
Abstract
The regenerative effects of stem cells derived from dental tissues have been previously investigated. This study assessed the potential of human tooth stem cells from apical papilla (SCAP) on nerve regeneration. The SCAP collected from nine individuals were characterized and polarized by exposure to interferon-γ (IFN-γ). IFN-γ increased kynurenine and interleukin-6 (IL-6) production by SCAP, without affecting the cell viability. IFN-γ-primed SCAP exhibited a decrease of brain-derived neurotrophic factor (BDNF) mRNA levels, followed by an upregulation of glial cell-derived neurotrophic factor mRNA. Ex vivo, the co-culture of SCAP with neurons isolated from the rat dorsal root ganglion induced neurite outgrowth, accompanied by increased BDNF secretion, irrespective of IFN-γ priming. In vivo, the local application of SCAP reduced the mechanical and thermal hypersensitivity in Wistar rats that had been submitted to sciatic chronic constriction injury. The SCAP also reduced the pain scores, according to the evaluation of the Grimace scale, partially restoring the myelin damage and BDNF immunopositivity secondary to nerve lesion. Altogether, our results provide novel evidence about the regenerative effects of human SCAP, indicating their potential to handle nerve injury-related complications.
Introduction
The past few decades have been marked by an expressive growth in studies employing cell therapy, by testing different types of stem cells. Their ability to expand and differentiate into specialized cells allows their application in the treatment of numerous diseases. Mesenchymal stem cells (MSCs) are among the most studied alternatives, due to the facility of obtaining, being isolated from tissues such as bone marrow, adipose tissue, umbilical cord, and dental tissues [1,2].
In vivo, MSCs play an important role in maintaining tissue homeostasis and in the repair and regeneration processes. These effects do not seem to rely on differentiation, but rather on the paracrine effects exerted by the cells. MSCs are capable of secreting anti-apoptotic, neoangiogenic, and pro-mitotic molecules. They also act on cells of the immune system, presenting important anti-inflammatory and pro-regenerative roles [3,4]. The modulation of the immune response involves their ability to regulate cytokine secretion patterns [3,5]. Nonetheless, this capacity depends on the signaling molecules and the microenvironment in which these cells are found, determining their therapeutic potential, as well as the outcomes of the regenerative process [6,7].
The presence of an inflammatory context seems to be important for the in vivo MSCs behavior, as they are capable of responding to pro-inflammatory stimuli, secreting molecules that control an exacerbated immune response [8]. Previous studies showed that pre-activation of MSCs increases their immunosuppressive potential, both in vitro and in vivo [6,9,10]. An exposure to pro-inflammatory cytokines induces an anti-inflammatory phenotype in MSCs, with the subsequent expression of immunomodulatory factors, such as chemokines [11]. The exposure to IFN-γ has been shown to induce the indoleamine (IDO) expression by MSCs [10,12]. IDO has been implicated in the immunosuppressive capacity of human MSCs, inhibiting T cell activation and increasing the number of Tregs [13,14]. IDO-mediated immunosuppression is related to the production of the metabolite kynurenine, derived from the action of IDO on tryptophan [14]. Thus, MSCs have therapeutic potential due to their immunoregulatory properties, being widely studied in the field of regenerative medicine [15,16].
The MSCs derived from a variety of human dental tissues have a high capacity of in vivo differentiation, leading to tooth and periodontal regeneration [16 –19]. Specifically, the stem cells from the apical papilla (SCAP) population have a high proliferation rate and plasticity [16]. It was demonstrated that transplanted SCAP regenerated to a typical dentin-pulp-like complex and functional tooth, in immunocompromised mice and mini-pigs, respectively [20,21].
Literature data showed that local implantation, intravenous or intraperitoneal administration of MSCs, from different sources, trigger potential regenerative effects in preclinical models of peripheral nerve injury [22 –26]. Ullah et al. demonstrated that transplantation of human dental pulp-derived stem cells into a fibrin glue scaffold ameliorated nerve regeneration in rats [27]. Other studies showed beneficial effects of dental pulp stem cells (DPSC), periodontal ligament stem cells, and stem cells from human exfoliated deciduous teeth on nerve injury models [28 –31]. Their effects are likely mediated by neurotrophic factors secreted by dental MSCs, such as brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT3), and neurotrophin-4/5 [28,31 –33]. However, data on the use of SCAP for the same purpose are still scarce [31,33,34].
The gold standard treatment for peripheral nerve injuries is the use of autologous nerve grafts, but this technique is associated with adverse physical and psychological effects for the patient. Thus, the search for new alternatives to this purpose still represents a matter of high interest [35,36]. Considering the evidence mentioned earlier, this study aimed at investigating the in vitro, ex vivo, and in a classical in vivo model of neuropathic pain induced by sciatic chronic constriction injury (CCI), the neural regenerative potential effects of SCAP.
Materials and Methods
Ethical concerns
The protocols for obtaining human teeth and for the isolation of MSCs from the apical papilla (SCAP) of third molars were approved by the Research Ethics Committee of PUCRS (#60389816.7.0000.5336). The apical papilla rich on stem cells was collected from young patients (18–21 years-old) with problems caused by impacted teeth, undergoing third molar surgery by indication, in the Oral Surgery Unit of the School of Health and Life Sciences (PUCRS). The confidentiality of identity was guaranteed, and all the patients signed the informed consent form for tooth donation.
For animal experiments, the procedures followed the current Brazilian guidelines for the care and use of animals for scientific and didactic procedures, from the National Council for the Control of Animal Experimentation (CONCEA, Brazil, 2014). The local Animal Ethics Committee evaluated and approved all the protocols (CEUA 9035). The animal studies are reported in compliance with the ARRIVE guidelines [37,38] for animal experiments. Adult male Wistar–specific-pathogen-free rats (5-weeks old, 150–200 g, N = 30) were obtained from the Centre of Experimental Biological Models (CeMBE, PUCRS, Porto Alegre, RS, Brazil).
The experimental n was determined based on previous literature data [23,39,40]. The n per group is indicated in legends. The experiments were independently replicated two to four times. We performed an analysis to select the sample size (GraphPad StatMate 2.00; by GraphPad Software, Inc.), considering a significance level alpha 0.05 (two-tailed) and power value >80%, considering Randall-Selitto test and neurite outgrowth quantification as outcomes. The analysis revealed an experimental n of 6 per group. An experimental n of 8 per group was used while considering the risks of surgical procedures and histological processing steps.
The rats were kept in micro-isolator cages (4 per cage), equipped with inlet/outlet air filters, under controlled temperature (22°C ± 1°C) and humidity (50–70%), and a light–dark cycle of 12 h (lights on at 7 a.m., lights off at 7 p.m.). The cages were filled with autoclaved wood chip bedding. The animals received pelleted feed and sterile water ad libitum. During the experimental procedures, the laboratory temperature was maintained at 22°C ± 1°C. A period of adaptation to the new environment of at least 1 h was used. All the experiments were performed between 7 a.m. and 7 p.m.
The animals were randomized to have a similar mean body weight in the different experimental groups. The rats were treated and assessed in the behavioral tests in the following order: SHAM group, CCI group, and SCAP-treated group, one rat from each group per turn. The investigators were blinded to the experimental groups in either in vivo or ex vivo assessments. The animals were euthanized by isoflurane inhalation. The collection of sciatic nerve for histochemical studies was performed after the behavioral assessments, to reduce the total number of animals included in the study.
