
Abstract
A growing body of work suggests that canine mesenchymal stromal cells (cMSCs) require additional agonists such as bone morphogenic protein-2 (BMP-2) for consistent in vitro osteogenic differentiation. BMP-2 is costly and may challenge the translational relevance of the canine model. Dexamethasone enhances osteogenic differentiation of human MSCs (hMSCs) and is widely utilized in osteogenic protocols. The aim of this study was to determine the effect of BMP-2 and dexamethasone on early- and late-stage osteogenesis of autologous and induced pluripotent stem cell (iPS)-derived cMSCs. Two preparations of marrow-derived cMSCs were selected to represent exceptionally or marginally osteogenic autologous cMSCs. iPS-derived cMSCs were generated from canine fibroblasts. All preparations were evaluated using alkaline phosphatase (ALP) activity, Alizarin Red staining of osteogenic monolayers, and quantitative polymerase chain reaction. Data were reported as mean ± standard deviation and compared using one- or two-way analysis of variance and Tukey or Sidak post hoc tests. Significance was established at P < 0.05. In early-stage assays, dexamethasone decreased ALP activity for all cMSCs in the presence of BMP-2. In late-stage assays, inclusion of dexamethasone and BMP-2 at Day 1 of culture produced robust monolayer mineralization for autologous cMSCs. Delivering 100 nM dexamethasone at Day 1 improved mineralization and reduced the BMP-2 concentrations required to achieve mineralization of the marginal cMSCs. For iPS-cMSCs, dexamethasone was inhibitory to both ALP activity and monolayer mineralization. There was increased expression of osteocalcin and osterix with BMP-2 in autologous cMSCs but a more modest expression occurred in iPS cMSCs. While autologous and iPS-derived cMSCs respond similarly in early-stage osteogenic assays, they exhibit unique responses to dexamethasone and BMP-2 in late-stage mineralization assays. This study demonstrates that dexamethasone and BMP-2 can be titrated in a time- and concentration-dependent manner to enhance osteogenesis of autologous cMSC preparations. These results will prove useful for investigators performing translational studies with cMSCs while providing insight into iPS-derived cMSC osteogenesis.
Introduction
Mesenchymal stromal cells (MSCs) are promising agents for bone regeneration and repair [1 –3]. For MSCs to successfully translate from proof-of-concept rodent studies to clinical reality, safety and efficacy must first be evaluated in large animal models such as the sheep, goat, pig, or dog [4 –6]. The dog represents an excellent translational model for cell-based orthopedic treatments. Compared to rodents, dogs are genetically diverse, experience similar biomechanical loads to human beings, and are amenable to post-treatment exercise regimens. Dogs can also be more readily evaluated with assessment tools that directly translate to human clinical trials [7]. Although canine MSCs (cMSCs) share many similarities to hMSCs [8 –16], cMSCs appear to respond differently to traditional hMSC osteogenic stimuli in the in vitro setting. For example, cMSCs exhibit minimal alkaline phosphatase (ALP) activity and monolayer mineralization when cultured using methods that induce robust early- and late-stage osteogenesis of hMSCs [8,17 –19]. However, when cMSC osteogenic media are supplemented with agonists such as bone morphogenic protein-2 (BMP-2) [8,17], IGF-1 [18], or NELL-1 [19], they exhibit robust ALP activity and monolayer mineralization.
As with other species, the osteogenic potential of different preparations of cMSCs varies remarkably and represents a major hurdle for cMSC-based translational studies. Canine MSC osteogenesis is influenced by donor age, health, tissue source, cell isolation and culture technique, cryopreservation, and passage number [8 –10,15,20]. The ideal cell source of cMSCs for bone regeneration remains unknown, although marrow- and adipose-derived cMSCs have received the most attention [9,11 –13,15,18,20]. For these reasons, reliable and cost-effective methods are needed to optimize the osteogenic performance of cMSCs isolated from different donors, tissues, or laboratories.
One notable limitation of autologous MSCs is their limited culture capacity. Once exhausted, new cMSCs must be isolated, characterized, and screened for use in specific studies. In an effort to overcome these challenges, induced pluripotent stem cell (iPS)-derived MSCs have emerged as a promising alternative source of MSCs [21 –23]. iPS-derived cMSCs have been differentiated from canine iPSs using methods similar to human iPS-derived MSCs [21,24,25]. iPS-derived cMSCs possess many attractive properties, such as a prolonged culture capacity and the elimination of donor and tissue variability [21,26 –28]. To date, few preparations of canine iPS-derived cMSCs have been produced. Moreover, the optimal culture conditions for iPS-derived cMSC osteogenesis remain to be determined. Given the reported differences between canine and human autologous MSCs, substantial differences may also exist between iPS-derived hMSCs and cMSCs. Thus, a thorough in vitro evaluation of iPS-cMSC osteogenesis is warranted.
