPSC-Neurons), and microglia-like cells (hiPSC-MGLs) from sporadic AD (sAD) patients, age-matched cognitive normal controls (CNCs), and young subjects to observe human neural cell-type response to H2O2 stimulation. Without H2O2 exposure, reactive oxygen species (ROS) cannot be detected in hiPSC-NSCs from all three groups, but the viability of hiPSC-NSCs from AD patients was significantly lower than those of CNCs and young subjects. There were no significant differences in ROS, viabilities, neurite length, and neurite branch points in hiPSC-Neurons among three groups. No significant differences in viabilities, phagocytosis, and secretion of cytokines were observed in hiPSC-MGLs among three groups, but higher ROS levels in sAD hiPSC-MGLs. Under H2O2 exposure, the viability, neurite length, and neurite branch points of hiPSC-Neurons from AD patients reduced more significantly accompanied by more ROS release. H2O2 exposure caused hiPSC-MGLs from AD patients to release more ROS, cytokines, and stronger phagocytosis. Nevertheless, H2O2 exposure had no effect on viability of hiPSC-NSCs. Our results showed hiPSC-Neurons and hiPSC-MGLs were more sensitive to H2O2 than hiPSC-NSCs, which indicated the different response styles of hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs to oxidative stress. HiPSC-derived neural cells from AD patients suffered more severe injury from H2O2 than those of CNCs and young subjects, indicating that the vulnerability to oxidative stress of AD patients can be recapitulated in hiPSCs.
Introduction
Alzheimer's disease (AD) is the most common neurodegenerative disorder, characterized by progressive decline of cognitive functions and memory loss [1,2]. While the pathological mechanism of AD is still not well elucidated. It is generally agreed that AD is not only a neuron-centric disease but likely due to complex interactions dysfunction among different brain cell types [3,4]. The most obvious pathological change of AD is the loss of neurons and synapses in the specific brain regions (cortex and hippocampus) [5,6]. Neural stem cells (NSCs), the only source of newborn neurons in the adult brain, have been found to be reduced or decreased neuronal regeneration in the brains of AD patients [7,8]. Meanwhile, microglia, the most important immune cells in the brain, were found to be overactivated and to consistently secrete large amounts of inflammatory cytokines and neurotoxic substances leading to neuronal injuries and accelerating the progression of AD [9,10].
Studies have shown that oxidative stress plays an important role in the pathogenesis of AD [11]. Release of reactive oxygen species (ROS) exceeds the body's antioxidant capacity, leading to the accumulation of oxidizing substances, functional attenuation of antioxidant systems, thereby inducing tissue damage [12]. However, the cell-type-specific oxidative stress injuries in relation to AD pathology remain understudied. The immortal neuronal cell lines and primary neuronal cells from animal models have been used to study AD over the past few decades [13]. Due to the limitations of genetic and physiological differences between human and rodent brains, the studies of AD were full of controversy [14]. It is extremely difficult to obtain human neural cells from patients' brain, therefore the studies on human neural cells injury caused by oxidative stress were limited. Recent advances in neural cells from human-induced pluripotent stem cells (hiPSCs) hold great promise for the study of neurodegenerative disease, especially AD [15 –19]. HiPSCs can be differentiated into different neural cell types, including NSCs, neurons, astrocytes, and microglia, and can then be used to establish in vitro model for studying their roles in the pathogenesis of AD [19 –23]. Actually, there have been some applications of hiPSCs-derived neural cells in oxidative-stress-related studies [15,24 –26]. Microglia differentiation from iPSCs has been very challenging because of the special origin and lack of suitable identification markers [27]. Based on published research, we optimized and established a stable microglia-like cells (hiPSC-MGLs) differentiation method in our previous study [17,22,28]. In addition, we found for the first time that hiPSC-MGLs from sporadic AD (sAD) patients exhibited significant inflammatory characteristics and enhanced phagocytosis compared with hiPSC-MGLs from cognitive normal controls (CNCs) when stimulated by lipopolysaccharide (LPS) [17]. However, how hiPSC-MGLs and other types of neural cells, such as hiPSC-NSCs and hiPSC-Neurons, respond to oxidative stress remains understudied.
To better understand how different neural cells respond to the oxidative stress, hiPSCs from sAD patients, age-matched CNCs, and young subjects were differentiated into three main neural cells; that is hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs, and then the intracellular ROS and cellular viabilities of hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs, neurite outgrowth of hiPSC-Neurons, the cytokine secretion and phagocytic function of hiPSC-MGLs were investigated in the presence and absence of H2O2 exposure. The research may help us to understand the more vulnerability of the neural cell types to oxidative stress injuries in AD, which may provide a new perspective for the prevention and treatment of AD.
Materials and Methods
Summary of the experimental process
The summary of the experimental process is provided (Fig. 1). HiPSCs derived from young subjects, CNCs, and sAD patients were first differentiated into three types of neural cells, hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs, then the specific markers of the neural cells were identified by immunofluorescence, and then the effects of oxidative stress (H2O2) on hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs were studied. ROS and cell viabilities of hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs were evaluated. Neurite outgrowth of hiPSC-Neurons and cytokine secretion and phagocytosis of hiPSC-MGLs were studied.
Summary of the experimental process.