In vivo and ex vivo protocols
Isolation and cell culture
The SCAP isolation was performed as previously described [41]. Tooth apical papilla was mechanically removed and minced by using a scalpel. Fragments were digested by using a 0.2% type I collagenase solution (Gibco, Grand Island, NY) for 40 min at 37°C. After centrifugation at 500 g for 10 min, the cell pellets were resuspended and seeded onto a 12-well plate. The cells were cultivated in low-glucose Dulbecco's modified Eagle's medium (DMEM) containing 10 mM HEPES and supplemented with 10% fetal bovine serum (FBS; Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco). When confluence was reached, the cells were trypsinized by using a 0.25% trypsin-EDTA solution (Sigma-Aldrich, Inc., St. Louis, MO) and transferred into new tissue culture flasks. On the fifth passage, the cells were characterized. The experiments were performed with cells between the 4th and 7th passages.
SCAP characterization
The expression of surface markers for cell characterization was evaluated by flow cytometry (FACSCanto; BD Bioscience, San Jose, CA). The cells were trypsinized and resuspended in phosphate-buffer solution (PBS) at 106 cells/mL. A total of 100,000 cells was incubated with the following fluorochrome-conjugated antibodies: anti-CD14-FITC, anti-CD34-FITC, anti-CD45-APC, anti-CD73-PE, anti-CD90-FITC, anti-CD105-APC, anti-CD271-Alexa Fluor 647, and anti-HLA-DR-FITC (BD Biosciences) for 30 min, at 4°C. The potential of differentiation of the osteogenic lineages was assessed by cultivation of cells with specific inductors for 4 weeks, as previously described [1].
Polarization protocols of the SCAP
In the first protocol, poly(i:c), which is a single-strand RNA analogue, and bacterial lipopolysaccharide (LPS) were used. These agents are known agonists of Toll-like receptors 3 and 4, respectively. SCAP were treated during 1 h with the agonists. The concentrations used for poly(i:c) (1 and 5 μg/mL) and LPS (0.01 and 0.1 μg/mL) were based on previous publications that induced the differentiation of human bone-marrow mesenchymal stem cells (BMSCs) in anti-inflammatory and pro-inflammatory phenotypes, respectively [8,42]. In the second protocol, SCAP were stimulated with poly(i:c) (1 and 5 μg/mL), and the concentrations of LPS were increased (0.1 and 1 μg/mL), with incubation times of 24 and 48 h. In the third protocol, SCAP were plated at 4 × 104 cells/cm2 on tissue culture plates and the pro-inflammatory cytokine IFN-γ (100 and 200 ng/mL) was used as an alternative protocol to stimulate SCAP polarization, after incubation for 24, 48, and 72 h.
MTT assay
MTT assay was performed on cultured SCAP (60% confluence) in the presence of poly (i:c) (1 and 5 μg/mL) or LPS (0.01 and 0.1 μg/mL) for 1 h at 37°C (first tested protocol), in a humidified incubator with 5% CO2. After 1 h of treatment with agonists, cells were washed 2 × with PBS and the effect of the agonists on cell viability was analyzed by MTT assay after 48 h, according to the manufacturer's instructions. The SCAP proliferation in the presence of agonists was calculated in percentage, in relation to the negative control data.
Assessment of live and dead cells
The SCAP were grown to 60% confluence, harvested, plated on tissue culture plates, and incubated with 0.01 and 0.1 μg/mL LPS or 1 and 5 μg/mL poly(i:c), for 1 h (first tested protocol). The cells were washed 2 × with PBS, and the effect of the agonists on live and dead cells was carried out after 48 h by using fluorescein diacetate (FDA) and propidium iodide (PI), respectively. Briefly, the cells were incubated for 30 min with 8 μg/mL FDA and 5 μg/mL PI in DMEM. Then, the cells were analyzed by fluorescence microscopy (Olympus IX71 inverted microscope). Fluorescent images were taken to qualitatively assess the cell viability and adhesion.
Cell viability
The number of metabolically active cells was determined based on the reduction of WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H tetrazolium, monosodium salt] by cellular dehydrogenases, as an indicative of cell viability, which is proportional to the absorbance reading at 450 nm. Cells were plated at 40,000 cells per well of 24-well plates (third protocol) and incubated with 100 or 200 ng/mL of IFN-γ, for 24 or 48 h.
Lactate dehydrogenase activity
Lactate dehydrogenase (LDH) contents were measured in supernatants of control or treated SCAP, as a biochemical marker of cell death [43]. The cells were treated with LPS, poly(i:c), or IFN-γ, as described earlier. The LDH activity was measured by using a commercially available kit, according to the manufacturer's recommendations (Labtest, Lagoa Santa, Minas Gerais, Brazil). The absorbance was read at 340 nm in a spectrophotometer (SpectraMax M2/M2e Microplate Readers; Molecular Devices, San Jose, CA). The protein content was determined by the biuret method (total protein monoreagent kit, Bioclin), and the values of LDH were normalized by the protein content of the same sample.
Kynurenine levels and IDO activity
IDO is the enzyme responsible for the transformation of tryptophan into kynurenine. It is possible to determine the biological activity of the enzyme by measuring kynurenine levels in the cell supernatant [44] or in cell lysates [45]. The cells were lysed through freeze-thaw (5 × ) cycles in 100 μL of culture medium. Subsequently, the cells were centrifuged at 2,000 rpm, for 5 min, and 80 μL of the supernatant were transferred to a new tube. Eighty microliters of 2 × IDO buffer (100 mM PBS, 40 mM ascorbate, 20 μM methylene blue, 200 μg/mL catalase, 800 μM L-Tryptophan, pH 6.5) was added to the supernatant, and it was incubated for 30 min at 37°C. The supernatant was mixed with 50 μL of 30% trichloroacetic acid to stop the reaction, and a further incubation of 30 min at 52°C was performed. After centrifugation at 8,000 g for 5 min, the supernatant (or standard curve) was mixed with an equal amount of Ehrlich's reagent (100 mg of p-dimethylbenzaldehyde in 5 mL of glacial acetic acid) and added to a straight-bottom 96-well plate. The absorbance reading was performed at a wavelength of 492 nm on a plate reader (SpectraMax M2/M2e Microplate Readers; Molecular Devices). Serial dilutions of L-kynurenine were used as standards (0, 6.25, 12.5, 25, 50, 100, 200, and 400 μM).
mRNA expression of Toll-like receptors
The mRNA expression of Toll-like receptors 3 and 4 was assessed by reverse transcriptase-polymerase chain reaction (RT-PCR) in lysates of SCAP (105 cells/well). The total RNA was isolated with TRIzol® Reagent (Thermo Fisher Scientific, Waltham, MA) in accordance with the manufacturer's instructions. RNA purity (Abs 260/280 nm ∼2.0) and concentrations were determined by NanoDrop Lite (Thermo Fisher Scientific). The samples were treated with Deoxyribonuclease I – Amplification Grade (Sigma-Aldrich, Inc.) to eliminate genomic DNA contamination in accordance with the manufacturer's instructions. The cDNA was synthesized with ImProm-II™ Reverse Transcription System (Promega, Madison, WI) from 1 μg of the total RNA, following the manufacturer's instructions. PCRs were performed in a volume of 20 μL by using 0.2 μM of specific primers for TLR-3 and TLR-4 and 0.25 U of Platinun Taq DNA Polymerase (Invitrogen, Carlsbad, CA) in the supplied reaction buffer. The PCR cycling conditions were: an initial polymerase activation step for 5 min at 95°C, 40 cycles of 15 s at 95°C for denaturation, 35 s at 60°C for annealing, and 15 s at 72°C for elongation. ACTB was used as an internal control for PCR analysis. HaCat keratinocyte cell line was used as a positive control for expression of TLR-3 and TLR-4. Negative controls were performed by substituting the templates for DNAse/RNAse-free distilled water in each PCR. The amplified PCR products (5 μL) were detected by agarose gel (2.0%) electrophoresis using GelRed™ (Biotium) as staining and visualized under ultraviolet light. The sequences of reverse and forward primers are in Supplementary Table S1.
