
Abstract
Cancer metastasis is the leading cause of mortality among breast cancer patients. Type 2 diabetes mellitus (T2DM) has been suggested as a risk factor of breast cancer; however, whether or not T2DM is associated with breast tumor metastasis remains unclear. In this study, we examined the involvement of T2DM with breast cancer metastasis by a combined approach of a meta-analysis and experimental research. The results of a systematic review and meta-analysis suggested that diabetes significantly increases the risk of lymph node metastasis by 1.10-fold (P < 0.01). Consistently, our data from experimental research showed that T2DM induced paracrine effects of mesenchymal stem cells (MSCs), a key contributor to cancer progression, to stimulate metastasis of breast cancer cells (BCCs) by two independent mechanisms. First, T2DM induced the excess secretion of interleukin 6 (IL6) from MSCs, which activated the JAK/STAT3 pathway in BCCs, thus promoting the metastasis of BCCs. Second, beside the EGR-1-/IL6-dependent mechanism, T2DM altered the functions of MSC-derived extracellular vesicles (EVs), which are highly associated with the metastasis of BCCs. Our present study showed that T2DM is a risk factor for breast cancer metastasis, and MSC-derived EVs might be useful for developing a novel anti-breast cancer therapy strategy.
Introduction
Breast cancer (BC) is one of the most common types of cancer among women, with cancer metastasis recognized as the primary cause of high mortality [1,2]. Metastasis is the process wherein cancer cells are disseminated from their original location to other organs [3].
Metastasis includes distinct sequential phases to establish a network for the spreading of cancer cells [4]. The priming phase is considered the starting point of metastasis, wherein the formation of the immature premetastatic niche is activated at secondary sites outside the primary tumor by the secretome derived from primary cancer cells [5]. The premetastatic niche matures in the licensing phase through the recruitment of immune cells, remodeling of the extracellular matrix at the secondary sites, and enrichment of the tumor-promoting molecular components that induce the epithelial-mesenchymal transition of cancer cells [5]. The initiation phase is recognized by the arrival of circulating tumor cells from vasculature and colonization inside the niche, resulting in micrometastasis [5]. Finally, in the progression phase, macrometastases develop due to the increasing number of tumor cells migrating from the primary site and then colonize, proliferate, and progress at the metastatic niche [5].
As metastasis is the main cause of cancer-related mortality, understanding the molecular mechanisms involved and the cell-cell interactions that take place inside the tumor microenvironment is essential for developing novel cancer therapy [3 –5].
The tumor microenvironment is composed of various cell types, with cancer and non-cancer cells interacting through a signaling network of cytokines, chemokines, growth factors, and extracellular vesicles (EVs) [6]. Numerous reports showed that cancer cells recruit mesenchymal stem cells (MSCs) as key members of tumor microenvironment to facilitate tumor progression [7]. MSCs support the cancer cells to avoid immune attack, chemotherapeutics, and anticancer drugs [8]. Furthermore, MSCs promote the invasion and metastasis of cancer cells [8]. Numerous reports have suggested the contribution of MSCs to the directional migration of invasive breast cancer cells (BCCs) in metastasis by secretomes, including cytokines, chemokines, growth factors (eg, TGF-β, CCL5, VEGF, and IL6 [9 –11]), and EVs. MSC-derived EVs (MSC-EVs) include exosomes, microvesicles, and apoptotic bodies [12], which have been reported to function as regulators of tumor cells in proliferation, angiogenesis, and metastasis in a paracrine manner [13 –15]. For instance, previous studies have shown that MSC-EVs mediate the metastatic niche by transferring miRNAs to cancer cells to activate several signaling pathways and induce the proliferation, migration, and invasiveness of BCCs [16 –18]. However, while numerous studies on the relationship between MSCs and BC have been reported, whether or not the functions of MSCs to facilitate the metastasis of BC are affected by a subject's disease history remains unclear.
We previously described the induction of altered characteristics and impaired wound healing functions of MSCs by insulin resistance, which is a complication of type 2 diabetes mellitus (T2DM), suggesting the effects of a history of disease on the MSC functions [19]. Notably, insulin resistance of cells and tissues has been reported to increase the risk of BC [20,21]. Insulin resistance, which is highly associated with obesity and T2DM, is the condition in which cells or tissues show a decreased response to insulin, resulting in an excess level of circulating insulin or hyperinsulinemia [22]. Indeed, obesity has long been recognized as an important determinant of the progression of BC and mortality [23]. In addition, a previous report suggested that premenopausal woman with diabetes tend to develop triple-negative and basal breast tumors [24]. Furthermore, a systematic review and meta-analysis reported a 20% increase in the incidence of BC in women with diabetes [25]. However, while these recent studies have suggested the association between T2DM and BC, whether or not T2DM affects the invasion and metastasis of BC remains unclear.
Since T2DM is recognized as a risk factor of BC [25], in this study, we conducted a meta-analysis and experimental research to analyze the association between T2DM and BC metastasis. The objectives were to investigate how T2DM affects adipose tissue-derived MSCs (AT-MSCs) to promote the metastasis of BCCs as evidenced by a conditioned medium and EVs derived from T2DM-AT-MSCs.
Materials and Methods
A meta-analysis of the association between T2DM and BC metastasis
For the systematic review, an electronic literature search was performed using PubMed and EMBASE. The search terms, which were chosen to be as inclusive as possible, were “breast cancer” AND “diabet*” AND “metastas*.” This search was completed in December 2018.
After duplicate articles were removed, a total of 377 articles were identified. The titles and abstracts were screened to exclude those that were not related to statistical studies focusing on metastasis in diabetic BC subjects. Non-English articles were excluded. A study was considered eligible if it met all of the following inclusion criteria: (1) outcomes in subjects who underwent BC treatment; (2) BC subjects with diabetes; (3) a comparison between diabetic and non-diabetic BC subjects; and (4) the number of subjects with lymph node metastasis was reported. Studies were excluded if they met any of the following exclusion criteria: (1) use of in vivo or in vitro experiments; (2) case report; (3) only about another kind of cancer; (4) <15 diabetic BC subjects; or (5) a review, abstract, or meeting abstract.
For the statistical analysis, articles identified by the systematic review were recorded by Elsevier Mendeley, and the extracted data were entered into a Microsoft Excel Office 365 (Microsoft, Redmond, WA) spread sheet for further analysis. The Review Manager (REvMan, version 5.3) software program (Cochrane Training, Copenhagen, Denmark) was used for the meta-analysis. The risk ratio was calculated using random effects. The meta-analysis was used to estimate the effect size.
Culture of BCCs and AT-MSCs
All experiments were performed according to the amended Declaration of Helsinki, and all of the experiments related to human samples were approved by the Ethics Committee of the University of Tsukuba. For BCC culture, MDAMB-231 was purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained and cultured in culture medium [Iscove's modified Dulbecco's medium (IMDM); Invitrogen, Waltham, MA] supplemented with 10% heat-inactivated fetal bovine serum (FBS; Invitrogen). Cells were maintained and cultured at 37°C in a 5% CO2 atmosphere.