Maintenance and culture of human pluripotent stem cells
The experiments including the stem cells were approved by Medical Ethics of Committee Chinese People's Liberation Army (PLA) General Hospital, and informed consent was obtained from the patients. The protocol was approved by the Institutional Review Board (IRB) of PLA General Hospital (no. 2019019D). Human peripheral blood mononuclear cells (PBMCs) were used for the reprogramming of hiPSCs. Blood samples were collected from the Department of Neurology in Chinese PLA General Hospital. AD patients were clinically diagnosed and characterized by neurologists and neuropsychologists based on the NINCDS/ADRDA criteria. Volunteers of age-sex-matched cognitively normal controls and young subjects without dementia (assessed by clinical evaluation) were used as controls. Generation and quality control of hiPSCs were carried out as described earlier [29,30]. In brief, PBMCs were programmed into hiPSC by Yamanaka factors OCT3/4, SOX2, KLF4, and C-MYC delivered by nonintegrating episomal vectors, and the pluripotency of all cell lines was confirmed by EB differentiation in vitro. HiPSCs were cultured in feeder-free conditions, coated with hESC-qualified Matrix Matrigel (BD Bioscience, Cat: #354277, San Jose, CA) in complete mTeSR™1 medium (STEMCELL Technologies; Cat: #05850, Vancouver, BC, Canada) under the condition of humidified incubator (5% CO2, 37°C). The medium was changed every day since the hiPSC line reached 60–70% confluence. Then, the hiPSC line was dissociated by Versene Solution (Thermo Fisher Scientific, Cat: #15040066, Waltham, MA) and passaged into clumps at a ratio of 1:3. The genders, phenotype, and origin of each line used in this study are enlisted in Table 1.
Induced Pluripotent Stem Cell Lines Used in This Study
Table summarizing demographic information with each line.
CNC, cognitive normal control; sAD, sporadic AD; iPSC, induced pluripotent stem cell.
NSCs generation from hiPSC and maintenance
Generation of hiPSC-NSCs was carried out based on a published protocol with modifications [29,31]. TryplE™ Express-dissociated hiPSCs were plated onto Matrigel (BD Biosciences)-coated six-well plates at a density of 0.25 × 105/cm2. To prevent cell death, 10 μM ROCK inhibitor (TORCIS, Cat: #Y27632, Bristol, United Kingdom) was added to the complete mTeSR™1 medium. Twenty-four hours after seeding, culture medium was changed by NSC induction medium (Shanghai IxCell Biotechnology Co., Ltd, Cat: #N170305, Shanghai, China) containing NSC induction basal medium supplemented with NSC induction supplement. During induction, NSC induction medium was changed every other day for 7 days when the cells reached confluence. To ensure sufficient nutrients in the later stage of induction, we made a minor modification for the method, namely double the amount of medium was replaced from day 4 to day 7. At day 7 of induction, primitive NSCs were dissociated and identified.
To maintain the expansion of NSCs, primitive NSCs were dissociated with Accutase (Thermo Fisher Scientific; Cat: #A1110501) and cultured on Matrigel-coated dishes in the NSCs expansion medium (Shanghai IxCell Biotechnology Co., Ltd; Cat: #N160505) containing NSC expansion basal medium supplemented with NSC induction supplement at a density of 1 × 105/cm2. To prevent cell death, 10 μM rock inhibitor (TORCIS; Cat: #Y27632) was added to NSCs expansion medium. The NSC was passaged every 4–5 days (reached 90% confluence).
Neuronal differentiation from NSCs
Generation of hiPSC-Neurons was carried out based on a published protocol [32]. Accutase-dissociated NSCs were plated onto 20 μg/mL Polo-L-ornithine (Sigma-Aldrich, Cat: #P4957, St. Louis, MO) and 10 μg/mL laminin (Life Technologies, Cat: #23017015, Gaithersburg, MD)-coated six-well plates in NSCs expansion medium at a density of 1.2 × 105/well. To prevent cell death, 10 μM rock inhibitor (TORCIS; Cat: # Y27632) was added to NSCs expansion medium. After 24 h, equal volume of complete neuronal differentiation medium was added to the existing medium in each well. The complete neuronal differentiation medium consists of BrainPhys™ Neuronal Medium (STEMCELL Technologies; Cat: #05790), 2% NeuroCult™ SM1 (STEMCELL Technologies; Cat: #05711), 1% N2 Supplement (Thermo Fisher Scientific; Cat: #17502048), 20 ng/mL BDNF (R&D Systems; Cat: #248-BDB-010/CF, Minneapolis, MN), 20 ng/mL GDNF (R&D Systems; Cat: #212-GD-010/CF), 1 mM cAMP (Sigma-Aldrich; Cat: #9501), 200 nM Ascorbic Acid (STEMCELL Technologies, Cat: #72132), and the spent medium was changed every 3 days.
Differentiation of hiPSCs into microglia
Generation of microglia was carried out as described earlier [17]. In brief, hiPSC-derived hematopoietic progenitors (iHPCs) were generated using the STEMdiff™ Hematopoietic Kit (STEMCELL Technologies; Cat: #05310) in accordance with the manufacturer's instructions. For microglia differentiation, isolated CD43+ iHPCs were seeded onto Matrigel-coated 12-well plates at 2.0 × 105 cells/well in 1 mL of differentiation media Microglia Medium (ScienCell Research, Cat: #1901, Carlsbad, CA), 2% B27 (Thermo Fisher Scientific; Cat: #17504044), 0.5% N2 (Thermo Fisher Scientific; Cat: #17502048), 1 × Glutamax (Thermo Fisher Scientific; Cat: #35050061), 1 × NEAA (Thermo Fisher Scientific; Cat: #11140050), 25 ng/mL GM-CSF (R&D Systems; Cat: #215-GM), 25 ng/mL M-CSF (R&D Systems; Cat: #216-MC), 50 ng/mL IL-34 (R&D Systems; Cat: #5265-IL), 50 ng/mL TGFβ-1 (R&D Systems; Cat: #240-B), and 25 ng/mL IGF-1 (R&D Systems; Cat: #291-G1). Each well was supplemented with 0.5 mL of differentiation media every 2 days.