mRNA expression of neurotrophins
Molecular analysis of BDNF, glial cell-derived neurotrophic factor (GDNF), nerve growth factor (NGF), and neurotrophin 3 (NTF3) gene expression in lysate of naive or IFN-γ (100 ng/mL)-primed SCAP (5 × 105 cells/well) was performed according to the Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE) Guidelines for RT-qPCR experiments. [46,47]. The total RNA was isolated with TRIzol® Reagent (Thermo Fisher Scientific) in accordance with the manufacturer's instructions. RNA purity (Abs 260/280 nm ∼2.0) and concentration were determined by NanoDrop Lite (Thermo Fisher Scientific) and after treatment with Deoxyribonuclease I – Amplification Grade (Sigma-Aldrich, Inc.) to eliminate genomic DNA contamination in accordance with the manufacturer's instructions. The cDNA was synthesized with ImProm-II™ Reverse Transcription System (Promega) from 1 μg of the total RNA, following the manufacturer's instructions. Quantitative PCR was performed by using SYBR® Green I (Thermo Fisher Scientific) to detect double-strand cDNA synthesis on the 7500 Real-time PCR System (Applied Biosystems, Foster City, CA).
The PCR cycling conditions were: an initial polymerase activation step for 5 min at 95°C, 40 cycles of 15 s at 95°C for denaturation, 35 s at 60°C for annealing, and 15 s at 72°C for elongation. At the end of the cycling protocol, a melting-curve analysis was included and fluorescence was measured from 60°C to 99°C to confirm the specificity of primers and absence of primer dimers; in all of the cases, one single peak was detected. All real-time assays were carried out in quadruplicate, and a reverse transcriptase negative control was included by substituting the templates for DNase/RNase-free distilled water in each PCR. GAPDH was used as a reference gene for normalization. The efficiency per sample was calculated by using LinRegPCR 2018.0 Software (
Isolation of dorsal root ganglion cells
This protocol was accomplished as described earlier [49], with minor modifications. Dorsal root ganglion (DRG) cells were obtained from six male Wistar rats (4–6-week-old, 180–200 g) after euthanasia by an overdose of inhaled isoflurane. The ganglia were manually isolated and transferred to Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. At least 16 ganglia were isolated per rat. Ganglia were dissociated with a scalpel and digested with 2 mg/mL collagenase type 1 (Sigma-Aldrich, Inc.) for 45 min at 37°C, followed by centrifugation at 900 rpm, for 10 min. An additional digestion step was carried out, by adding a 0.1% trypsin-EDTA solution for 20 min at 37°C. After centrifugation at 900 rpm for 10 min, the cells were transferred to a 15-mL conical tube with 5 mL of DMEM medium without fetal bovine serum (FBS). Additional dissociation was performed mechanically by using a sterile plastic Pasteur pipette. The cell suspension was placed over 3 mL of 15% BSA solution (dripping down the wall with a Pasteur pipette), without mixing the phases, and centrifuged at 200 g for 10 min. The pellet containing mainly neurons was resuspended in Neurobasal medium and centrifuged at 900 rpm for 10 min. The pellets were resuspended in Neurobasal medium supplemented with 2% B27 and containing 100 U/mL penicillin and 100 μg/mL streptomycin. The neurons were grown on SCAP.
Co-culture of neurons and SCAP
Neurons were plated on Geltrex™–treated 24-well plates containing naive or IFN-γ (100 ng/mL)-primed SCAP. NGF-7S (100 ng/mL) was added to the culture medium as a positive control for axonal ramification. Co-cultures were maintained for 1 or 3 days, with medium change 24 h after treatment with IFN-γ. Supernatants were collected for ELISA assay, and cells were fixed for immunofluorescence.
BDNF and cytokine levels
Supernatants of control or treated SCAP were used to determine the levels of IL-6, CCL3 [Chemokine (C-C motif) ligand 3] or MIP-1α, CCL4 [Chemokine (C-C motif) ligand 4] or MIP-1β and CCL5 [Chemokine (C-C motif) ligand 5] or RANTES. The chemokines CCL3, CCL4, and CCL5 control macrophage and NK cell migration and participate in interactions between T cell and dendritic cells, recruiting and guiding innate immune effectors to tissue injury sites [50]. The cells were treated with LPS, poly(i:c), or IFN-γ, as described earlier. The supernatants of neurons exposed to naive or IFNγ-primed SCAP were used to determine the levels of BDNF. For this purpose, ELISA (sandwich enzyme-linked immunosorbent assays) kits were used according to the manufacturer's recommendations (Thermo Fisher Scientific). The results were expressed in pg/mL.
Neurite quantification
The DRG cultures were fixed in 10% formaldehyde (PFA) for 30 min. The cells were washed three times with PBS, after fixation. Next, the cells were blocked and permeabilized with a PBS solution containing 1% BSA and 0.1% Triton X-100, for 30 min at room temperature. The cells were then incubated with a mouse anti-B-III-tubulin antibody for 2 h, at 37°C (dilution 1:500; Sigma-Aldrich, Inc.). The corresponding Alexa 488-conjugated secondary antibody (dilution 1:1000; Invitrogen) was added to visualize the cells. Images were acquired in an inverted microscope (Olympus IX71 inverted microscope) with 10 × objective lens. The analysis of neurite growth was performed by using the NeurphologyJ Plugin [51] of NIH ImageJ 1.36b Software. The neurite length, neurite outgrowth (neurite area/soma), total soma area, and soma counts were analyzed according to the software instructions. Images were examined with a Zeiss AxioImager M2 light microscope (Carl Zeiss, Gottingen, Germany). For this purpose, the images were captured in × 100.
In vivo assessments
CCI model in rats and treatment with SCAP
CCI was performed according to Austin et al. [52], with minor modifications. Briefly, rats were anesthetized with an intraperitoneal administration of ketamine plus xylazine (100 and 10 mg/kg, respectively). The intraperitoneal injection of ketamine plus xylazine is a common anesthesia technique used for experimental procedures performed in the rats, producing good analgesia, dissociation, and muscle-relaxing effects [53,54]. The surgery site was shaved and disinfected with 2-% chlorhexidine gluconate. The sciatic nerve of the right hindlimb was exposed at the mid-thigh level, below the femur, and four loose ligatures using 4-0 nylon suture were placed around the exposed sciatic nerve, with about 2 mm between ligatures [52]. Sham-operated animals underwent the same procedures but without placement of ligatures to the sciatic nerve. The animals were monitored daily after surgery.
The CCI animals received SCAP topically (106 cells reconstituted in 200 μL of PBS; treated group), or 200 μL of PBS (control group) at the surgery site, around the sciatic nerve on day 0. The dose and route of administration of SCAP were determined based on prior publications [22 –25]. Mechanical hyperalgesia, radiant heat hyperalgesia, and cold allodynia were assessed before (day -3; basal) and at days 3, 7, 14, and 21 after surgery.
Behavioral tests
Mechanical hyperalgesia
The mechanical hyperalgesia was analyzed by using a Randall–Selitto apparatus (Ugo Basile, Comerio, Italy), according to the method described by Farghaly et al. [55]. The animals were gently restrained and nociceptive threshold, expressed in grams, was measured after the application of an incremental pressure onto the dorsal surface of the right hind paw, by means of an automated gauge. The cut-off pressure of 200 g was adopted to avoid tissue damage.
Heat hyperalgesia
A plantar test apparatus (Ugo Basile) was used to determine the thermal hyperalgesia, as described by Rodriguez-Gaztelumendi [39], with minor modifications. The rats were individually placed in clear acrylic cubicles positioned on a stand with Plexiglas surface. The intensity of the radiant heat stimulus was maintained at 25°C ± 0.1°C and applied to the plantar surface, with a cut-off time of 20 s to prevent any tissue damage. The average of three measurements was calculated for each paw at 2-min intervals. The percentage of relative difference in paw withdrawal latency (ΔPWL) between the right (CCI operated) and the left hind paws was calculated as: ΔPWL = (right - left)/left × 100.