Human adipose tissue was obtained with the informed consent of subjects from the Department of Cardiovascular Surgery, University of Tsukuba Hospital (Tsukuba, Japan), including non-diabetic subjects (n = 10, female, average age of 80 years, HbA1c<6) and T2DM donors (n = 10, female, average age of 80 years, HbA1c>7). AT-MSCs were isolated according to the previously described method [19]. In brief, AT was minced and digested with 0.1% collagenase (Invitrogen) in phosphate-buffered saline (PBS) at 37°C for 1 h followed by centrifugation to harvest the cell pellets and cultured in culture medium of IMDM (Invitrogen) supplemented with 10% heat-inactivated FBS (Invitrogen), 2 mg/mL
Collection of AT-MSC-conditioned medium and AT-MSC-derived EVs
EV-depleted FBS was used in the culture of AT-MSCs for the collection of AT-MSC-conditioned medium (CM) and EV isolation. AT-MSCs were cultured in IMDM fresh medium containing 0.25% EV-depleted FBS, which was obtained following the previous report [26]. Briefly, the EV-depleted FBS was obtained by centrifuging of FBS (Invitrogen) using the Amicon ultra-15 centrifugal filters (100 kDa; Merk Milipore, Burlington, MS) for 55 min at 3,000 g and the flow-through was collected as the EV-depleted FBS.
For the collection of AT-MSC-CM, a total number of 106 AT-MSCs were seeded in 10 cm dishes and cultured for 12 h when cells reach 70% confluency. The culture medium was replaced with fresh IMDM containing 0.25% EV-depleted FBS, and incubation was continued for a further 48 h. The AT-MSC-CM was then collected by centrifuging at 1,000 rpm for 5 min, followed by centrifugation at 2,100 rpm for 20 min to remove the cell debris.
For AT-MSC-derived EV isolation, AT-MSC-CM was ultracentrifuged at 37,000 rpm (rotor 70Ti; Beckman Counter) for 70 min at 4°C as previous reports [27,28]. The supernatant was then removed and the pellet was resuspended in 500 μL Diluent C (PKH26 Red Fluorescent Cell Linker Kits for General Cell Membrane Labeling; Sigma Aldrich, St. Louis, MO). Next, 500 μL of Diluent C containing 4 μL of PKH26 red (Sigma Aldrich) was added in the EV suspension, mixed, and incubated for 5 min at room temperature. The staining reaction was stopped by adding 1 mL EV-depleted FBS and incubated for 1 min at room temperature. Then, 30 mL PBS was added and the EV pellet was collected by ultracentrifugation at 37,000 rpm for 70 min at 4°C, followed by a washing step with PBS and further ultracentrifugation at 37,000 rpm for 70 min at 4°C to collect the PKH26-stained EVs. The pellet was then resuspended in PBS, and the protein concentration was examined by a Bradford assay.
In addition, the morphology of EVs was analyzed using the transmission electron microscopy and the size of the EVs was measured using a Particle Size Analyzer (FDLS3000; Shimadzu Corporation, Kyoto, Japan). The EV markers were examined by Western blotting.
The cell survival assay
BCCs were seeded at 5 × 104 cells/well in a 96-well plate and incubated in culture medium for 6 h until the cells attached to the culture plate. The culture medium was then replaced with IMDM as the control or either non-diabetic AT-MSC-CM (nCM) or T2DM AT-MSC-CM (dCM) as the treated samples and incubated for 48 h. After 48 h, 10 μL of the cell counting kit CCK-8 (Dojindo, Kumamoto, Japan) was added to each well, which were incubated for a further 2 h. The number of live cells was measured by colorimetric reading using a microplate reader (Vaioskan; ThermoFisher Scientific, Waltham, MA) at 450 nm.
The cell apoptotic assay
The cell apoptotic assay was performed as our previously described method [29]. A total number of 106 BCCs were seeded onto 10 cm dishes and cultured in the cultured medium until 80% confluency, and then cells were collected by treatment with trypsin and centrifuged at 1,000 rpm for 5 min. For staining, cells were resuspended in 100 μL of 2% FBS/PBS and stained with 5 μL of PE annexin V and 7-AAD (BD Biosciences, San Jose, CA) at 4°C in the dark for 30 min. After that, 300 μL of 2% FBS/PBS (2%) was added, and the apoptotic cells were analyzed by flow cytometry.
The in vitro migration assay
The in vitro migration assay was performed as the previously described method [30]. A total of 3 × 105 BCCs were seeded onto four-well plates in culture medium and incubated overnight until they reached 100% confluency. BCCs were then treated with mitomycin C at 10 μg/mL at 37°C in a 5% CO2 atmosphere for 3 h. A 1-mm-wide scratch was created, followed by gently washing with PBS, and IMDM was added as the control or either nCM or dCM as the treatment. Pictures of the migration area were taken every 6 h over the 24-h period postscratching at 40 × magnification (10 × objective and 10 × eyepiece) under a microscope (Keyence, Itasca, IL). The cell migration was determined using the ImageJ software program (NIH, Bethesda, MD).
Treatment of BCCs with human recombinant interleukin 6
For the gene and protein analysis, BCCs were seeded (at a total number of 105 BCCs for gene analysis and 106 BCCs for protein analysis) in the cultured medium containing human recombinant interleukin 6 (IL6) (Sigma-Aldrich) at a concentration at 600 pg/mL. After 48 h, BCCs were collected and the analysis was performed. For the in vitro migration assay, a total of 3 × 105 BCCs were cultured in cultured medium overnight; then inhibited the proliferation by mitomycin C at 10 μg/mL and scratched. BCCs were gently washed with PBS, and IMDM containing IL6 at 600 pg/mL was added. The BCCs cultured under culture medium without IL6 were used as control.
Treatment of BCCs with human anti-IL6 antibody
For in vitro migration assay, a total of 3 × 105 BCCs were seeded onto four-well plates in cultured medium overnight. After being treated with mitomycin C at 10 μg/mL, a 1-mm-wide scratch was created and BCCs were gently washed with PBS, and then changed to dCM containing IL6 antibody (I2143; Sigma Aldrich) at 10 μg/mL. For protein analysis, BCCs were seeded at a total number of 106 in dCM containing IL6 antibody at 10 μg/mL for 48 h, and then, the cells were collected for Western blot.
Establishment of shegr1 dAT-MSCs (dshEGR-1)
The anti-EGR1 shegr1 vector (NM_001964, TRCN0000273850; Sigma-Aldrich) was used to establish the dAT-MSCs with knockdown of EGR-1 according to instruction from the manufacturer. Briefly, dAT-MSCs were plated in complete culture medium until 60% confluency. shegr1 vector was thawed slowly on ice and gently added to the culture containing infant AT-MSCs at an MOI 10 and incubated at 37°C overnight. The following day, the viral particle-containing medium was removed and replaced with fresh complete culture medium containing 4 μg/mL puromycin. The medium was replaced every 3 days until resistant colonies were identified. The effect of shRNA of SOD1 or SOD3 was assessed by quantitative reverse transcription–polymerase chain reaction (qRT-PCR) and Western blot analysis. Scrambled control transduction particles (Sigma Aldrich) were used as the control.