Immunofluorescence staining
Cells were fixed in 4% paraformaldehyde (PFA) in PBS for 15 min at room temperature (RT). Endogenous peroxidase of the cells was quenched in 1% hydrogen peroxide (H2O2) for 30 min before immunofluorescence staining. Then, the fixed cells were permeabilized with 0.1% Triton X-100 (Sigma Aldrich; Cat: #X100RS) for 10 min and blocked with 3% bovine serum albumin for 30 min at RT. Thereafter, cells were stained for 2 h at RT with primary antibodies. These included the following: Mouse Anti-Nestin antibody (Abcam, Cat: #ab22035, 1:500, RRID: AB_446723, Cambridge, MA), Sox2 (D6D9) XP® Rabbit mAb (CST, Cat: #3579, 1:500, RRID: AB_2195767, Danvers, MA), Rabbit Anti-MAP2 antibody (Abcam; Cat: #ab32454, 1:1000, RRID: AB_776174), goat Anti-Iba1 antibody (Abcam; Cat: #ab5076, 1:500, RRID: AB_2224402), and Rabbit Anti-TMEM119 antibody (Abcam; Cat: #ab185333, 1:500, RRID: AB_2687894). After repeated wash with TBST for three times, the cells were incubated with secondary antibodies for 60 min at RT. These included the following: peroxidase-conjugated goat antirabbit IgG (ZSGB-BIO, Cat: #ZB-2301, 1:1000, RRID: AB_2747412, Beijing, China), peroxidase-conjugated goat antimouse IgG (ZSGB-BIO, Cat: #ZB-2305, 1:1000, RRID: AB_2747415); then incubated with Cy3-Tyramide (Ty-Cy3) or FITC-Tyramide (Ty-FITC) (PerkinElmer, Cat: #NEL748001KT, Waltham, MA) at RT for 10 min. The nuclei were finally stained with DAPI for 5 min at RT before observation under a laser scanning confocal microscopy (Nikon A1, Tokyo, Japan).
Time-lapse microscopic imaging
For long-term, continuous, imaging analysis, cells plated in six-well plates were incubated in the InCuCyte Zoom live-cell imaging instrument (Essen Bioscience, WGC, United Kingdom). Images of 16 fields per well were acquired every 4 h after seeding. To obtained cell growth curve, basic analysis software (Essen Bioscience) was used to automatically define cell confluence that reflects cell number. For label-free quantitation of neurite dynamics in real time during neuronal differentiation, NeuroTrack Assays analysis software (Essen Bioscience) was used to automatically define neurite length, neural cell body clusters as well as neurite branch points.
Measurement of intracellular ROS levels
Cells were plated onto 96 wells at a density of 3.0 × 104 cells/well at 37°C in a 5% CO2 incubator. After 24 h, the medium was aspirated, and 100 μL of fresh medium containing different concentrations of H2O2 was added to the well. After 24 h, ROS levels were measured using ROS Assay Kit (Beyotime, Cat: S0033M, Shanghai, China) based on 1 × 2′, 7′-dichlorofluorescein-diacetate (DCFH-DA) fluorescent probe in accordance with the manufacturer's instructions. Then, the cells were observed using laser scanning confocal microscopy (Nikon A1), and measured at 488 nm excitation and 525 nm emission by an EnSpire Multilabel Reader (PerkinElmer 2300). The data of ROS were normalized with Young-Con.
Cell viability assays
Cells were plated onto 96 wells at a density of 3.0 × 104 cells/well at 37°C in a 5% CO2 incubator. After 24 h, the medium was aspirated, and 100 μL of fresh medium containing different concentrations of H2O2 was added to the well. After 24 h, living cells and dead cells were detected using Calcein/PI Cell Viability/Cytotoxicity Assay Kit (Beyotime; Cat: C2015M) in accordance with the manufacturer's instructions. Then, the cells were observed under a laser scanning confocal microscopy (Nikon A1). Then, cell viability was calculated using the following formula: cell viability (%) = (number of Calcein-AM+ cells) / (number of Calcein-AM+ cells + number of PI+ cells) × 100.
Luminex analysis of cytokines
HiPSC-MGLs were plated onto 96 wells at a density of 3.0 × 104 cells/well at 37°C in a 5% CO2 incubator. After 24 h, the medium was aspirated, and 100 μL of fresh medium containing different concentrations of H2O2 was added to the well. After 24 h, the cell culture supernatant was collected. Then, the concentrations of human IL-10, IL-6, IL-1β, TNF-α, CCL-2, and CXCL10 in cell culture supernatant were determined using Luminex assay kits (Millipore; Cat: #HCYTOMAG-60K, Billerica, MA) in accordance with the manufacturer's instructions.
Phagocytosis assays
HiPSC-MGLs were plated onto 96 wells at a density of 3.0 × 104 cells/well in microglia medium at 37°C in a 5% CO2 incubator. After 24 h, the medium was aspirated, and 100 μL of fresh medium containing different concentrations of H2O2 was added to the well. After 24 h, the medium with H2O2 was aspirated, and 100 μL of PBS containing 0.05% neutral red was added, and the cells were cultured for an additional 30 min. Thereafter, cell culture supernatant was aspirated, and the cells were washed thrice with PBS. Then, 100 μL of cell lysate [V (glacial acetic acid): V (ethanol) = l: 1] was added to each well, and the plates were incubated at 4°C for 2–3 h, followed by measurement of the optical density at 540 nm using EnSpire Multilabel Reader (PerkinElmer 2300).
Statistical analysis
All experiments were performed in triplicate independently, and statistical analyses were performed using Graph Pad Prism 8.0 (San Diego, CA). Differences with P < 0.05 were considered statistically significant. All data were presented as mean ± standard error of the mean (SEM). Data was analyzed using one-way ANOVA, followed Dunnett's multiple comparisons test or two-way ANOVA, followed by Sidak's multiple comparisons test.