Cold allodynia
Cold allodynia was assessed by using the acetone droplet method, as described by Tegeder et al. [56]. The rats were placed on the top of a wire mesh grid, allowing access to the hind paws. Acetone (100 μL) was applied onto the plantar surface of the right hind paw. Cold sensitive reaction with respect to either paw lifting, licking, shaking, or rubbing was registered for 2 min after acetone application.
Grimace scale
The Grimace Scale was scored as described by Sotocinal et al. [57]. Briefly, orbital tightening, nose/cheek flattening, ear changes, and whisker changes were assigned a score of 0, 1, or 2, based on degree of presentation. The whisker changes were scored in real time, whereas the other parameters were recorded for posterior video analysis. Observations began 30 min after placing the rat in the observation chamber and facial expressions were analyzed every 3 min, with a total of 10 scores, for each time point, over a 30-min period. The Grimace Scale was scored at 14 and 21 days after CCI.
Histochemical analysis
The sciatic nerves were excised (21st day after CCI) and immediately fixed in 10% formalin solution. Paraffin sections were stained by hematoxylin–eosin (H&E), or Luxol Fast Blue (LFB) as described by Silva et al. [58]. In addition, the immunopositivity for BDNF (1:100; Santa Cruz Biotechnology, Dallas, TX; Catalog Number sc-546) was also assessed in sciatic nerve sections. Images were examined with a Zeiss AxioImager M2 light microscope (Carl Zeiss). The images were captured in × 400 magnification. H&E-stained slides were used for qualitative assessment of nerve fiber vacuolization and cellularity.
LFB staining was quantitatively assessed as an indicative of myelin damage. The ratio of color intensity/area on each section was calculated by using the NIH Image J 1.36b software. A specific macro was created to quantify the blue color intensity in sciatic nerve sections. For this purpose, an image from the SHAM group, without CCI induction, was chosen, and this macro was applied for all images from the different experimental groups. To present the data acquired from demyelinating areas in percentage, the size of lesion regions was divided by the total area of the section × 100. BDNF immunopositivity was quantified manually by counting the number of cells positive for BDNF brown staining through the entire slide of the sciatic nerve. Mouse skin was used as a positive control for BDNF immunopositivity (data not shown).
Statistical analysis
The results were expressed as the mean ± standard error of the mean (SEM). Brown Forsythe and Bartlett tests were used for checking normality of data. Kruskal– Wallis (non-parametric data) and one-way ANOVA (parametric data) were used for comparison of the SHAM-operated group versus CCI control group versus CCI-treated group. Two-way ANOVA was used to compare data at the baseline and on 3, 7, 14, and 21 days. When the interaction of factors was significant (P values <0.05), pairwise comparisons were conducted by using Dunnett's or Tukey's post hoc tests, after Kruskal–Wallis and ANOVA, respectively. To minimize the problem of multiple comparisons, we calculated the area under the curve (AUC) for each individual animal and each performed the behavioral test. All tests were carried out by using GraphPad Software® version 5.0 (GraphPad Software, Inc., San Diego, CA).
Results
Characterization and differentiation of SCAP
The SCAP used in this study were obtained from nine patients. The cells were expanded in vitro until the 5th passage; they were then frozen, differentiated, and characterized. The cells were able to differentiate into osteoblasts by culturing them with specific inducers for 4 weeks. The cells were stained with alizarin red, which positively marked calcium-enriched extracellular matrix (Supplementary Fig. S1A–C). In addition, the analyzed cell lines were positive for CD73, CD90, and CD105; whereas they did not express CD14, CD34, CD45, CD271, and HLA-DR (Supplementary Fig. S1D–K).
Polarization of SCAP with TLR3 and TLR4 receptor agonists
The protocol 1 with the exposure to poly(i:c) (1 and 5 μg/mL) or LPS (0.01 and 0.1 μg/mL) for 1 h did not induce the expression of IL-6 (Fig. 1A) or CCL5 (Fig. 1B) by SCAP. To confirm that treatment with agonists did not interfere with the cell viability, LDH activity was measured in the cell supernatant (Fig. 1C), indicating the absence of cell death in this protocol. These results were confirmed by live and dead (Supplementary Fig. S2A–J) and MTT (Supplementary Fig. S2K) techniques.

Cytokine and chemokine levels (IL-6, CCL5) and LDH activity in the supernatant of naive and primed SCAP, treated with polarization protocol 1
In protocol 2, the cell response was quite variable among the different treatments, with a trend to increased IL-6 levels in cells polarized with LPS (0.1 and 1 μg/mL) and poly(i: c) (1 and 5 μg/mL) (Fig. 1D). The presence of CCL5 in the supernatant was observed only for the highest concentration of poly(i:c) (5 μg/mL), after 24 h of incubation (Fig. 1E). Regarding the LDH activity, no differences were observed among the groups (Fig. 1F), demonstrating that agonists did not induce cell death. The positive control for cell death 0.2% Triton X-100 led to a marked increase of LDH activity (Fig. 1C, F).
To better understand the lack of response to LPS and poly(i: c), an end-point PCR assay was performed to evaluate the mRNA expression of TLR-3 and TLR-4 in the different SCAP cell lines. It was not possible to obtain the amplification for TLR3 and TLR4 genes, suggesting the absence of their expression in these cells, whereas the endogenous control (β-actin) was amplified. Alternatively, the human keratinocyte cell line HaCat showed amplification products for both TLR3 and TLR4 (Fig. 1G).
Polarization of SCAP with IFN-γ
As an alternative to the inefficiency of SCAP polarization by TLR3 and TLR4 agonists, a third protocol was performed to determine the ability of IFN-γ to stimulate IDO activation. Tests carried out after 24, 48, and 72 h demonstrated that cells stimulated with IFN-γ (100 and 200 ng/mL), or IFN-γ plus poly(i:c) (10 and 50 μg/mL) led to a similar increase of kynurenine levels in the supernatant of SCAP. In contrast, LPS (0.1 and 1 μg/mL) failed to induce the conversion of tryptophan to kynurenine (Fig. 2A–C). The incubation of poly(i: c) (10 and 50 μg/mL) alone induced the production of kynurenine, although at significantly lower levels (Fig. 2A–C), in comparison with IFN-γ, at the three tested experimental periods. Of note, the stimulation of SCAP with IFN-γ (100 ng/mL) resulted in a significant increase of IDO activity after 24 h, an effect that persisted for up to 72 h, even after the stimulus withdrawal (Fig. 2D). The incubation with IFN-γ (100 and 200 ng/mL), for 24 h or 48 h, did not affect the cell proliferation or viability, as indicated by Wst-8 (Fig. 2E) and LDH activity (Fig. 2F) assays.