RT-PCR and quantitative RT-PCR
Total RNA from BCCs was isolated using Sepasol-RNA I Super G (Nacalai Tesque, Kyoto, Japan). One microgram of total RNA was used to synthesize the cDNA using an RT-PCR kit (Toyobo, Osaka, Japan) according to the previously described method [19]. The expression of target genes was examined by quantitative PCR of cDNAs with the reaction mixtures prepared using SYBR Green Real-time PCR master mix (Toyobo) and amplified using a GeneAmp 7500 Fast Real-time PCR System (Life Technologies, Camarillo, CA). The sequences of primers used for PCR are listed in Table 1. The experiments were performed in triplicate, and data were calculated by the double delta cycle number of threshold (ΔΔCt) method.
Primer Sets Used for Quantitative Polymerase Chain Reaction
Western blotting
BCC pellets were collected, and nuclear proteins were extracted according to the previously described method [19]. The protein concentration was measured by Bradford reagent (Biorad, Hercules, CA) and then mixed with sodium dodecyl sulfate (SDS) loading buffer (Wako, Osaka, Japan) and denatured at 95°C for 3 min. Equal amounts of 30 μg protein extracted from BCCs were applied to the SDS–polyacrylamide gels for electrophoresis and transferred onto polyvinylidene fluoride (PVDF) membranes (Merck Millipore). The membranes were immune blotted with primary antibodies overnight at 4°C, including rabbit anti-pan-STAT3 (8204S; Cell Signaling Technology, Danvers, MA) and anti-phosphorylated-STAT3 (pSTAT3) (8204S; Cell Signaling Technology) at 1:1,000 dilution. Goat anti-Lamin B antibody (sc-6216; Santa Cruz Biotechnology, Dallas, TX) was used as the internal control at the same dilution rate of 1:1,000.
To examine the expression of EVs' markers, 20 μg protein extracted from EVs or AT-MSCs was applied for electrophoresis and transferred onto PVDF membranes followed by the incubation with primary antibodies at 1:1,000 dilution rate at 4°C overnight, including rabbit anti-CD63 (CSB-PA006039; Cusabio Tachnology LLC, Houston, TX), rabbit anti-TSG101 (CSB-PA060017; Cusabio Technology LLC), and goat anti-actin (sc-1615; Santa Cruz Biotechnology). Horseradish peroxidase (HRP)-conjugated rabbit anti-goat IgG (Invitrogen) and HRP-conjugated goat anti-rabbit IgG (Invitrogen) were used as secondary antibodies at a dilution of 1:10,000. The target protein expression was detected by an enhanced chemiluminescence HRP substrate (EMD Millipore) for 1 min and visualized using an Image Quant LAS 4000 System (GE Health care, Chicago, IL).
The in vivo cancer metastasis assay
In this study, all experiments related to animals were approved by the Animal Care Committee of the University of Tsukuba. Male C57BL/6 and db/db mice were purchased from Charles River Japan, Inc., (Yokohama, Japan), given food and water ad libitum, and maintained on a 12-h light/12-h dark cycle in the Animal Research Center of the University of Tsukuba. Eight-week-old mice were used in the experiments. The in vivo metastasis assay was conducted based on a previously described study with some modifications [31]. In brief, BCCs were injected into mice at 105 cells/mouse by tail-vein injection. Immunosuppression was induced by the intraperitoneal injection of cyclosporin A (20 mg/kg; Wako) every 2 days. After 14 days, lungs were collected and fixed in 4% paraformaldehyde overnight and then washed with PBS followed by soaking in sucrose 10% for 2 h and sucrose 20% overnight before paraffin embedding. The paraffin-embedded lung samples were then sectioned for hematoxylin and eosin (H&E) staining to identify the tumor foci.
Statistical analyses
Data were analyzed using the Mann–Whitney U-test with the GraphPad Prism 5 software program (GraphPad Software, San Diego, CA). Data are presented as the mean ± standard deviation. P < 0.05 was considered significant.
Results
Relationship between T2DM and BC metastasis according to a meta-analysis
To determine the presence of a relationship between T2DM and BC metastasis in humans, we first conducted a systematic review and meta-analysis. The systematic review flow is described in Fig. 1A. In this study, reports containing only abstracts were excluded because of the lack of detailed data, such as the number of subjects who were positive for lymph node metastasis. In addition, two studies written in languages other than English were excluded since we were unable to determine the subjects' outcomes, and one study was excluded because the number of subjects was <15. The remaining 12 studies were used for the meta-analysis.
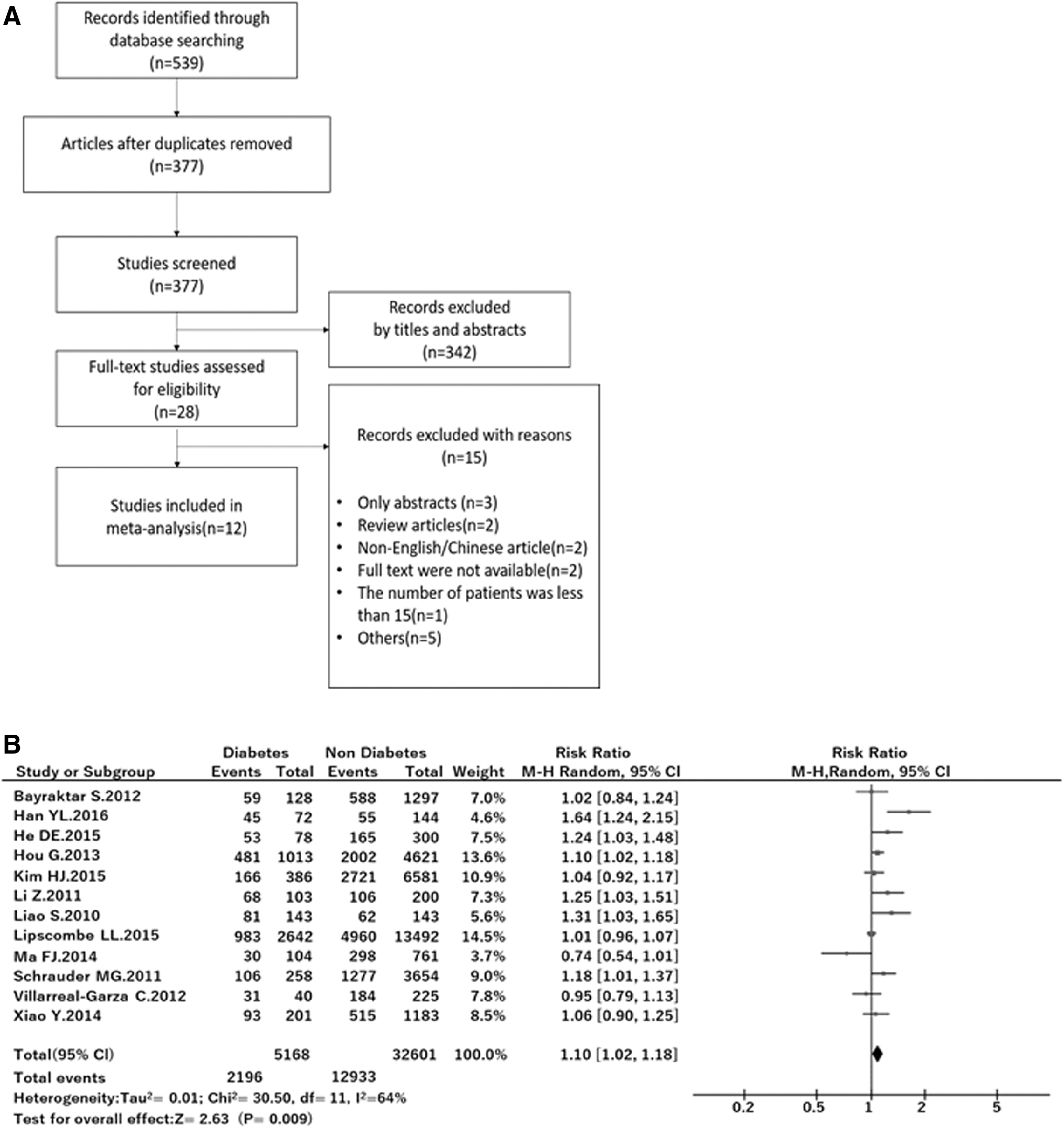
A meta-analysis of the association between T2DM and breast cancer lymph node metastasis.