Results
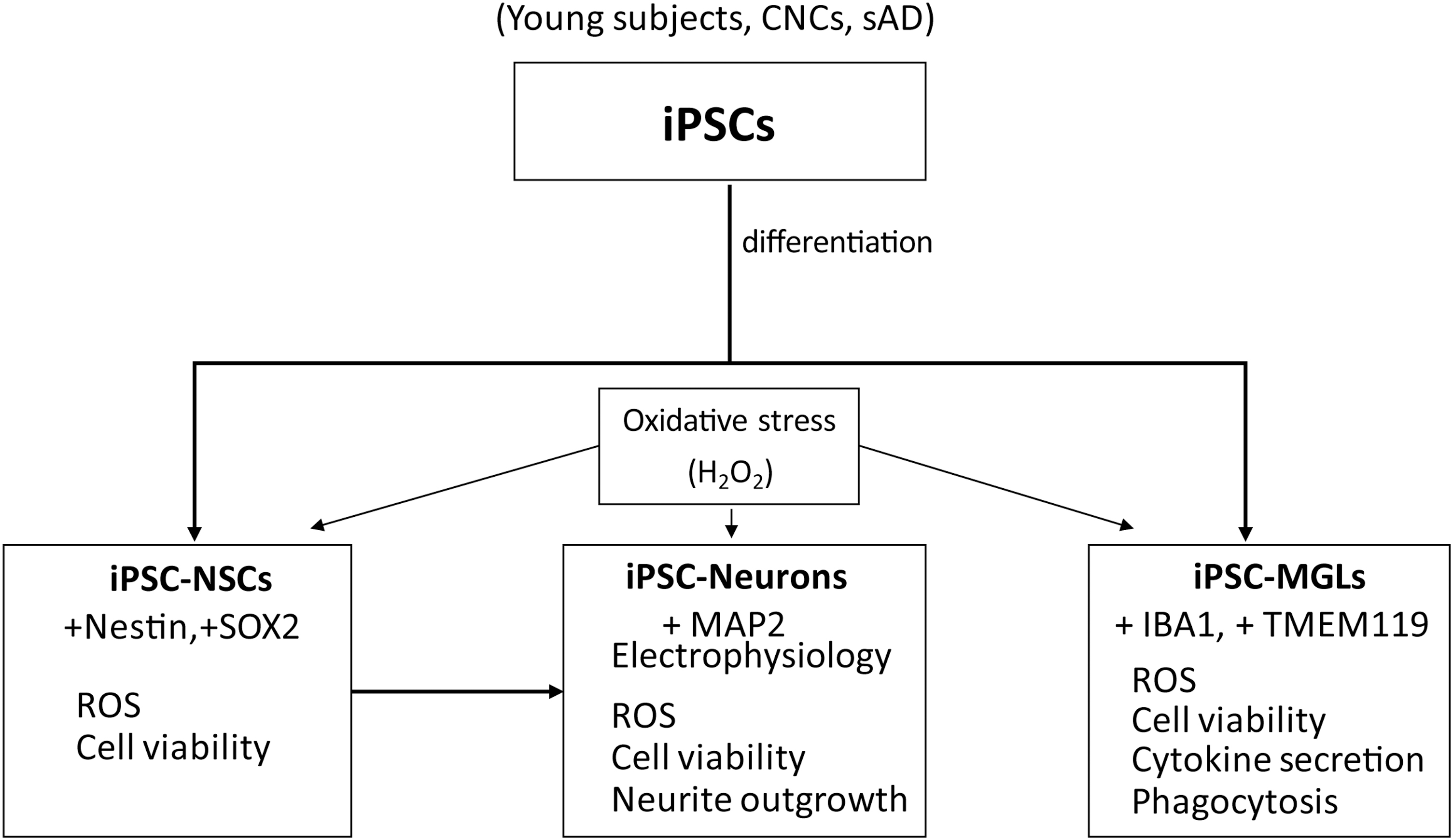
The viability of hiPSC-NSCs from sAD was lower, but not further affected by H2O2 exposure
HiPSCs were differentiated into NSCs according to published methods [29,31]. HiPSC-NSCs were identified by immunofluorescence. The immunostaining results showed that hiPSC-NSCs derived from young subjects, CNCs, and sAD patients all strongly expressed NSC markers, such as Nestin and SOX2 (Fig. 2A). Statistical analysis showed that the proportions of Nestin+ cells and SOX2+ cells in the differentiated cultures were >90%, and there were no significant differences in Nestin+ cells and SOX2+ cells among young subjects, CNCs, and sAD patients (Fig. 2B). ROS cannot be detected in hiPSC-NSCs from all the young subjects, CNCs, and sAD patients without or with H2O2 exposure (Fig. 2C). The viability of hiPSC-NSCs from sAD patients was significantly lower than that of hiPSC-NSCs from CNCs without H2O2 exposure. When treating the hiPSC-NSCs with increasing doses of H2O2, the viability of hiPSC-NSCs from sAD patients was not significantly affected, neither were those from young subjects and CNCs (Fig. 2D, E).

Characterization of iPSC-NSCs and effects of H2O2 exposure on ROS and viabilities of iPSC-NSCs from young subjects, CNCs, and sAD patients.
Effects of H2O2 on the ROS, viability, and neurite outgrowth of hiPSC-neurons
More ROS release and decrease of viability of hiPSC-neurons from sAD after H2O2 exposure
To study the effects of oxidative stress on hiPSC-Neurons, hiPSC-NSCs were first differentiated into hiPSC-Neurons continuously for 35 days, then intracellular ROS and viability of hiPSC-Neurons were studied. The differentiated hiPSC-Neurons displayed typical morphological feature of neurons after 35 days differentiation and strongly expressed neuronal marker microtubule-associated protein 2 (MAP2) (Fig. 3A). Electrophysiology recordings revealed that hiPSC-Neurons differentiated from hiPSC-NSCs exhibited strong electrophysiological activity (Fig. 3B). Statistical analysis showed that the proportion of MAP2+ cells in the differentiated cultures was ∼80%. There was no significant difference of MAP2+ cells among young subjects, CNCs, and sAD patients (Fig. 3C). Without exposure of H2O2, there were no significant differences in ROS and viabilities of hiPSC-Neurons among three groups (Fig. 3D, E). However, treatment with increasing doses of H2O2 significantly increased ROS levels in hiPSC-Neurons from all three groups, of which hiPSC-Neurons from sAD were enhanced to a greater extent (Fig. 3D, E). Meanwhile, increasing doses of H2O2 significantly suppressed viability of hiPSC-Neurons from sAD patients, while only 625 μM H2O2 caused significant decrease of viabilities of hiPSC-Neurons from young subjects and CNCs (Fig. 3F, G).