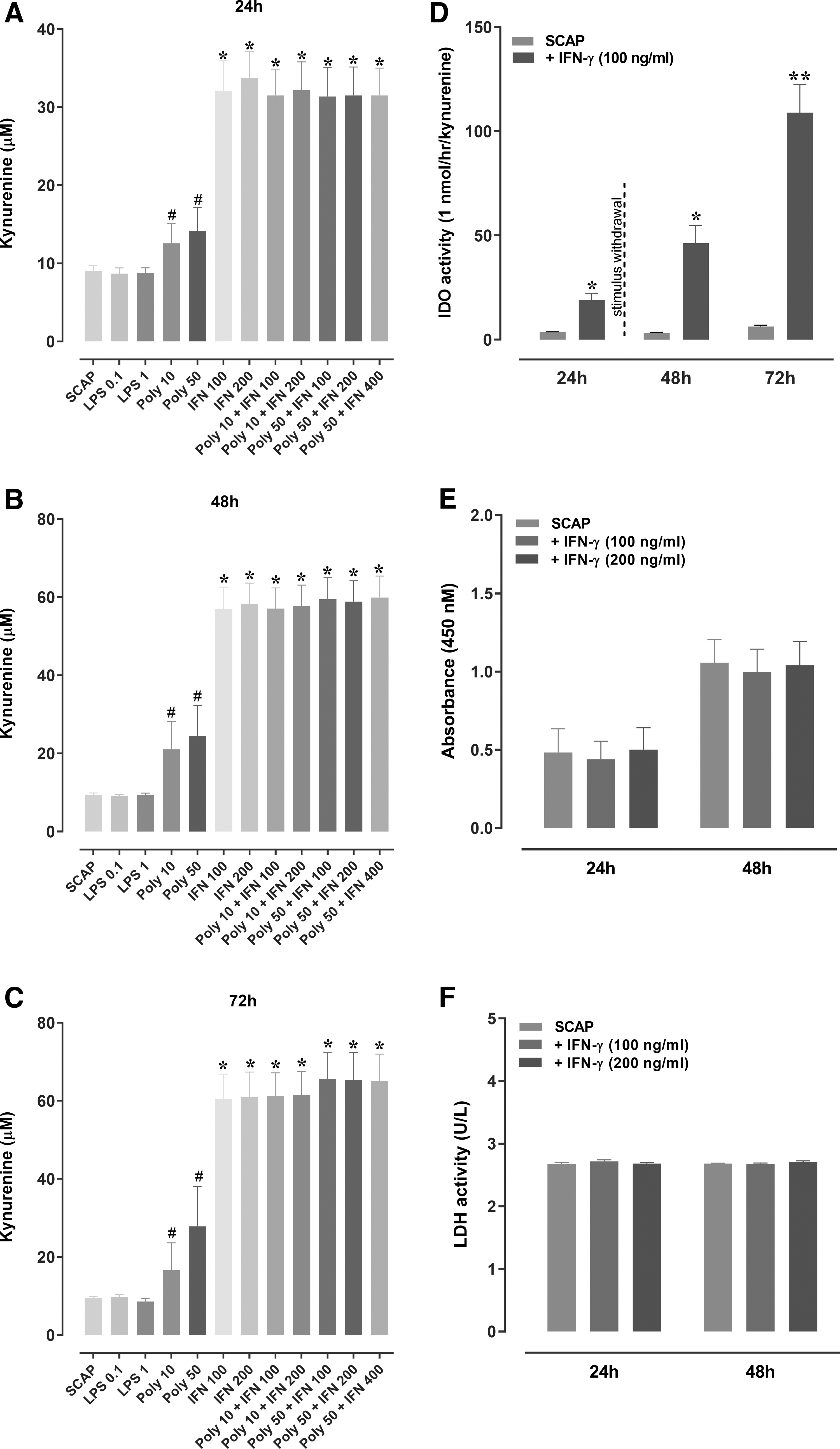
Kynurenine levels in the supernatant of SCAP stimulated with different inducers
Effects of IFN-γ on SCAP inflammatory profile
The levels of chemokines and cytokines were measured in the SCAP supernatant, after priming with IFN-γ (100 and 200 ng/mL). The exposure to IFN-γ led to a marked enhancement of IL-6 levels (with a five-fold increase) (Fig. 3A), despite the absence of detectable levels of the chemokines CCL3, CCL4, and CCL5 (Fig. 3B–D).

Cytokine and chemokine levels (IL-6, CCL3, CCL4, and CCL5) in the supernatant of naive and primed SCAP, treated with IFN-γ. ELISA assay for
BDNF, GDNF, NT3, and NGF mRNA expression
Considering the similar effects of IFN-γ at 100 and 200 ng/mL, the lower concentration of this cytokine was used for the next protocols. IFN-γ treatment (100 ng/mL) led to a decrease of BDNF mRNA expression at 24 h (Fig. 4A), whereas GDNF mRNA expression was increased at 72 h (Fig. 4C), according to the evaluation of SCAP lysates. The mRNA expression of NT3 (Fig. 4B) or NGF (Fig. 4D) was not significantly affected at any evaluated time-point.

mRNA expression of neurotrophic factors in lysates of SCAP.
SCAP effects on DRG neurons
Subsequently, functional immunofluorescence studies were performed to demonstrate the ability of SCAP to induce the growth of neurites in a co-culture system with rat DRG neurons (Fig. 5A–F and Supplementary Fig. S3). The treatment with naive SCAP significantly increased the length and growth of neurites at 24 and 48 h. However, a tendency to neurite growth decrease was observed for IFN-γ-primed SCAP, at 24 h (Fig. 5A, D and Supplementary Fig. S3). Regarding the soma counts and area, the co-culture with SCAP led to a significant reduction of total soma areas, regardless of IFN-γ priming, at 48 h (Fig. 5K). The other tested co-culture conditions did not elicit any significant alteration of soma features (Fig. 5G–J, L). Of note, co-culture of rat DRG neurons with IFN-γ-treated SCAP triggered a significant increase of BDNF levels, as assessed at 24 and 48 h (Fig. 6), when compared with the SCAP-free control group.
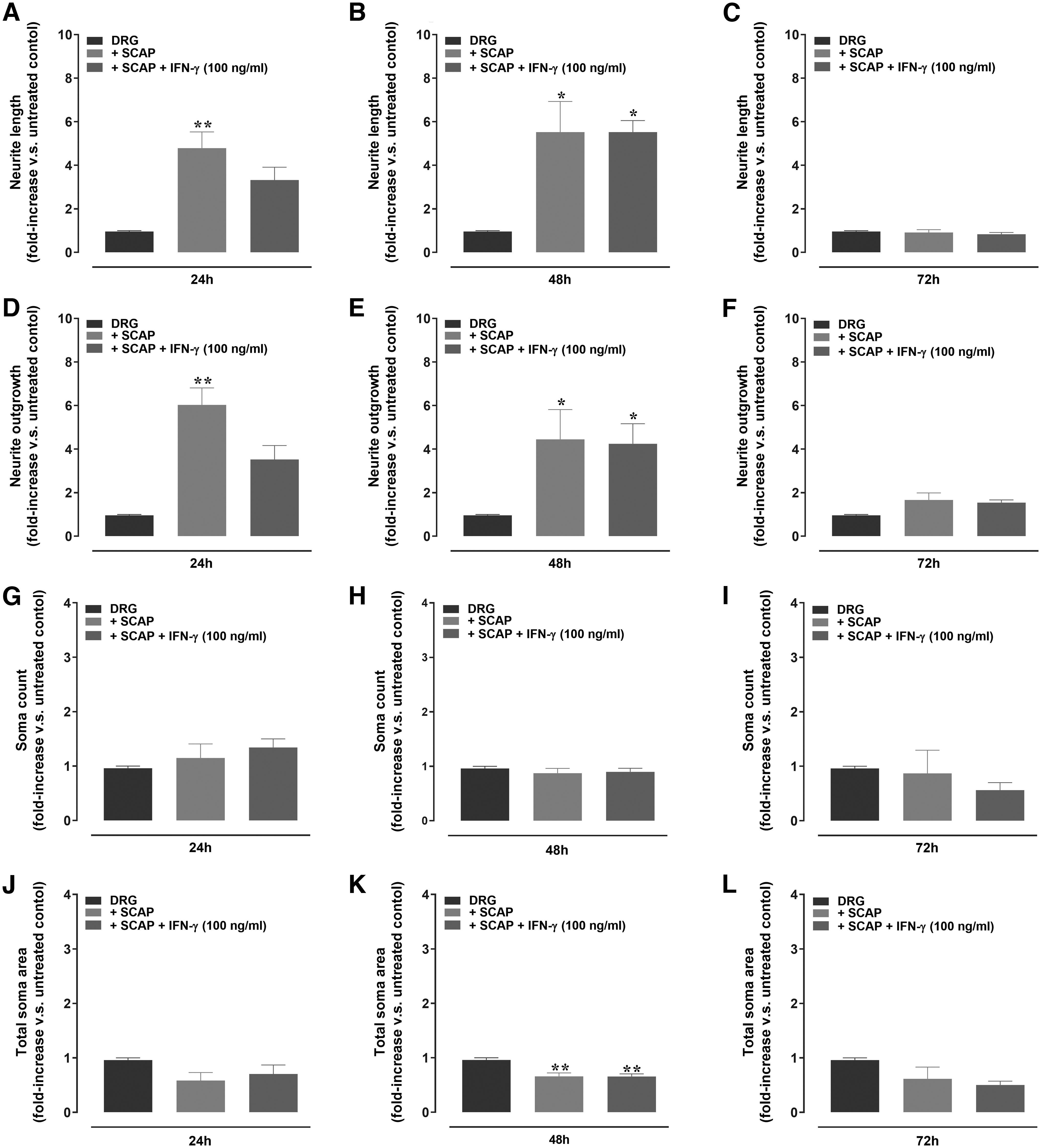
Quantitative analysis of neurite outgrowth, the total number and area of soma in DRG neurons, cultured with SCAP, with or without IFN-γ (100 ng/mL).