The effect of diabetes on lymph node metastasis in BC subjects is summarized in a forest plot (Fig. 1B), and the characteristics of the researches included in this meta-analysis are summarized in Table 2. The studies included in this study were conducted in the United States, Mexico, Canada, Germany, Korea, and China. Among them, two studies focused on triple-negative BC (TNBC), two evaluated invasive BC, one focused on luminal BC, and the others included multiple kinds of BC. The number of participants in each study ranged from 216 to 38,407. Almost all studies mentioned the body mass index (BMI; 9/12), menopausal status (8/12), and overall survival (OS; 9/12). Eight studies specified the follow-up time after surgery (32 months to 5 years). The point of time at which subjects were diagnosed with diabetes was clarified in only six studies. A random effect meta-analysis showed that the total risk ratio for lymph node metastasis of T2DM subjects was 1.10 (P < 0.01), suggesting a significant positive relationship between T2DM and BC metastasis.
Characteristics of the Articles Included in the Meta-analysis
BC, breast cancer; U.S., United States.
T2DM induced paracrine effects of AT-MSCs to promote the mobility of BCCs
The tumor microenvironment includes a variety of mesenchymal cell types that contribute to the tumor development [7]. Of note, several previous reports have shown that MSCs induced the metastatic potency of weakly metastatic human breast carcinoma cells [9 –11], suggesting the vital role of MSCs in the progression and metastasis of BC. Despite the known relationship between T2DM and BC [24,25], whether or not MSCs derived from AT of T2DM patients (dAT-MSCs) exerted greater inducible effects on BCCs than those from healthy donors (nAT-MSCs) has been unclear. Therefore, we next compared the paracrine effects of nAT-MSCs and dAT-MSCs in the high metastatic BCC line MDA-MB-231 using AT-MSC- CM.
First, the paracrine effects of nAT-MSCs and dAT-MSCs on the survival and apoptosis of BCCs were examined. BCCs cultured in nCM and dCM showed a similar survival ability and apoptotic rate to the control BCCs (Fig. 2A, B). Of note, treatment with AT-MSC-CM significantly increased the migration of BCCs, and interestingly, dCM showed greater induction effects than nCM (1.5-fold higher, Fig. 2C). Consistent with the migration, dCM dramatically upregulated the expression of genes related to migration in BCCs, such as CXCR4 and VEGF-C (Fig. 2D), and metastasis, such as IL6, IL8, TGFβ, bFGF, and EGF (Fig. 2E), compared to those treated with nCM.
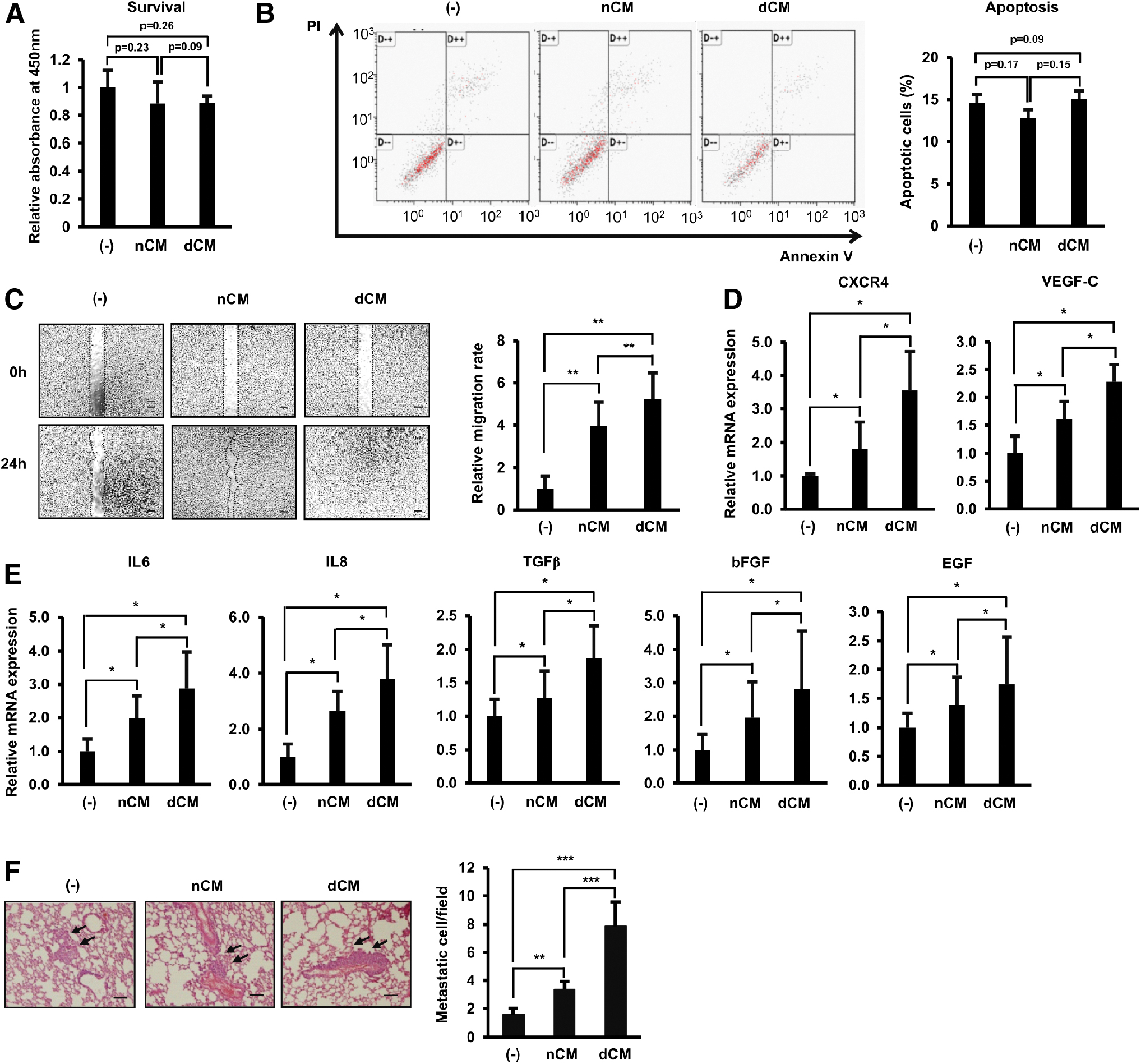
T2DM induced the paracrine effects of dAT-MSCs to promote the mobility of BCCs.