Dexamethasone is a classic in vitro osteogenic induction agent for MSCs across species [11,12,14,17,29 –38], yet its mechanism of action is incompletely understood [35,38 –40]. Dexamethasone has varying effects on osteogenic differentiation of MSCs based on concentration and duration of supplementation [39,41 –43]. In contrast to recombinant growth factors (e.g., BMP-2), dexamethasone is inexpensive, safe, and is commonly used in clinical practice. While the effective concentration and temporal delivery of dexamethasone are well established for human MSCs (hMSCs) [29 –31,33], the optimal dexamethasone delivery for either autologous or iPS-derived canine MSCs is yet to be determined. One of our laboratory's main goals is developing improved osteogenic differentiation methods for cMSCs. We have anecdotally observed that the timing and concentration of dexamethasone supplementation in osteogenic media have dramatic effects on cMSC osteogenesis and monolayer mineralization (unpublished observations).
Therefore, the objective of the present study was to evaluate both early- and late-stage in vitro osteogene_sis of representative autologous and iPS-derived cMSCs cultured with varying concentrations of BMP-2 and dexamethasone to identify optimal osteogenic culture conditions. We hypothesized that increasing concentrations of BMP-2 and dexamethasone would improve early- and late-stage osteogenesis of both autologous and iPS-derived cMSCs. Second, we hypothesized that supplementation of osteogenic cultures with dexamethasone early in the differentiation process would enhance osteogenesis.
Methods
Cell isolation, preparation, and characterization (autologous cMSCs)
All procedures were performed under the supervision of the Institutional Animal Care and Use Committee (IACUC) [Animal Use Protocol (AUP) 2011-149]. Autologous cMSCs were obtained from bone marrow aspirates of the iliac crest or humerus of two donor dogs (IC018 and H012, Table 1). These preparations of cMSCs were selected based on historic in vitro osteogenic performance using previously established canine osteogenic protocols [8]. IC018 cells exhibited excellent osteogenesis, while H012 cells exhibited marginal osteogenesis. Nucleated cells were isolated using gradient centrifugation (Ficoll-Paque Plus, GE Health Care Biosciences, Piscataway, NJ) and plastic adherence [8,33]. Nucleated cells were plated at 3 × 104 cells/cm2 on tissue culture dishes in complete culture medium (CCM) containing α-Minimum Essential Medium (MEM), 100 U/mL penicillin, 100 μg/mL streptomycin, 29.2 mg/mL glutamate (Invitrogen, Carlsbad, CA), and 10% premium select fetal bovine serum (PS-FBS; Atlanta Biologicals, Inc., Flowery Branch, GA).
Canine Mesenchymal Stromal Cell Donor Characteristics
iPS, induced pluripotent stem cell.
Cells were incubated at 37°C and 5% humidified CO2 for 24 h. Plates were washed with phosphate buffered saline (PBS) to remove nonadherent cells, and medium was exchanged daily for 3 days. Cultures were subsequently monitored for expansion of the primary cell population (passage 0; P0) with media exchange every other day. At 70% confluence (5–12 days), cells were lifted with 0.5% trypsin/EDTA (Invitrogen) and replated at 100 cells/cm2 for expansion of passage 1 (P1) cells. Media was exchanged every other day until cells reached 70% confluence, at which point P1 cells were cryopreserved in α-MEM containing 30% PS-FBS (Atlanta Biologicals, Inc.) and 5% dimethyl sulfoxide (DMSO; Sigma, St. Louis, MO). Passage 2 canine autologous bone marrow cells were characterized and confirmed to be cMSCs using established criteria for cMSCs [8,16,44] (Supplementary Data and Supplementary Figs. S1–S4; Supplementary Tables S1 and S2).
Preparation of iPS-derived cMSCs
Transgene integration-free canine iPS was generated from canine skin fibroblasts at the Colorado University Denver, Charles C. Gates Center for Regenerative Medicine and Stem Cell Biology iPSC Core [21]. Canine dermal fibroblasts were isolated using skin biopsy of a 6-year-old male standard poodle and incubated overnight with CytoTune iPS Reprogramming vectors (Life Technologies Corp. Grand Island, NY). Cells were cultured for 7 days and transferred to irradiated mouse embryonic fibroblast feeder cells (Global Stem, Gaithersburg, MD). iPS-derived cMSCs were then generated by supplementation of iPS culture medium with 10 μM of the transforming growth factor beta inhibitor SB 431542 as described [45]. Cells were allowed to differentiate for 10 days with daily media changes [21]. Canine iPS-derived cMSCs were previously characterized at the time of their creation [21] and were recharacterized as passage 21 iPS-derived cMSCs in conjunction with the autologous cell characterization [8,44,46] (Supplementary Data and Supplementary Figs. S1–S4).