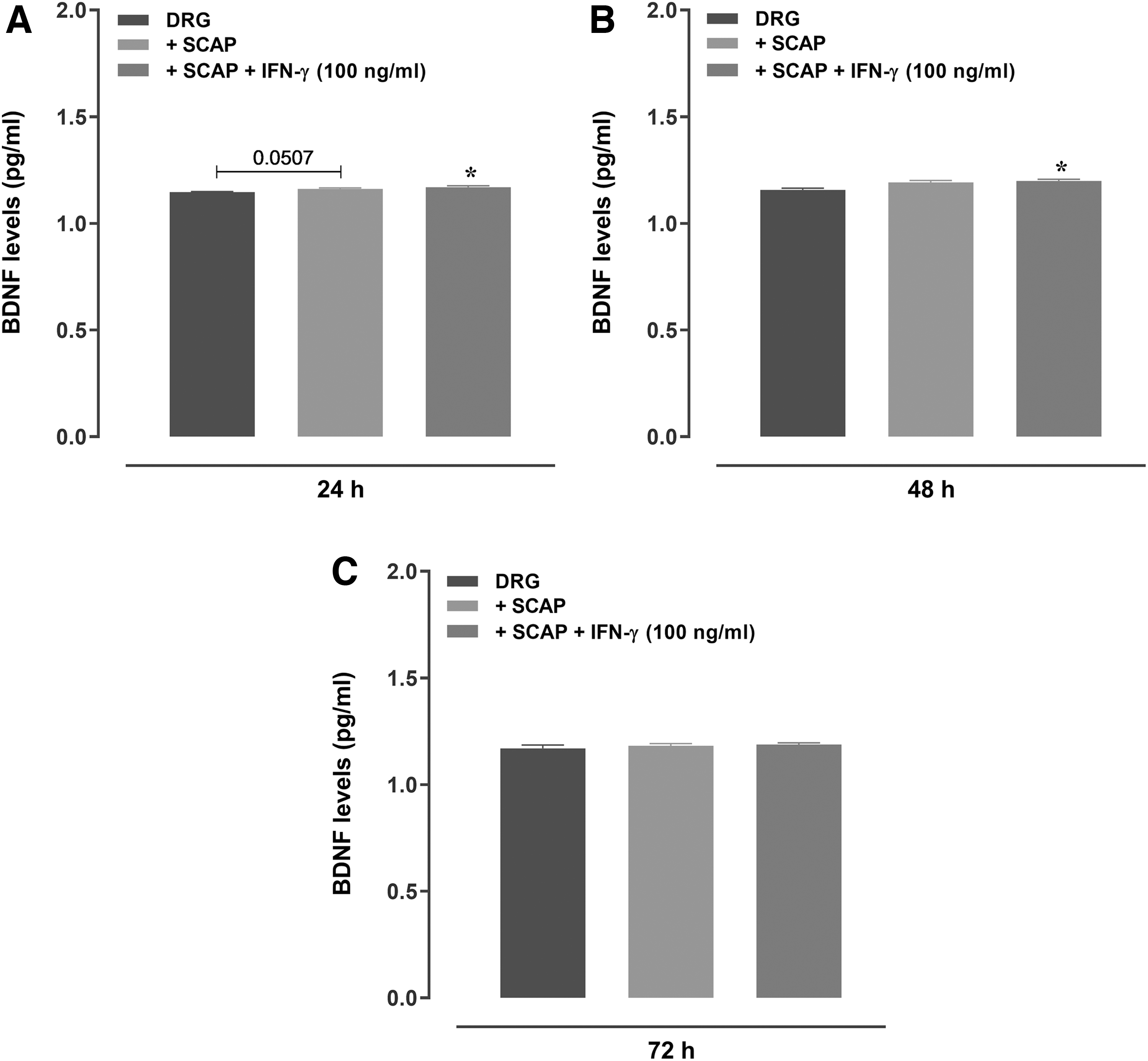
Analysis of BDNF levels in the supernatant of DRG neurons cultured with SCAP, with or without IFN-γ (100 ng/mL). BDNF levels after
SCAP treatment in CCI model in rats
This part of the study evaluated the in vivo effects of treatment with SCAP (acute dose of 106, applied locally) on nociceptive changes in rats submitted to the CCI model (Fig. 7A–H). The local treatment with SCAP significantly reduced the mechanical hyperalgesia, according to assessment at the 21st day (Fig. 7A). An analysis of the AUC revealed a trend toward recovery in SCAP-treated CCI rats (Fig. 7B). The local application of SCAP attenuated the thermal hyperalgesia along the different evaluated time-points (Fig. 7C). The AUC for thermal hyperalgesia in the SCAP-treated group was significantly lower, when compared with the CCI control group (Fig. 7D). A partial, but not significant reduction of cold allodynia was observed in SCAP-treated animals when compared with the CCI group, toward the values observed in the SHAM-group value (Fig. 7E, F).