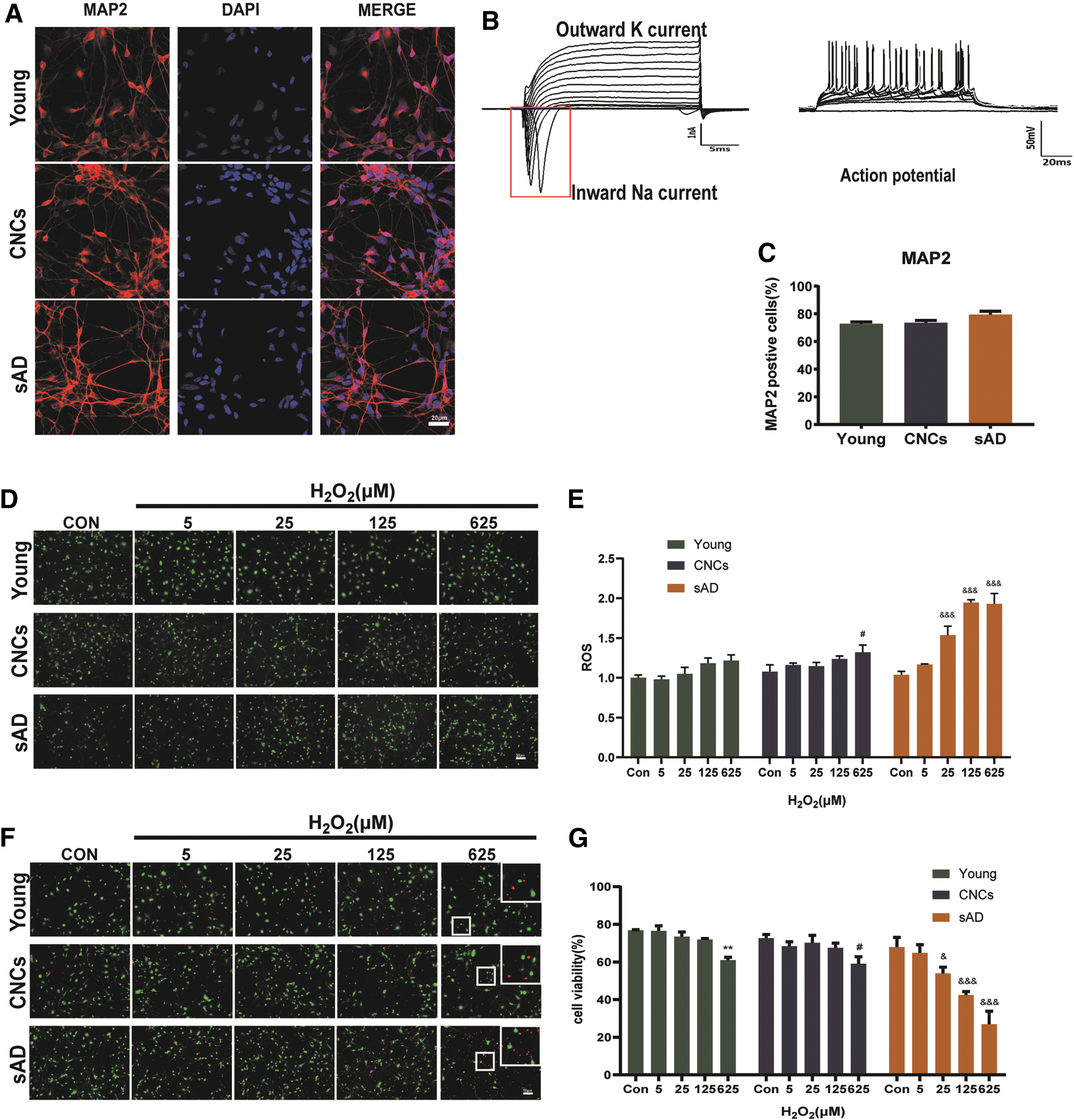
Characterization of iPSC-Neurons and effects of H2O2 on ROS and viabilities of iPSC-Neurons from young subjects, CNCs, and sAD patients.
Neurite outgrowth of hiPSC-Neurons from sAD was inhibited more severely after H2O2 exposure
Neurite outgrowth and the formation of synaptic connections are the basis for the realization of nervous system functions and key steps in the formation of memories. The neurite outgrowth can be determined by the parameters of neurite length and neurite branch points. To better understand the effects of oxidative stress on neurite outgrowth of hiPSC-Neurons, live-cell imaging was performed. The NeuroTrack module was used to measure neurite length and neurite branch points based on phase-contrast imaging. Representative bright field images marked by label-free NeuroTrack Assays analysis of hiPSC-Neurons from young subjects, CNCs, and sAD patients treated with 625 μM H2O2 are shown in Fig. 4A and B. Statistical analysis showed that there were no significant differences in neurite length and neurite branch points among young subjects, CNCs, and sAD patients without H2O2 exposure (Fig. 4D, F). The neurite length and neurite branch points began to decrease after H2O2 exposure (Fig. 4C, E). Statistical analysis of neurite length showed that H2O2 caused a dose-dependent decrease in neurite length of hiPSC-Neurons from young subjects, CNCs, and sAD patients after 24 h of H2O2 exposure. HiPSC-Neurons from sAD displayed ∼47% reduction of neurite length compared with 14% reduction in CNCs hiPSC-Neurons for 625 μM H2O2 exposure (Fig. 4D). Statistical analysis of neurite branch points after 24 h H2O2 exposure showed that increasing doses of H2O2 caused significant loss of neurite branch points only in hiPSC-Neurons from sAD patients in a dose-dependent manner. No such effect was observed on hiPSC-Neurons from young subjects and CNCs (Fig. 4F).