Next, to examine the paracrine effects of nAT-MSCs and dAT-MSCs on the metastasis of BCCs in vivo, we administered tail vein injections of BCCs to mice and examined the number of metastatic BCCs at the lung. Treatment with AT-MSC-CM induced the metastasis of BCCs to the lung, and notably, these inducible effects were higher in BCCs treated with dCM than in those treated with nCM (average number of metastatic cells/fields: control BCCs, 1.59; BCCs cultured in nCM, 3.37; and BCCs cultured in dCM, 7.86; Fig. 2F).
Taken together, these data showed that although both nAT-MSCs and dAT-MSCs possess similar paracrine effects to control BCCs, dAT-MSCs had a significantly greater inducing effect for the migration of BCCs, suggesting the involvement of dAT-MSCs in BC metastasis.
IL6 induced the migration of BCCs through the activation of the JAK/STAT3 pathway
The cytokines and growth factors secreted by MSCs, such as VEGF and IL6, are reportedly involved in the induced migration of several types of BCCs [11,32]. In addition, we previously reported the high secretion of IL6 from dAT-MSCs due to the upregulation of EGR-1 [19]. Therefore, we hypothesized that the strong induced paracrine effects of dAT-MSCs on the migration of BCCs were due to the high level of IL6 in dCM. To test this hypothesis, we first examined the direct effects of IL6 on the migration of BCCs by culturing BCCs under normal conditions without AT-MSC-CM and treating BCCs with IL6 at 600 pg/mL, which is similar to the concentration of IL6 secreted by dAT-MSCs. As expected, treatment with IL6 significantly induced the migration of BCCs (3.09-fold increase, Fig. 3A). Consistently, treatment with IL6 also upregulated the expression of genes related to the migration ability of BCCs, such as CXCR4 (1.7-fold increase, Fig. 3B) and VEGF-C (2.4-fold increase, Fig. 3B).
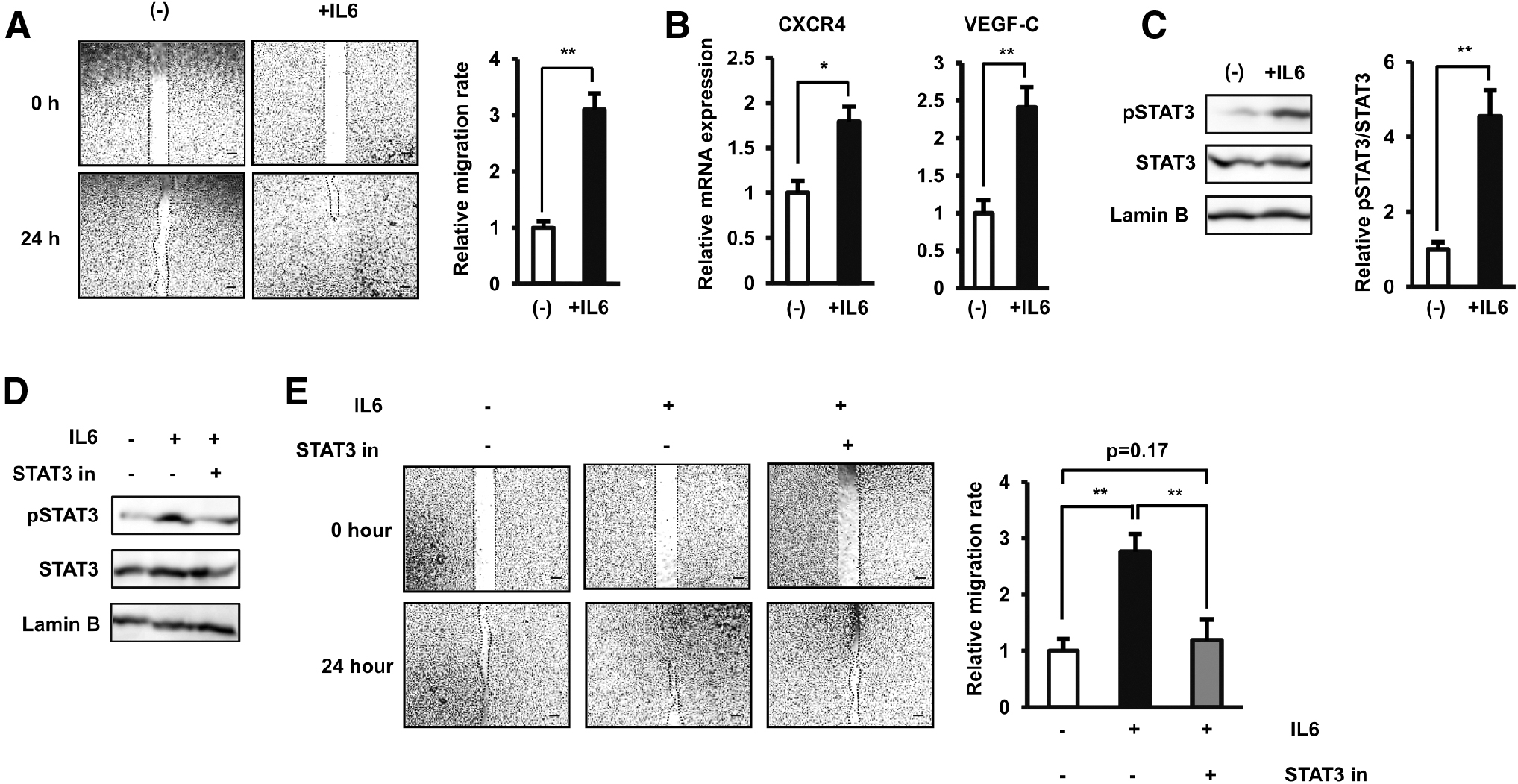
IL6 directly induced the migration of BCCs by the activation of the JAK/STAT3 pathway.
We next examined the signaling pathways involved in the induced migration of BCCs by IL6 and found that treatment with IL6 significantly activated the phosphorylation of STAT3 in the JAK/STAT3 pathway (4.56-fold increase, Fig. 3C). To examine the role of IL6-induced STAT3 activation in the migration of BCCs, we added a STAT3 inhibitor to the culture system. This addition impaired the activation of STAT3 induced by IL6 (Fig. 3D), which resulted in a reduction in the migration ability of BCCs (2.3-fold decreased migration ability, Fig. 3E). These data suggested the direct role of the JAK/STAT3 signaling pathway in the migration of BCCs.
Taken together, these data confirmed the direct role of IL6/STAT3 signaling pathways in the migration of BCCs, which supported our hypothesis concerning the relationship between a high level of dAT-MSC-secreted IL6 and the induced effects on the migration of BCCs.