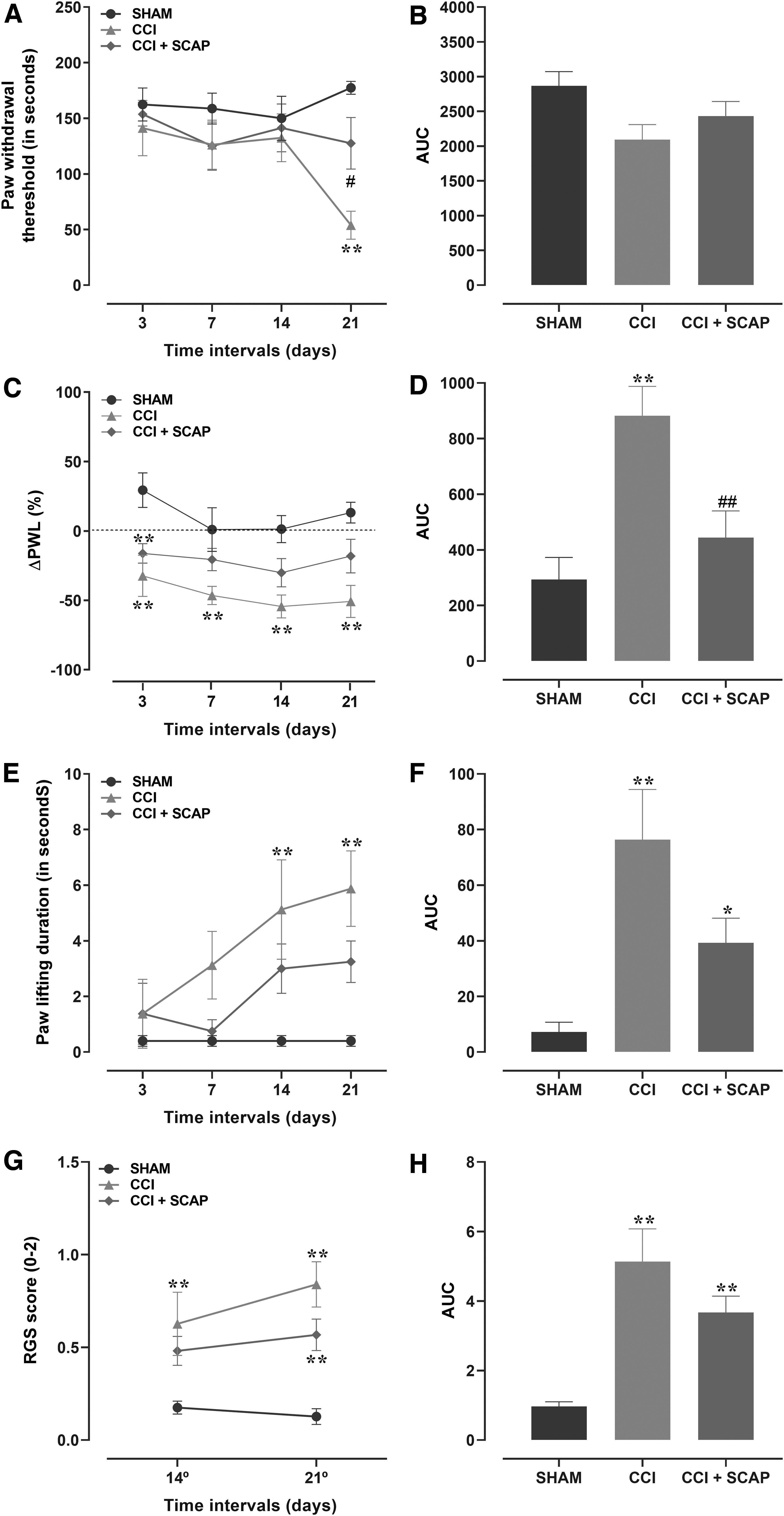
Effects of local treatment with SCAP (106) on nociceptive changes in rats submitted to the CCI model. The effects of SCAP were evaluated on
The pain scores were assessed by using the Grimace scale, on days 14 and 21 after CCI surgery. The scores were significantly increased in the CCI group at 14 and 21 days, when compared with the SHAM group (from 0.17 ± 0.03 to 0.63 ± 0.17 and from 0.13 ± 0.04 to 0.84 ± 0.12), respectively. The SCAP-treated group displayed inferior scores when compared with the CCI group (from 0.63 ± 0.17 to 0.48 ± 0.57 at 14 day; from 0.84 ± 0.12 to 0.57 ± 0.08 at 21 day), although the differences were not statistically significant (Fig. 7G–H). There were no significant differences among the groups regarding the body weight gain (Supplementary Fig. S4).
SCAP attenuated the pathological changes in the CCI model
A qualitative analysis of H&E-stained nerve sections was carried out. In the SHAM group, the myelinated nerve fibers were dense, round, and uniform (Fig. 8A). In the CCI group, the myelinated nerve fibers exhibited vacuolar-like defects, with the presence of inflammatory cells (Fig. 8B). The vacuolar degeneration of nerve fibers was partially restored in the SCAP-treated group, when compared with the CCI group (Fig. 8C).

Effects of treatment with SCAP (106) on histochemical aspects of injured nerves. Hematoxylin-eosin-and LFB staining was used to evaluate myelination and immunohistochemistry to determine the number of BDNF-positive cells on sections of excised sciatic nerves on postoperative 21st day in the SHAM
Regarding Luxol-stained sections, the CCI group presented numerous lipid vacuoles with different myelin sheath shapes, when compared with the SHAM group (Fig. 8D, E). The SCAP-treated group exhibited partial myelin lysis and uniform myelin sheath shapes, when compared with the CCI group (Fig. 8E, F). The blue color intensity was found to be lower in the CCI group than in the SHAM group, demonstrating higher demyelination (Fig. 8J). In this regard, the SCAP-treated group showed a significantly higher color density than the CCI group, showing partial nerve sciatic myelination recovery (Fig. 8J). The numbers of BDNF-positive cells in the sciatic nerve were significantly higher in the CCI group, in comparison with the SHAM group. We also detected an increase of inflammatory infiltrate in CCI and SCAP-treated groups when compared with the SHAM group. There was no statistically significant difference in BDNF immunopositivity between the CCI and the SCAP-treated groups (Fig. 8G–I, K). However, it was possible to observe an increase of Schwann cells positive for BDNF, with sprouting axons induced by SCAP treatment (red arrows).
Discussion
This study provides novel evidence on the characterization of SCAP, investigating their potential on nerve regeneration by using in vitro, ex vivo, and in vivo strategies. The lineages of SCAP analyzed in this study expressed the membrane markers CD73, CD90, and CD105 and they did not express the proteins CD14, CD34, CD45, CD271, and HLA-DR. In an attempt to further characterize the MSC population and purity after the in vitro expansion of primary cultures for 5 passages, a panel of recommended immunophenotyping antibodies was used.
The use of negative markers was used to distinguish MSCs from hematopoietic cells, such as CD14 (monocytes and macrophages), CD34 (primitive hematopoietic cells and endothelial cells), CD45 (leucocytes), and HLA-DR (antigen-presenting cells and lymphocytes). In addition, we used the positive MSC markers CD73, CD90, and CD105 to verify mesenchymal identity. The investigation of the CD271 expression was performed, because it is a marker of neural crest stem cells that can differentiate into peripheral neurons. This pattern of surface markers is in accordance with the criteria established for MSCs by the International Society for Cell Therapy [59].
As a first step, we evaluated the capacity of SCAP polarization by TLR-4 and TLR-3 agonists, namely LPS and poly(i:c), respectively. The protocol 1 used in this study failed to polarize SCAP, contrasting somewhat with that observed for BMSC [8,42]. It was concluded that SCAP do not respond in the same way as BMSC, with 1 h of stimulation, according to the literature protocol. The evaluation of long-term exposure to both agonists showed that only the higher concentration of poly(i:c) (5 μg/mL) led to a significant increase of CCL5 production, with a trend to enhanced IL-6 levels for both agonists. Distinct results were found in a previous study, showing that treatment with poly(i:c) (1 μg/mL) and LPS (10 ng/mL) for 1 h led to an increase of CCL5 and IL-6 contents, respectively, as assessed in the conditioned medium of human multipotent mesenchymal stromal cells (hMSCs) [8]. The absence of TLR-3 and TLR-4 mRNA expression might explain the inability of LPS and poly(i:c) to induce SCAP polarization in our study, which also contrasts with previous data for BMSC. Indeed, it was found that BMSC can be polarized by priming with TLR4 and TLR3 agonists into two phenotypes, with increased levels of pro-inflammatory mediators and immunosuppressive molecules, respectively [8].