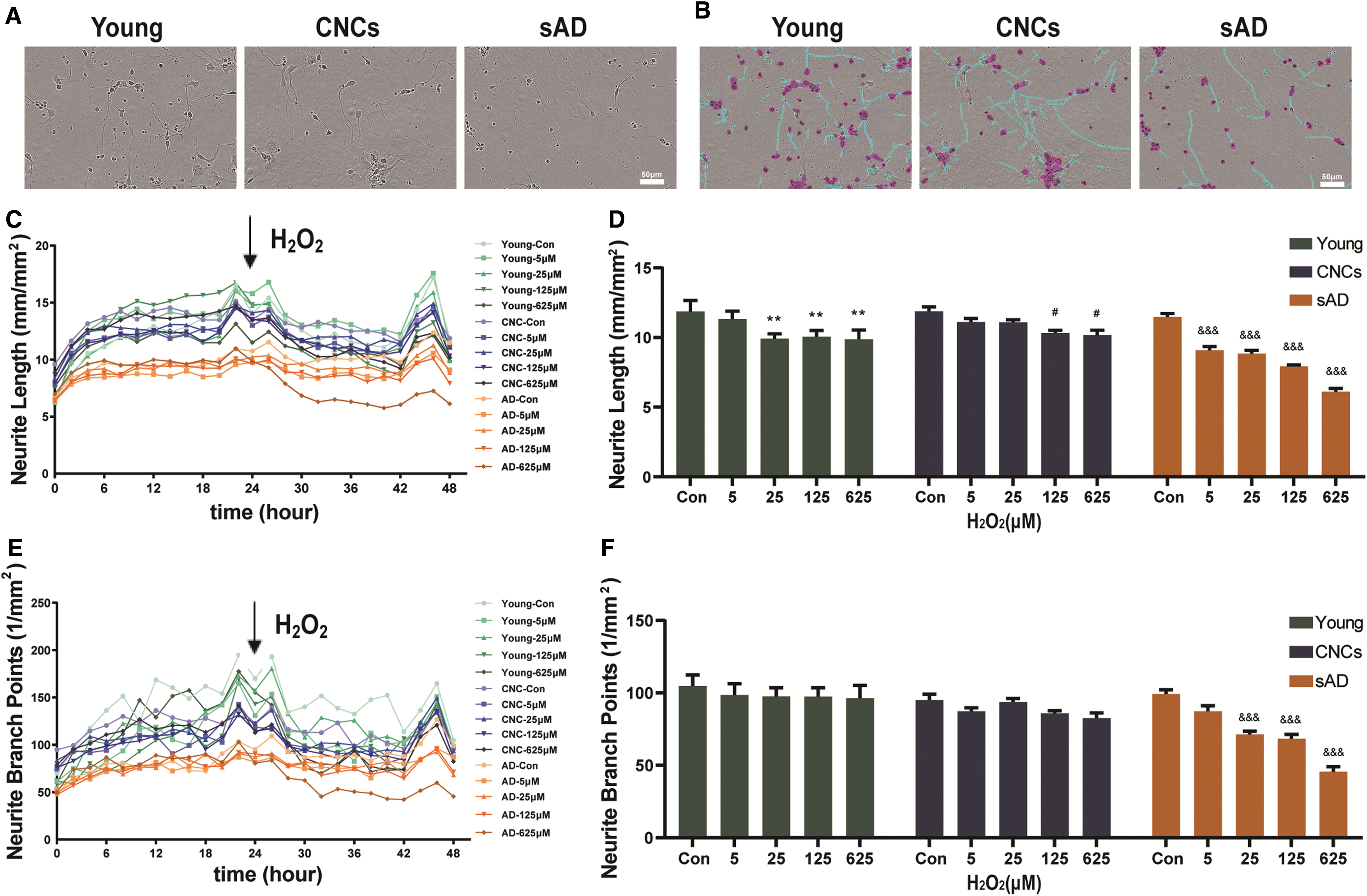
Effect of H2O2 on the neurite outgrowth of iPSC-Neurons.
Effects of H2O2 on the ROS, viability, cytokine secretion and phagocytosis of hiPSC-MGLs
More ROS release and decrease of viability of hiPSC-MGLs from sAD after H2O2 exposure
HiPSCs were differentiated into hiPSC-MGLs based on a method described earlier [17]. The differentiated cells displayed typical morphological feature of microglia after 25 days differentiation and strongly expressed microglia markers, IBA1 and TMEM119 (Fig. 5A). Statistical analysis showed that the proportions of IBA1+ cells and TMEM119+ cells in the differentiated cultures were ∼90%. There were no significant differences in IBA1+ cells and TMEM119+ cells among young subjects, CNCs, and sAD patients (Fig. 5B). Without exposure of H2O2, the level of ROS in hiPSC-MGLs from sAD was significantly higher than that found in young subjects and CNCs. 5, 25 μM H2O2 increased the levels of ROS of hiPSC-MGLs from young subjects, CNCs, and sAD patients, while 625 μM H2O2 caused a decrease of ROS for all three groups, of which the level of ROS in sAD hiPSC-MGLs was decreased to a greater extent (Fig. 5C, D). Without exposure of H2O2, there were no significant differences in viabilities of hiPSC-MGLs among three groups. 5, 25, and 125 μM H2O2 had no significant effect on the viabilities of hiPSC-MGLs from three groups, while 625 μM H2O2 caused a decrease of viabilities for all three groups, of which the viability of sAD hiPSC-MGLs was decreased to a greater extent (Fig. 5E, F).