Effect of dexamethasone and BMP-2 on early-stage osteogenesis
Passage 2 autologous and passage 21 iPS-derived cMSCs were plated at 1 × 104 cells/cm2 in 12-well plates (n = 3 wells/condition). The following day (Day 1), cells were treated with control (CCM) or osteogenic medium [α-MEM with 10% FBS, 10 μg/mL β-glycerophosphate (Sigma), 50 mg/mL ascorbate-2-phosphate (Sigma)]. Dexamethasone (Sigma) was supplemented at 0, 1, 10, or 100 nM at Day 1. Cells were also treated with 50, 100, or 150 ng/mL recombinant human BMP-2 (rhBMP-2) (R&D Systems, Minneapolis, MN) at Day 1 [8,17,30] (Fig. 1). Media exchange was performed twice weekly. At Day 7, wells were washed twice with PBS and incubated with 500 μL ALP activity buffer (4°C) containing 1 mM magnesium chloride (Sigma), 100 mM Tris-HCl (Sigma), and 100 mM sodium chloride (Sigma) in PBS [30]. The ALP substrate p-nitrophenylphosphate (PNPP; Thermo Fisher, Waltham, MA) was added at 4°C to each well in 500 μL volumes to initiate the reaction. Absorbance for each well was determined at 405 nm in 1-min intervals for 20 min at 37°C using an automated plate reader (Cytation 5; BioTek, Winooski, VT) and Gen5Bio software [30]. Kinetic curves representing the rate of p-nitrophenolate accumulation were generated for each well, and ALP activity was calculated from the slope of each curve using the linear curve fit method (Supplementary Fig. S5). Finally, ALP activity was normalized to number of cells per well using DNA quantification [8,30] (Supplementary Fig. S6).
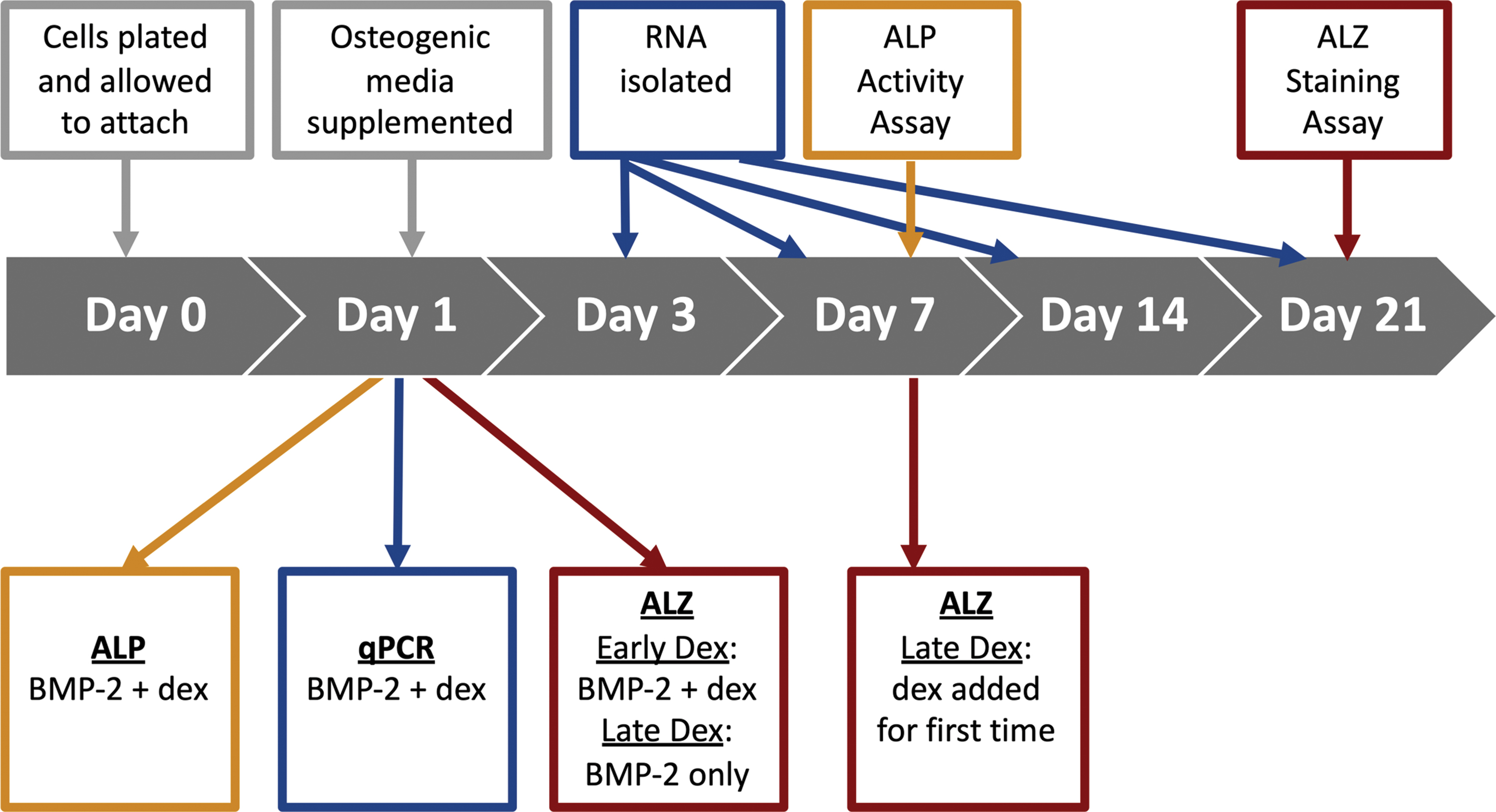
Overall experimental approach and design. On Day 0, passage 2 (autologous) and passage 21 (iPS-derived) cMSCs were seeded at 1 × 104 cells/cm2 and allowed to attach overnight. On Day 1, osteogenic media were supplemented. For ALP and qPCR assays, dexamethasone was supplemented beginning at this Day 1 timepoint. For ALZ assays, dexamethasone was supplemented beginning at this Day 1 timepoint only for the Early Dex treatment groups. On Day 7, dexamethasone was introduced for the first time to the Late Dex ALZ treatment groups. Media exchange for all treatment groups was performed twice weekly. RNA was isolated for qPCR at Days 3, 7, 14, and 21. All ALP assays were performed at Day 7. All ALZ assays were performed at Day 21. ALP, alkaline phosphatase; ALZ, Alizarin red stain; cMSCs, canine mesenchymal stromal cells; iPS, induced pluripotent stem cell; qPCR, quantitative polymerase chain reaction. Color images are available online.