EGR-1-induced excess IL6 in dAT-MSCs promoted the mobility of BCCs by activating the JAK/STAT3 pathway
Next, we examined the activation of the JAK/STAT3 pathway in BCCs cultured in dCM compared to control BCCs without any treatment. Under culture in dCM, BCCs showed the significant activation of the phosphorylation of STAT3 compared to the control BCCs (7.3-fold increase, Fig. 4A). As expected, the addition of STAT3 inhibitor to the culture of BCCs with dCM impaired the phosphorylation of STAT3 (Fig. 4B), thereby reducing the migration of BCCs (twofold decrease compared to BCCs cultured in dCM without STAT3 inhibitor, Fig. 4C). Next, to show that IL6 is one of the key mediators of the migratory effects in dCM, we performed the analysis of BCCs cultured under dCM with the addition of human IL6 antibody. As the results, the addition of IL6 antibody in dCM decreased the migration of BCCs compared to those under control dCM in a scratch assay (Fig. 4D). Also, the addition of IL6 antibody in dCM decreased the expression of pSTAT3 in BCCs compared to those under control dCM (Fig. 4E).
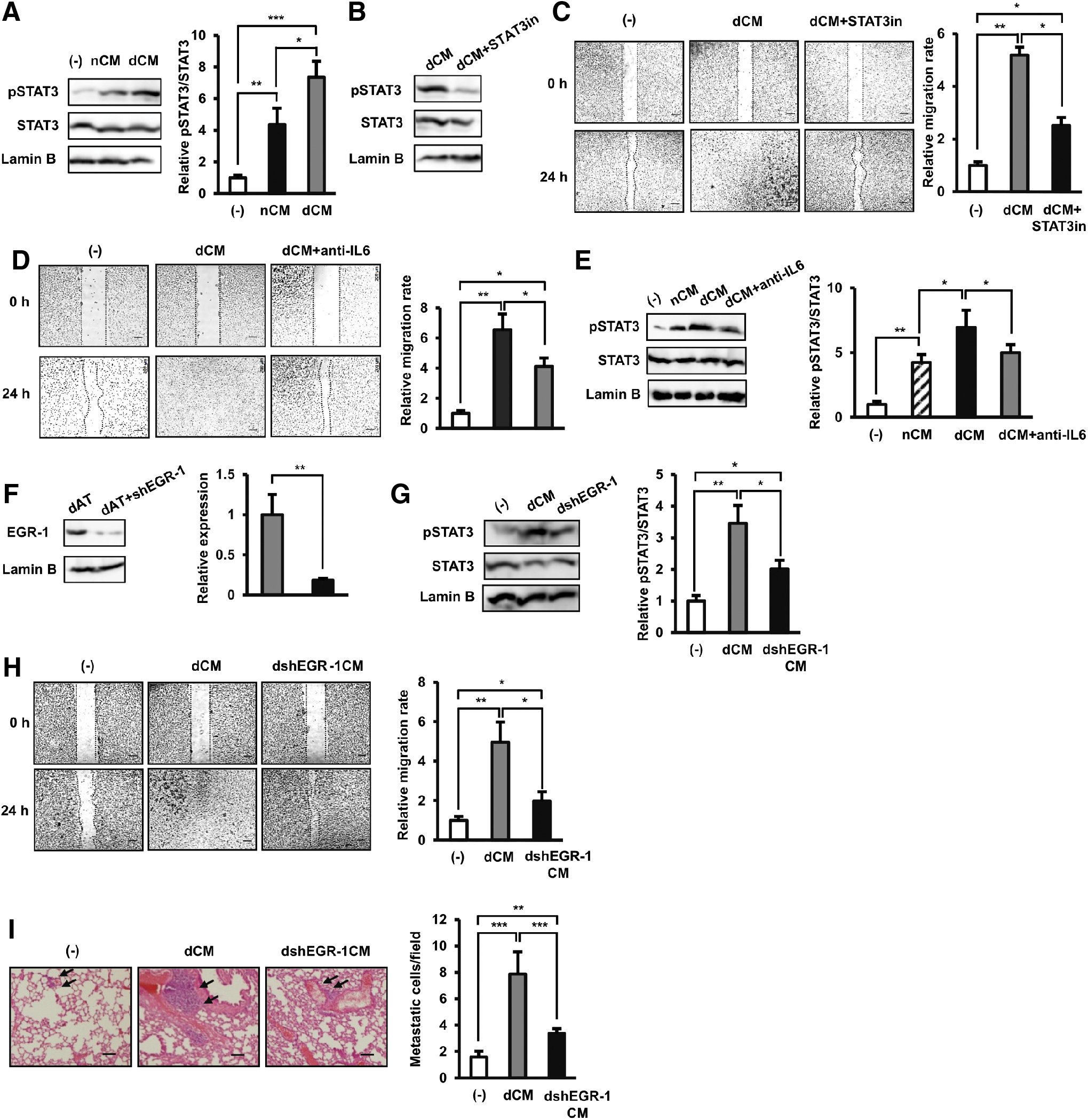
The EGR-1-induced excess IL6 in dAT-MSCs promoted the mobility of BCCs by activating the JAK/STAT3 pathway.
We previously reported the high secretion of IL6 in dAT-MSCs is due to the upregulation of EGR-1 [19]. Therefore, in this study, we examined whether or not the impairment of EGR-1 in dAT-MSCs, which has been reported to reduce the secretion of IL6, might cause any paracrine effect on the migration of BCCs. The impairment of EGR-1 expression in dAT-MSCs with shEGR1 was confirmed by Western blot (Fig. 4F). Next, BCCs were treated with CM either derived from wild-type dAT-MSCs (dCM) or dAT-MSCs with the knockdown of EGR-1 (dshEGR-1CM). Interestingly, BCCs cultured in dshEGR-1CM showed less activation of STAT3 (Fig. 4G) than those cultured in dCM, which is consistent with the reduced migration ability in vitro (2.57-fold decreased migration ability compared to BCCs cultured under dCM, Fig. 4H). In addition, the in vivo metastasis assay also confirmed that the impairment of EGR-1 reduced the paracrine effects of dAT-MSCs to induce the metastasis of BCCs (average metastatic cells/fileds: BCCs: 1.59, BCCs cultured under dCM: 7.86, and BCCs cultured under dshEGR-1CM: 3.39, Fig. 4I) compared with BCCs cultured in dCM, suggesting the involvement of a high expression EGR-1 in the induced mobility of BCCs.
Taken together, these data suggested that the high expression of EGR-1 in dAT-MSCs induced the elevated secretion of IL6, thereby activating the JAK/STAT3 pathway in BCCs, which was involved in cancer metastasis.
dAT-MSC-EVs markedly induced the mobility of BCCs
Given that BCCs cultured in the dshEGR-1CM still exhibited greater mobility than those without treatment (Fig. 4H), we hypothesized that, in addition to IL6, other factors in dAT-MSC-conditioned medium also contributed to the induced migration of BCCs. In addition to growth factors, cytokines, and chemokines, AT-MSC-CM contains EVs, which have been reported to be tools for cell-to-cell communication and play typical roles in the modulation of recipient cells [33]. We therefore next focused on the effects of EVs on the migration of BCCs.