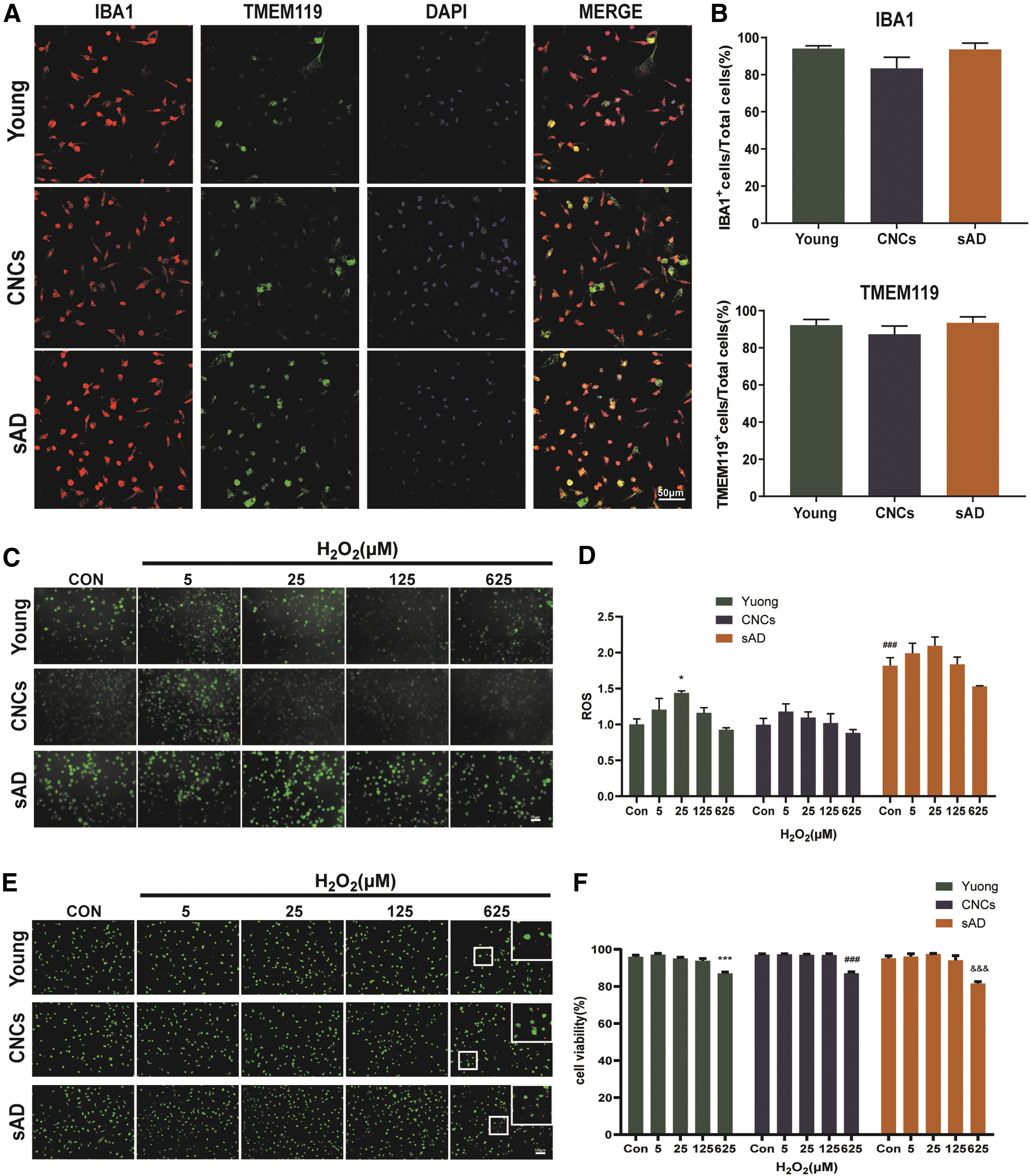
Characterization of iPSC-MGLs and effect of H2O2 on ROS and viabilities of iPSC-MGLs from young subjects, CNCs and AD patients.
HiPSC-MGLs from sAD showed stronger cytokine secretion and phagocytosis after H2O2 exposure
To better understand the effects of oxidative stress on the function of hiPSC-MGLs, the abilities of cytokine secretion and phagocytosis were investigated. There were no significant differences in the levels of cytokines (IL-10, IL-6, IL-1β, TNF-α, CCL-2, and CXCL10) secreted by hiPSC-MGLs among young subjects, CNCs, and sAD patients without H2O2 exposure (Fig. 5A). The secretion of IL-10, IL-6, and CXCL10 in hiPSC-MGLs from all young subjects, CNCs, and sAD patients was significantly increased with the exposure of H2O2, while hiPSC-MGLs from sAD secreted more IL-10 and CXCL10 with 625 μM H2O2 exposure and more IL-6 with 25 μM H2O2 exposure. Interestingly, hiPSC-MGLs from young subjects and CNCs secreted more IL-10 and CXCL10 with 5 μM H2O2 exposure (Fig. 6A). Then, the phagocytosis of hiPSC-MGLs was evaluated. There were no significant differences in hiPSC-MGLs phagocytosis among young subjects, CNCs, and sAD patients without H2O2 exposure. However, H2O2 exposure increased the phagocytosis of hiPSC-MGLs from young subjects, CNCs, and sAD patients. Phagocytosis of hiPSC-MGLs from sAD increased ∼51%, while 45% increase was observed in hiPSC-MGLs from CNCs and 36% increase observed in hiPSC-MGLs from young subjects with 5 μM H2O2 exposure (Fig. 6B).

Effects of H2O2 on the cytokine secretion and phagocytic ability of iPSC-MGLs.