Effect of dexamethasone and BMP-2 on late-stage osteogenesis
Detachment of high-density monolayers has been described in late-stage mineralization assays using cMSCs [8,11,12,15,17]. To avoid monolayer detachment, the periphery of each well of a 12-well plate was scored using a sterile Dremel bit (Dremel, Racine, WI) to create a circular etching [8]. Wells were coated with human fibroblast-derived fibronectin (Corning, Corning, NY) at 5 mg/mL in PBS for 30 min at 37°C [8]. Excess fibronectin solution was removed, and passage 2 autologous and passage 21 iPS-derived cMSCs were seeded at 1 × 104 cells/cm2 (n = 3 wells/condition). Cells were treated with either CCM or various osteogenic media. Dexamethasone was supplemented at 0, 1, 10, or 100 nM according to two groups: (1) Early Dex where dexamethasone was introduced beginning at Day 1 of the assay and (2) Late Dex where dexamethasone was introduced at Day 7 of the assay (Fig. 1). Media exchange was continued twice weekly. In addition, media were also supplemented with 0, 100, or 200 ng/mL rhBMP-2 at Day 1 [8,15,17,30]. At 21 days, cells were washed with PBS, fixed with 500 μL 10% neutral buffered formalin, and stained in 40 nM Alizarin Red stain (ALZ; Sigma) to identify calcium deposition within the monolayers. Wells were photographed, and ALZ was extracted using acetic acid for quantification through spectrophotometry [8,30].
Real-time quantitative polymerase chain reaction
Passage 2 autologous and passage 21 iPS-derived cMSCs were plated at 1 × 104 cells/cm2 in 12-well plates (n = 3 wells/condition) and treated with either control (CCM) or osteogenic media. Dexamethasone was supplemented at 0, 10, or 100 nM at Day 1 of the assay and continued twice weekly (Fig. 1). Media were also supplemented with 0 or 200 ng/mL rhBMP-2 at Day 1 [8,15,17,30]. At Days 3, 7, 14, and 21, total RNA was extracted using the High Pure RNA Isolation Kit (Roche Life Science, Penzberg, Germany). Samples were treated with DNase and quantified using a NanoDrop™ 1000 Spectrophotometer (Thermo Fisher). Complimentary DNA (cDNA) was synthesized from 125 ng RNA (normalized across all samples) using the SuperScript III RT Kit (Invitrogen) according to the manufacturer's instructions.
Canine quantitative polymerase chain reaction (qPCR) primers for known osteogenic genes were commercially prepared (Sigma) as outlined in Table 2. Based on previous work assessing optimal canine housekeeper genes for qPCR [47], RPL32 was selected as the housekeeping gene.
Real-Time Polymerase Chain Reaction Genes and Primer Sequences
PCRs (10 μL) were prepared with 5 μL SYBR Green Master Mix (Thermo Fisher), 0.5 μL of forward and reverse primer, 3 μL water, and 1 μL cDNA. Reactions were performed using a CFX96 Real-Time System (Bio-Rad, Hercules, CA) with an initial SYBR Green PCR Master Mix enzyme activation at 95°C for 10 min followed by 40 cycles of: denature at 95°C for 15 s, anneal/extend at 60°C for 1 min. A melt curve was performed immediately following qPCRs. Cycling conditions were 55°C for 30 s followed by 55°C for 5 s with a temperature increase of 0.5°C per cycle per second for 80 cycles.
Two independent cDNA preparations were used for qPCRs, assessing relative expression of the three osteogenic genes of interest with RPL32 as the housekeeping gene. Mean expression was determined between the two independent qPCRs. Threshold (CT) levels were normalized to the housekeeping gene and the control (CCM) condition for each time point to provide relative gene expression using the 2−ΔΔCt method [48].
Statistical analyses
Descriptive statistics was reported as mean ± standard deviation for all data. Analytical statistics was generated using one- or two-way analysis of variance with Tukey's or Sidak's post hoc tests. All statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software, La Jolla, CA). Significance was established at P ≤ 0.05.