Different strategies have been tested to increase the therapeutic capacity of MSCs. Thus, another approach was carried out to induce an anti-inflammatory phenotype in SCAP, with the incubation of IFN-γ. IFN-γ triggered a marked increase of IDO levels in the supernatant of SCAP. It was previously demonstrated that IFN-γ priming (200 IU/mL) induced IDO expression in MSCs obtained from umbilical cord blood, adipose tissue, Wharton's–Jelly (WJ-MSCs), and bone marrow [10], corroborating our data. Wada et al. [60] investigated the effects of IFN-γ (25 ng/mL) on different types of MSCs, showing that IDO mRNA expression was upregulated after stimulation with IFN-γ, for 48 h. The authors also demonstrated that IFN-γ did not induce any deleterious effects on MSCs, being considered a safe protocol for the infusion of non-activated cells. In another study, hMSCs exposed to IFN-γ showed a viability of ≥90% [61]. Herein, we demonstrated that IFN-γ stimulation (either at 100 or 200 ng/mL) did not affect the cell proliferation or viability, upholding its low toxicity on SCAP.
The exposure to IFNγ led to a marked increase of IL-6 levels, without any effect on the production of the chemokines CCL3, CCL4, or CCL5, according to the evaluation in the supernatant of SCAP. An increased expression of IL-6 in the presence of IFN-γ has been demonstrated for other cell types, such as monocytes [62], somewhat supporting our data. We also tested whether SCAP priming by IFN-γ might modulate the production of neurotrophic factors. First, we demonstrated that mRNA expression of both GDNF and BDNF was increased in SCAP lysates. Similar data were demonstrated by Kolar et al. in a study also evaluating SCAP [31]. Herein, the treatment with IFN-γ was associated with a decrease of BDNF mRNA expression at 24 h, whereas GDNF mRNA expression was increased at 72 h. Koh et al. demonstrated that MSCs derived from the human umbilical cord secrete higher levels of vascular endothelial growth factor (VEGF), GDNF, and BDNF in comparison with BMSCs. However, both cell types showed measurable amounts of secreted neurotrophic factors [63]. The production of BDNF and GDNF was also increased in human DPSC co-cultivated with Schwann cells [64]. In another study, DPSC secreted neurotrophic factors related to the subsequent differentiation of neural stem cells [65]. In our study, the exposure to SCAP increased the neurite outgrowth and the length of DRGs, which paralleled with enhanced levels of BDNF in the supernatant of these co-cultures. Similarly, the exposure of differentiated human neuroblastoma SH-SY5Y cells to conditioned medium of SCAP led to an increase of the percentage of cells producing neurites and the total neurite outgrowth length, an effect that was dependent on BDNF secretion [31].
To extend in vitro and ex vivo evidence, we tested the effects of SCAP in a rat model of CCI. The CCI model is reliable and generates symptoms of causalgia or complex regional pain syndrome in patients [66]. This experimental set showed that local administration of SCAP reduced both mechanical and thermal hyperalgesia, in this classical model of neuropathic pain. Corroborating our data, the local administration of ADMSC significantly reduced the mechanical allodynia and reversed the thermal hyperalgesia in the CCI model in rats [24]. In addition, our results demonstrated that SCAP treatment partially ameliorated the pain scores in the CCI model by using the Grimace scale. In agreement with our results, Kim et al. (2020) showed that local treatment with ADMSC improved Grimace scores in a rat model of interstitial cystitis [67].
The regenerative potential of SCAP was demonstrated in vivo, with a higher distance of regeneration in animals exposed to nerve surgery and treated with conduits seeded with SCAP [31]. In addition, the local application of BMSC improved sciatic nerve regeneration, with higher mean axon counts, a trend toward significantly higher axon density, and an increase in the diameter of the axon and fiber, in comparison with the control group [25]. Interestingly, we demonstrated that local treatment with SCAP partially diminished the nerve degeneration, with fewer cells displaying extensively vacuolated cytoplasm. We found that BDNF mRNA and GDNF mRNA expression was increased in the supernatant of culture and lysate of SCAP, respectively, supporting the improvement of nerve regeneration in rats treated with SCAP in the CCI model. A previous publication demonstrated that conduits filled with WJ-MSCs enhanced nerve regeneration, likely by modulation of neurotrophins and neurite guidance proteins, in the nerve of rats submitted to sciatic nerve injury [68]. In addition, an increased expression of NGF and BDNF was observed in injured nerves, after local treatment with BMSCs in a sciatic nerve crush injury model [69].
Regarding the possible mechanisms underlying the analgesic effects of SCAP in the CCI model, it is tempting to propose the involvement of paracrine signaling through BDNF modulation. The cell-to-cell contact mediated by the release of neurotrophic factors might favor the recovering effects of SCAP on demyelization induced by CCI in this study. Naturally, this study has limitations and further studies with alternative models as pathways for cell treatment are still required.
The role of BDNF in pain control is controversial and complex [70,71]. In our study, both CCI groups, irrespective of treatment with SCAP, presented higher numbers of BDNF-positive cells, according to the evaluation of sciatic nerve sections. According to Xu and cols. (2013), an intrathecal transplantation of neural stem cells alleviated the neuropathic pain by modulation of GDNF release in the spinal dorsal horn and DRG, without any changes of BDNF expression [72], somewhat supporting our data. Of interest, despite us not detecting any significant differences regarding the number of cells presenting immunopositivity for BDNF, when comparing the CCI and the SCAP-treated groups, the topical application of SCAP was associated with an increased number of Schwann cells with a regenerative phenotype, as indicated by a qualitative analysis.
Indeed, we demonstrated that BDNF was released in SCAP supernatants, suggesting that mesenchymal cells can modulate the interface between Schwann cells and axons, likely stimulating a proregenerative profile, promoting axonal growth, and ameliorating persistent pain, indirectly. This repair profile featured by Büngner band appearance was observed after the local treatment with adipose tissue-derived stem cells in the crushed sciatic nerve model [73]. After the peripheral nerve injury, the mature Schwann cells dedifferentiate to a stem cell-like state, proliferating and migrating to form the Büngner bands [74].
The therapeutic use of stem cells to stimulate the differentiation of Schwann cells in the sciatic nerve transection model has been previously demonstrated [75]. A recent study showed that treatment with human adipose-derived stem cell exosomes accelerated sciatic nerve regeneration, optimizing Schwann cell functioning. In this case, the exosomes induced migration, proliferation, and myelination of Schwann cells via internalization processes [76]. The SCAPs might also boost a massive infiltration of inflammatory cells (macrophages), besides the phagocytosis of Schwann cells to remove degenerated myelinic induced by the CCI model [77].
Conclusion
Collectively, this study shed new light on the regenerative potential of SCAP, showing their ability to promote neurite outgrowth and to increase the length of DRGs ex vivo, likely via BDNF and GDNF modulation. As for the in vivo protocols, the local application of SCAP alleviated the pain symptoms in the rat model of CCI, leading to a recovery of nerve vacuolization and myelinization levels in this experimental paradigm, with an increased number of regenerative BDNF-positive Schwann cells.
Footnotes
Acknowledgments
The authors thank Mrs. Janaína Pasetti Nunes for her valuable technical assistance in histological processing and Dr. Daniel Rodrigo Marinowic for sharing anti-BDNF, which was essential for our work. The authors are grateful to the staff of the Centre of Experimental Biological Models (CeMBE/PUCRS) for animal care.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES) Finance Code 001, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), and Pontifícia Universidade Católica do Rio Grande do Sul (PUCRS), in addition to a Financiadora de Estudos e Projetos (FINEP) research grant “Expansão da Infraestrutura Multiusuária de Pesquisa na PUCRS (MULTIPUCRS)” # 01.13.0292-00 (Brazil). A.P.A.D. and L.W.K. are recipients of the PNPD/CAPES fellowship. M.M.C. (CNPq, 304042/2018-8) and M.R.B. (CNPq, 303776/2013-7) are Research Career Awardees of the National Research Council of Brazil (CNPq). J.B.S. received a grant from Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul (FAPERGS, 17/2551-0001031-0).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.