Discussion
The development of in vitro models of the human nervous system disorders, especially AD, has always been a great challenge because of the specificity of central nervous system. Over the years, owing to the limitations of accessibility to the relevant cell types, primary neuronal cells or immortal neuronal cell lines from animal models were used for the study of AD [33]. But due to the species differences, results from animal models are often restricted to translate directly to humans [34]. In recent years, iPSCs provide new tools for studying neurodegenerative diseases and have been widely used in this field. Moreover, the contribution of different neural cell types to the pathology of AD and how different types of neural cells react to pathogenic factors of AD, especially oxidative stress, have attracted the interest of researchers. However, the cell-type-specific responses of neural cells in brain to oxidative stress injuries in relation to AD pathology remain understudied. Therefore, in this study, we used hiPSCs derived from sAD patients, age-matched CNCs, and young subjects, and differentiated them into hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs, then studied the different responses of oxidative stress to hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs.
In this study, hiPSC-NSCs and hiPSC-Neurons from sAD patients exhibited lower viabilities when compared with those of CNCs and young subjects, while hiPSC-MGLs derived from sAD patients exhibited increased inflammatory characteristics and phagocytosis. Similar findings have been shown in clinical and animal research. In the brains of AD patients, NSCs in the dentate gyrus of the hippocampus were found to show reduced neuronal regeneration [7,8]. And results from animal models also showed that NSCs isolated from hippocampus of PS1▵E9 transgenic AD model mice exhibited significantly lower viability, and the self-renewal ability was also decreased [35]. Significant neuron loss was observed in both clinical patient and animal models with AD [36 –38]. In the brain of AD patients, both aggregated Aβ and overphosphorylated tau proteins can directly cause microglia overactivation, thereby secreting a large number of inflammatory factors and increasing the phagocytic ability to eliminate toxic substances [39 –42], which was concurrent with our results that hiPSC-MGLs from sAD patients also showed stronger inflammatory characteristics and phagocytosis, suggesting an already active state of sAD-hiPSC-MGLs. The results above suggested that hiPSCs-derived neural cells, including hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs, demonstrated pathological features of AD to some extent, which may serve as suitable models for the pathological mechanism investigations and drug screening of AD.
As a key risk factor for AD, oxidative stress has long been known to cause the damage to brain tissue and play an important role in the development of AD [43 –45]. It has been widely recognized that AD patients are more susceptible to the risk factors of AD, but the relevant data support is indeed very limited. In our study, we found that neuronal viability and neurite outgrowth of hiPSC-Neurons from sAD patients were more susceptible to be inhibited by H2O2, when compared with those of young subjects and CNCs. And the secretory capacity of cytokines and phagocytic ability of hiPSC-MGLs from sAD patients were significantly higher than those of young subjects and CNCs with H2O2 exposure. A recent study has reported that iPSCs differentiated neurons from AD patients have significantly reduced cell viability when exposed to H2O2 and Aβ stimulation [24], which was consistent with our findings. Furthermore, our results found enhanced secretory capacity of cytokines and phagocytic ability of hiPSC-MGLs from sAD patients, which were consistent with our previous study that hiPSC-MGLs from sAD exhibited stronger inflammatory characteristics and phagocytic ability stimulated by LPS [17]. These results indicated that neural cells from sAD patients were more sensitive to oxidative stress, providing evidence that AD patients are more vulnerable to oxidative stress. In addition, we found that hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs responded differently to oxidative stress. Under H2O2 exposure, neuronal viability and neurite outgrowth were significantly inhibited, and hiPSC-MGLs were activated for secreting more cytokines and enhanced phagocytosis. However, the viability of hiPSC-NSCs was unaffected. The above results indicated that the responses of hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs to oxidative stress were cell-type specific, and hiPSC-Neurons and hiPSC-MGLs were more sensitive to the oxidative stress. Similar findings were reported in previous research. Thiamine deficiency (TD) will cause chronic oxidative stress leading to selective neuronal death. A recent study found that only in neurons did TD induce apoptosis, while it had no significant effect on the survival of other cell types, such as microglia, astrocytes, and neuroblastoma, indicating that neurons are more susceptible to oxidative stress [46], which was consistent with our results. Possible reasons for the different responses to oxidative stress of hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs may be as follows. First, oxidative damage is associated with mitochondrial abnormalities [45]. Mitochondria are not only the main source of ROS production but also important target organelle damaged by oxidative stress [47,48]. Mitochondrial DNA copy number and mitochondrial mass increased dramatically during neuronal differentiation, which may account for a greater sensitivity to H2O2 in neurons than NSCs [49]. Our research also confirmed that ROS production was almost undetectable in hiPSC-NSCs. In addition, glutathione, as an important antioxidant in the body, can combine with ROS, thereby accelerating the excretion of ROS and combating damage to the body by ROS. Studies have found that glutathione in glial cells was more than twice that of neurons [50,51], which may further explain why hiPSC-Neurons were more likely to die when injured by oxidative stress, while hiPSC-MGLs exhibited greater resistance.
In summary, our results showed that neural cell types of sAD patients differentiated by hiPSCs can reflect pathological features of AD to a certain degree, providing evidence that neural cell types of sAD patients are more vulnerable to oxidative stress. Furthermore, the responses of hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs to oxidative stress were cell-type specific; hiPSC-Neurons and hiPSC-MGLs are more sensitive to oxidative stress than hiPSC-NSCs. However, there are still some limitations in this study. Human brain is clearly much more complex than the separate cell culture models, research on coculture of hiPSC-NSCs, hiPSC-Neurons, and hiPSC-MGLs will help us understand AD pathology while taking into account the interactions between cells in the brain. As the largest number of glial cells in the brain, astrocytes play an important role in the process of the onset of AD [52]. Further investigations of astrocytes to oxidative stress will help us have a more comprehensive understanding of different brain cell types' contribution of AD pathology. We believe that hiPSC-derived neural cells from sAD patients may serve as good tools for studying the pathological mechanism and screening new treatment strategies for AD.
Footnotes
Author Disclosure Statement
The authors have no conflict of interest to report.
Funding Information
This work was supported by National Key Research and Development Program (2016YFC1306300).