Results
Effect of dexamethasone and BMP-2 on early-stage osteogenesis
As previously described, autologous marrow-derived cMSCs exhibited minimal ALP activity when cultured in osteogenic medium lacking BMP-2 [8,17] (Fig. 2a,b). Similarly, the iPS-derived cMSCs did not exhibit detectible ALP activity in response to traditional osteogenic medium (Fig. 2c). All three preparations of cMSCs exhibited a marked increase in ALP activity in response to increasing concentrations of BMP-2.
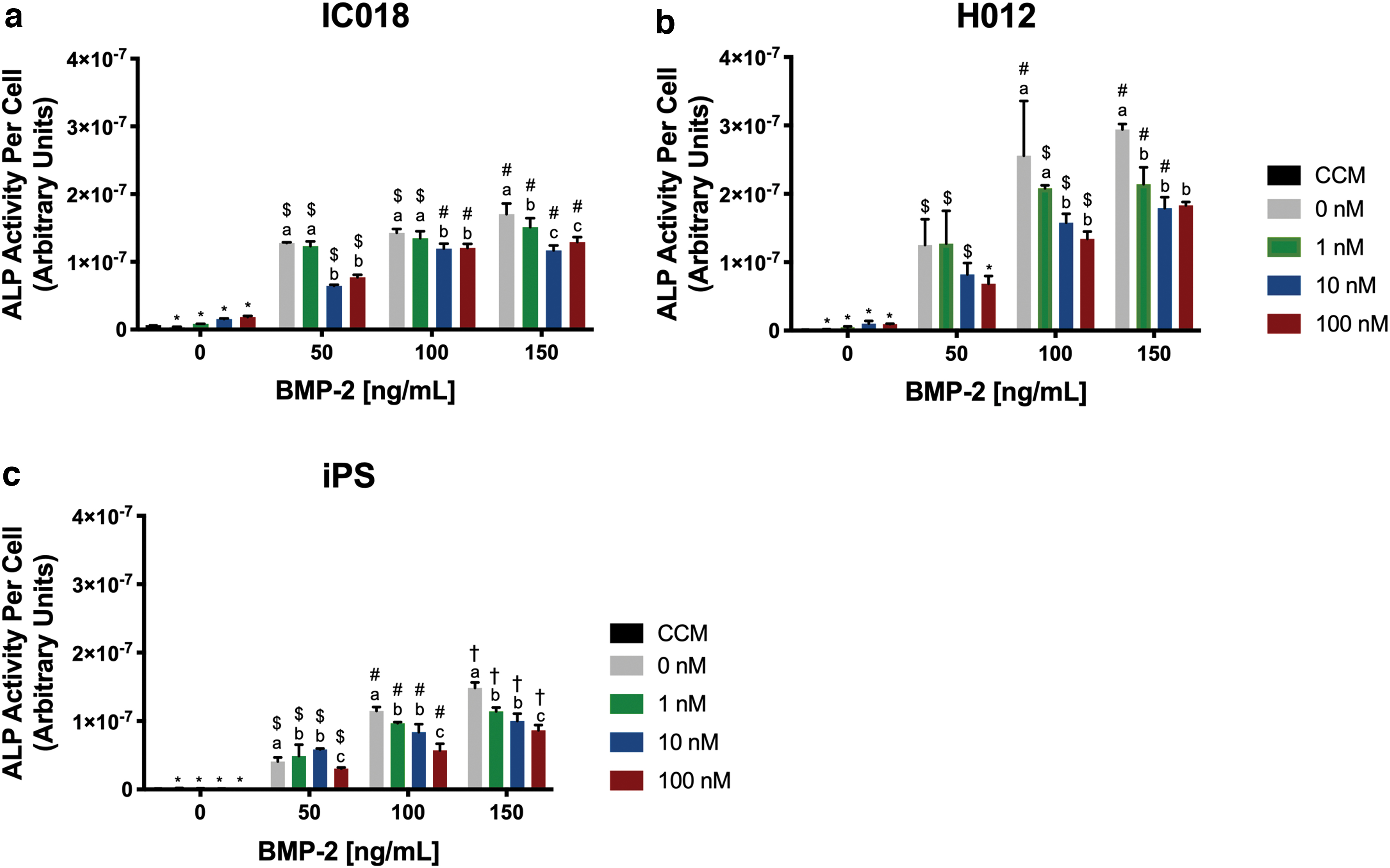
Early-stage osteogenesis of marrow-derived and iPS-derived cMSCs. Osteogenesis was evaluated using the ALP activity assay. Passage 2 (autologous) and passage 21 (iPS-derived) cMSCs were cultured in CCM or osteogenic media supplemented with varying concentrations of dexamethasone and rhBMP-2 (n = 3 wells/condition). At Day 7, ALP activity was determined by measuring the colorimetric conversion of PNPP to PNP and normalizing to cell number through DNA quantification. Statistical analysis was performed using two-way ANOVA and Tukey's post hoc test. Letters denote significant differences in ALP activity between different concentrations of dexamethasone within individual rhBMP-2 treatment groups (P < 0.05). Symbols denote significant differences in ALP activity between different concentrations of rhBMP-2 within individual dexamethasone treatment groups (P < 0.05).