EVs were isolated from the CM of nAT-MSCs (nEVs) or dAT-MSC (dEVs) by serial ultracentrifugation. We noted no significant difference in the level of EVs secreted from nAT-MSCs and dAT-MSCs (Fig. 5A). The images of nEVs and dEVs under the transmission electron microscopy clearly showed that both nEVs and dEVs are oval particles (Fig. 5B), with diameter from 70 nm to 600 nm (Fig. 5C). In addition, both nEVs and dEVs showed the expression of EV markers, such as CD63 and TSG101, and showed no expression of actin (Fig. 5D), which is consistent with previous reports [34,35]. The PKH-26-labeled EVs then were incorporated into BCCs. The presence of EVs inside BCCs was examined by a fluorescence-activated cell sorting analysis for the PKH-26 signal (Fig. 5E).
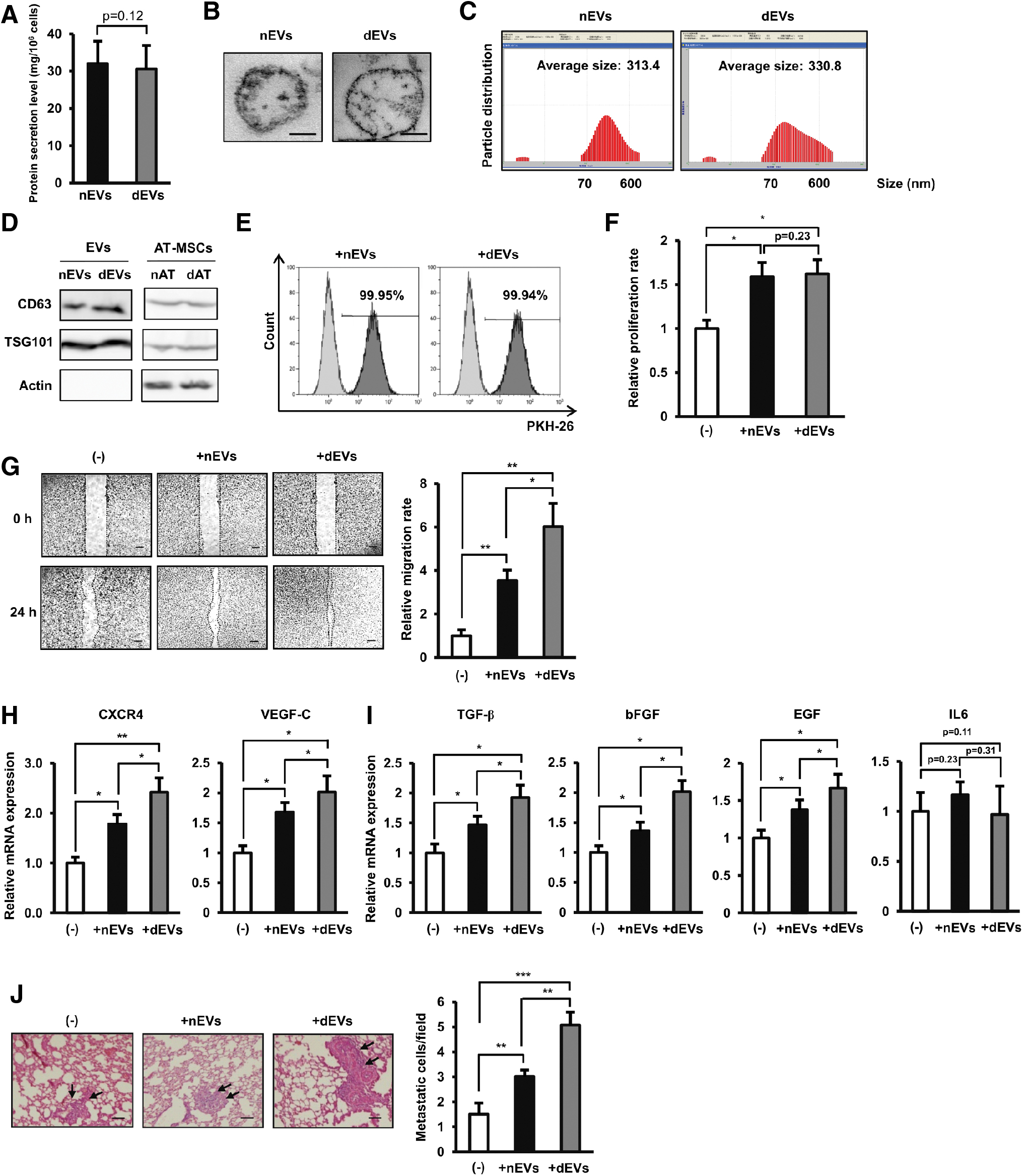
Extracellular vesicle derived from dAT-MSCs markedly induced the mobility of BCCs.
After most BCCs in the population were determined to be positive for PKH-26 signal, the effects of nEVs and dEVs on BCCs were examined. We found that both nEVs and dEVs induced the survival of BCCs after 48 h to a similar degree (Fig. 5F). Next, the migration of nEV- or dEV-incorporated BCCs was compared to that of control BCCs without the incorporation of EVs. Interestingly, consistent with the effects of CM, although treatment with both nEVs and dEVs induced the migration of BCCs, the effects of dEVs were significantly greater than those of nEVs (nEV-incorporated BCCs: 3.5-fold increased migration ability and dEV-incorporated BCCs: sixfold increased migration ability, Fig. 5E). In addition, compared to nEVs, dEVs also showed a greater induction of the upregulation of genes related to BCC migration, such as CXCR4 and VEGF-C, as well as genes related to BCC metastasis, such as TGF-β, bFGF, and EGF (Fig. 5G, H). Notably, treatment with neither nEVs nor dEVs showed any significant upregulating effect on the expression of IL6 in BCCs (Fig. 5I). Consistent with the in vitro results, the in vivo study showed that dEV-incorporated BCCs exhibited the highest rate of metastasis to the mouse lung (average metastatic cells/field: BCCs: 1.51, nEV-incorporated BCCs: 3.02, and dEV-incorporated BCCs: 5.07, Fig. 5J).
Taken together, these data showed that, in addition to the upregulation of the IL6-dependent pathway, T2DM altered the phenotype of dEVs to induce the paracrine effects of dAT-MSCs on BC metastasis.
Discussion
In this study, using a systematic review and meta-analysis, we showed that diabetes significantly increased the risk for lymph node metastasis by 10% (P < 0.01). These meta-analytic results were the basis of our hypothesis, concerning our experimental research that T2DM might alter the phenotype of cells present in the tumor microenvironment to facilitate the progression of BC metastasis. Focusing on MSCs as the key contributors to cancer progression, we found that T2DM accelerated the paracrine effects of AT-MSCs to induce the migration of BCCs and upregulate the expression of migration and EMT-related factors in BCCs. Of note, our data suggested that dAT-MSCs promoted the migration of BCCs by the combined effects of overexpression of EGR-1/IL6 activating JAK/STAT3 pathway and the functions of EVs (Fig. 6).