In the absence of BMP-2, there was a slight increase in ALP activity when IC018 cells were cultured with dexamethasone at 10 and 100 nM; however, this increase was not significant (Fig. 2a). When BMP-2 was included in the osteogenic media, dexamethasone at these same concentrations (10 and 100 nM) significantly decreased ALP activity. Similar results were obtained for H012 (Fig. 2b). iPS-derived cMSCs exhibited lower overall ALP activity compared to the autologous cMSCs (Fig. 2c). We were unable to detect measurable ALP activity levels in iPS-derived cMSCs cultured in traditional osteogenic induction medium lacking BMP-2, regardless of the concentration of dexamethasone. Consistent with autologous cMSCs, ALP activity increased in response to BMP-2 although the BMP-2 response was more modest. Within each BMP-2 group, ALP activity significantly decreased in response to dexamethasone in a concentration-dependent manner. Collectively, these results demonstrate that both autologous and iPS-derived cMSCs require an additional agonist such as BMP-2 to exhibit consistent ALP activity and that dexamethasone is inhibitory to early-stage ALP activity.
Late-stage osteogenesis
In contrast to the early-stage osteogenesis results where autologous and iPS-derived cMSCs responded to dexamethasone similarly, dexamethasone led to markedly different results in late-stage osteogenesis assays. In the absence of dexamethasone, monolayers of both autologous cMSCs failed to mineralize, which is consistent for cMSCs in this assay (Figs. 3 and 4) [8,14,15]. Addition of dexamethasone at Day 7 is an accepted technique to generate monolayer mineralization in human and canine MSCs [8,29,30,49]. When high concentrations of dexamethasone (10 and 100 nM) were added to cultures at Day 7, mineralization was improved (Figs. 3a and 4a, bottom right panels). Addition of dexamethasone (10 and 100 nM) to osteogenic media at Day 1 led to the greatest monolayer mineralization for the autologous cMSC preparations (Figs. 3a and 4a, top right panels). In fact, mineralization of the historically marginal cMSC preparation H012 was improved to levels similar to IC018 by treatment with 100 nM dexamethasone (Day 1) and 200 ng/mL BMP-2. For the highly osteogenic IC018 cMSCs, supplying 10 nM dexamethasone at Day 1 with 200 ng/mL BMP-2 led to robust monolayer mineralization. However, simply increasing dexamethasone to 100 nM at Day 1 and providing one-half the concentration of BMP-2 (100 ng/mL BMP-2) resulted in monolayer mineralization equivalent to that produced by 200 ng/mL BMP-2. The late-stage osteogenesis results for the autologous cMSCs demonstrate that by modulating the concentration and temporal delivery of dexamethasone and BMP-2, weakly osteogenic cells may be stimulated to produce monolayer mineralization that approaches that of highly osteogenic cells [Fig. 3b (Day 1, 200 ng/mL BMP-2, 100 nM Dex) vs. Fig. 4b (Day 1, 100 ng/mL, 100 nM Dex)]. It is important to note that the effect of dexamethasone on the late-stage mineralization is in sharp contrast to early-stage osteogenesis, where high concentrations of dexamethasone inhibited ALP activity. For the autologous cMSCs, dexamethasone supplementation was required for robust late-stage osteogenesis.

Late-stage osteogenesis of IC018 marrow-derived cMSCs. Passage 2 cMSCs were cultured in CCM or osteogenic media supplemented with varying concentrations of dexamethasone and rhBMP-2 (n = 3 wells/condition). At Day 21, monolayers were formalin fixed, stained with ALZ, and photographed. Statistical analysis was performed using two-way ANOVA and Tukey's post hoc test with P < 0.05.

Late-stage osteogenesis of H012 marrow-derived cMSCs. Late-stage osteogenesis was performed as described.
The late-stage osteogenesis results for iPS-derived cMSCs were in stark contrast to the autologous cMSCs. The greatest monolayer mineralization for the iPS cMSCs was achieved at 200 ng/mL BMP-2 in the absence of dexamethasone (Fig. 5). Addition of dexamethasone to osteogenic media at either Day 1 or 7 inhibited monolayer mineralization in a concentration-dependent manner. Conditions that enhanced mineralization of autologous cMSCs (high concentrations of dexamethasone at Day 1) had the greatest inhibitory effect on the iPS-derived cMSCs. Quantification data support these observations and highlight the differential response of iPS-derived cMSCs to dexamethasone in the late-stage osteogenesis assay.
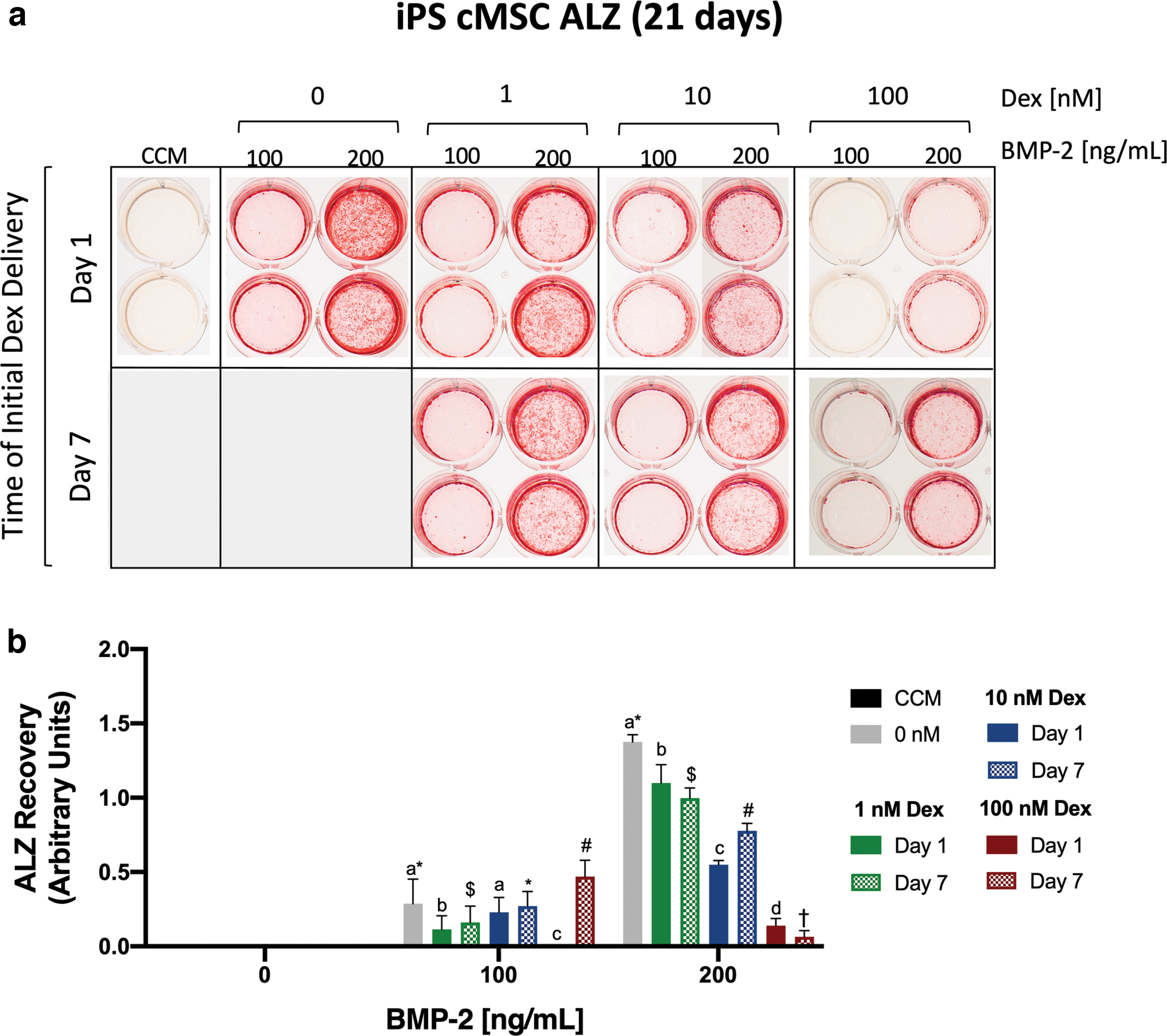
Late-stage osteogenesis of iPS-derived cMSCs. Late-stage osteogenesis was performed as described in Fig. 4.
Real-time quantitative PCR
In both autologous cell preparations, osteocalcin and osterix were markedly upregulated in response to BMP-2 at all time points (Fig. 6a,b), confirming the importance of BMP-2 in both early- and late-stage cMSC osteogenesis. There was a time-dependent increase in expression of these genes in the autologous cells, with higher fold changes appreciated at later time points. Supplementation of osteogenic media with dexamethasone led to a subtle dose-dependent increase in osteocalcin expression for both the IC018 and H012 cells. Increasing concentrations of dexamethasone resulted in increased osterix expression for the H012 cMSCs; however, the IC018 cells exhibited a dose-dependent decrease in osterix expression in response to dexamethasone (Fig. 6b). In both autologous cell preparations, Runx2 was upregulated in response to BMP-2 (Fig. 6c). However, the fold increase of Runx2 was more modest than osteocalcin or osterix. There was a similar time-dependent increase in Runx2 expression. While supplementation with dexamethasone did not appear to affect Runx2 expression in the IC018 cells, high concentrations of dexamethasone (100 nM) resulted in a significant increase in Runx2 expression at Day 21 for the H012 cells (Fig. 6c).