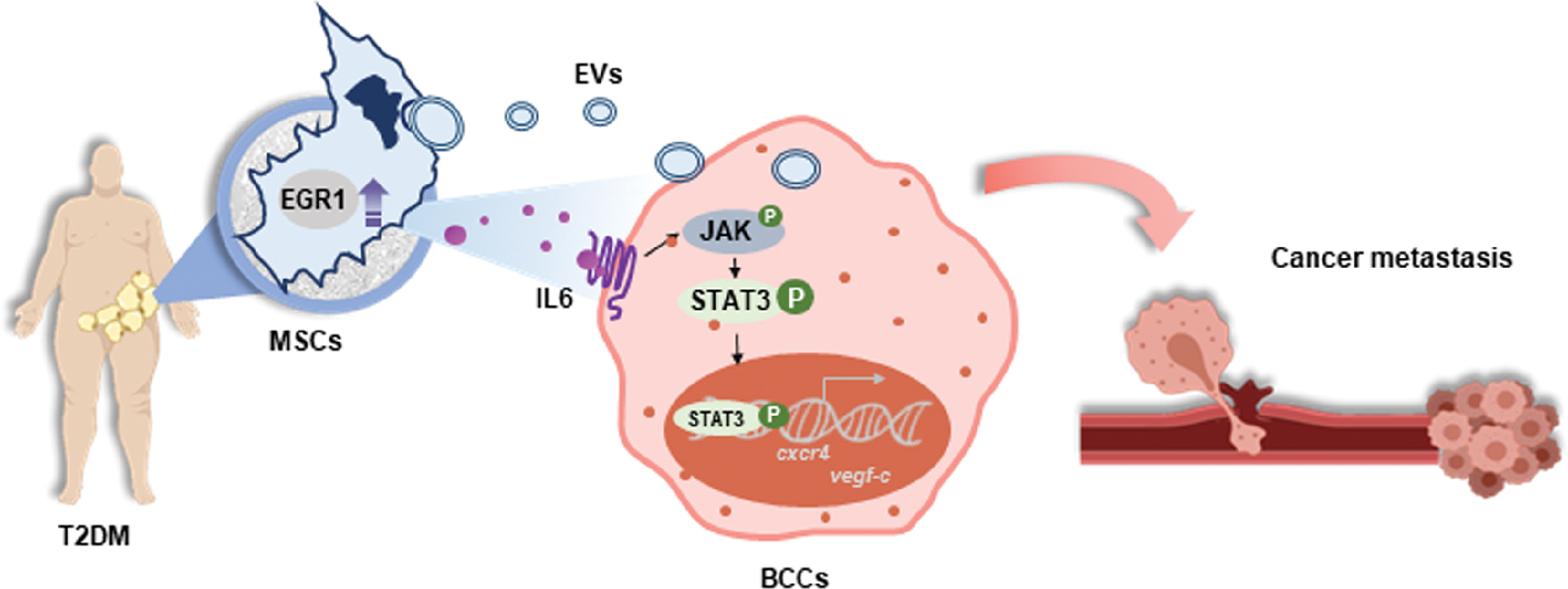
Proposed model: T2DM induced the paracrine effects of dAT-MSCs to promote breast cancer metastasis. The strong paracrine effects of dAT-MSCs to induce breast cancer metastasis were attributed to the elevated secretion of IL6, which activated JAK/STAT3 signaling and the functions of extracellular vesicles. Color images are available online.
We previously reported the changes in the phenotype and healing functions of AT-MSCs induced by T2DM [19]. In this study, we found that, compared to nAT-MSCs, dAT-MSCs significantly upregulated the expression of migration-related factors in BCCs, including CXCR4 and VEGF-C. The chemokine receptor CXCR4 has long been recognized as a prognostic marker of BC. CXCR4 is responsible for the migration of BCCs toward the signal of SDF-1 [36]. In addition, VEGF-C is a lymphangiogenic factor that is upregulated in highly metastatic BCCs and plays an autocrine role to promote cellular mobility through VEGF-C-binding receptors, including VEGFR-1, VEGFR-3, NRP-1, NRP-2, and integrin α9β1 [37]. dAT-MSC-CM also induced the expression of genes involved in the induction of premetastatic niche formation and EMT, such as IL6, IL8, TGFβ, , and EGF, which facilitate the dissemination of cancer cells from their original tumor sites to distant organs [5].
Our previous study also reported the induced secretion of IL6 in dAT-MSCs due to the high expression of EGR-1 in T2DM subjects [19]. As the role of IL6 to induce the migration of BCCs has been suggested in previous reports [38,39], we speculated that the strongly induced paracrine effects of dAT-MSCs on the migration of BCCs might be due to the excess secretion of IL6. Indeed, our data showed that treating BCCs with either IL6 at a concentration similar to that secreted by dAT-MSCs (600 pg/mL) or dCM directly promoted the migration of BCCs by the activation of the JAK/STAT3 pathway. In addition, the addition of anti-IL6 antibody to dCM resulted in the decreased migration and phosphorylation of STAT3 in BCCs. These data are consistent with those of a previous report showing that an IL6/JAK signal transducer and activator of STAT3 pathway enhance breast tumor progression through the stroma and metastatic niche [40]. Of note, the impairment of EGR-1 expression in dAT-MSCs, which was reported to suppress the secretion of IL6, decreased the paracrine effects of dAT-MSCs to induce the activation of STAT3 in BCCs and the migration of these cancer cells. This study suggested that the EGR1/IL6/STAT3 pathways should be considered a potential target for reducing metastasis in T2DM subjects with BC.
In our data, BCCs with impairment of IL6-activated STAT3 still showed higher mobility than control BCCs, suggesting the contribution of other factors in dAT-MSC-CM for BCC migration. Therefore, we next targeted the functions of EVs, the communication tool for cell-to-cell interaction [41]. Several studies have examined the effects of MSC-EVs on BCCs; however, their findings were conflicting, so the role of MSC-EVs in cancer metastasis has remained controversial [42 –44]. In this study, we found that treatment with AT-MSC-CM exerted no effect on the survival of BCCs; however, treatment with nEVs or dEVs promoted the survival of BCCs. Since the survival of BCCs is dependent on the concentration of EVs (data not shown), these data suggested that the amount of EVs in AT-MSC-CM might not be sufficient to support the survival of BCCs. In addition, together with the elevated secretion of IL6, dAT-MSC-EVs also contributed to the induced mobility of BCCs, which might facilitate BC metastasis in T2DM subjects. Notably, the incorporation of EVs in BCCs showed no effect on the expression of IL6, suggesting that EVs affected the metastasis of BCCs in a different manner from the EGR1/IL6 signaling pathway (Fig. 5F). Because dEVs exert their activities through the transfer of their genetic content, such as proteins, miRNA, and mRNA, to BCCs, the genetic content of dEVs might implicate underlying related pathways involved in the metastatic pathway. Future studies should examine the mechanisms underlying how T2DM alters the role of AT-MSC-EVs in the progression of BC.
In conclusion, this study suggested that T2DM promoted the paracrine effects of AT-MSCs on the metastasis of BCCs, suggesting that follow-up to detect metastasis in the early stage is required for BC patients with T2DM. Of note, this study demonstrated the novel role of AT-MSC-EVs to induce the metastasis of BCCs, exacerbated by T2DM, suggesting that AT-MSC-EVs might be a novel target for inhibiting BC metastasis. Future studies evaluating the effects of AT-MSC-EVs on the progression of other cancers will be needed to develop new anticancer therapies.
Footnotes
Acknowledgment
We thank the Japanese Ministry of Education, Culture, Sports, Science & Technology (MEXT) for their support.
Author Disclosure Statement
The authors declare no conflicts of interest in association with this study.
Funding Information
There funding was received for this study.