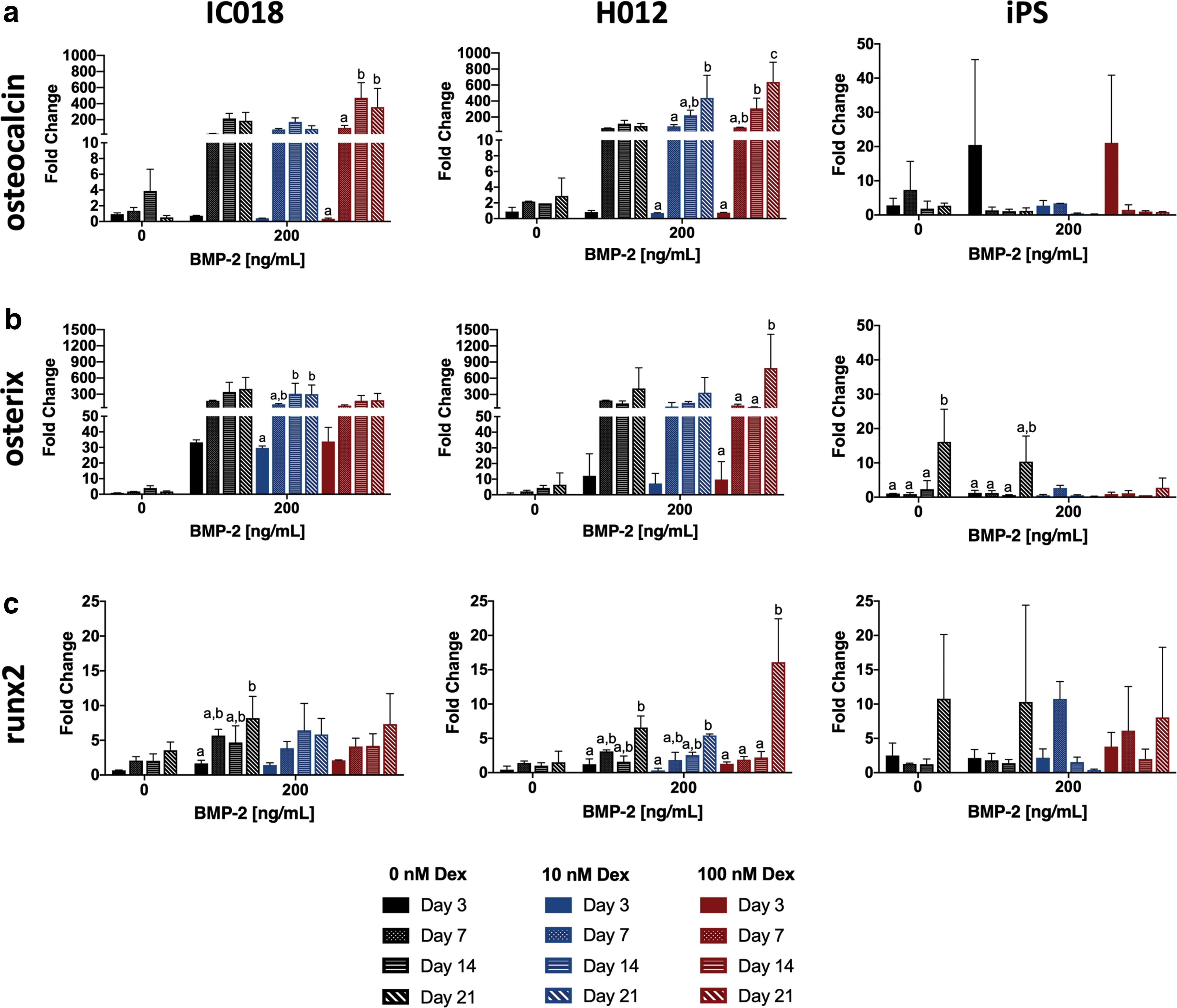
Quantitative assessment of gene transcription in bone marrow-derived and iPS-derived cMSCs. cMSCs were cultured as described in Fig. 1 for 3, 7, 14, and 21 days. At each time point total RNA was isolated and normalized to 125 ng to generate cDNA. Real-time qPCR was performed for three canine osteogenic genes of interest. All genes were normalized to a housekeeping gene (RPL32). Relative gene expression is provided for
In contrast to the autologous cells, the iPS-derived cMSCs exhibited more modest fold changes for all genes of interest. In the absence of BMP-2, cultures containing dexamethasone exhibited a modest upregulation of osteocalcin and osterix (Fig. 6a,b). However, in sharp contrast to the autologous cells, expression of these genes was markedly decreased at later time points and higher concentrations of dexamethasone (Fig. 6a,b). These findings are in support of the late-stage mineralization results in which inclusion of dexamethasone was inhibitory to monolayer mineralization for the iPS-derived cMSCs. Similar to the autologous cells, the iPS-derived cMSCs exhibited modest changes in Runx2 expression (Fig. 6c). Runx2 expression generally increased over time for the iPS cMSCs (with exception of 10 nM dexamethasone).
Discussion
MSCs have received considerable attention as bone regenerative agents due to their ease of procurement, ability to undergo differentiation and form de novo bone, and their immunomodulatory potential [8,46,50]. While the ideal source of MSCs for bone regeneration remains to be determined, an equally important barrier for MSC-based therapies is the variability in osteogenic performance of MSCs from different tissues and donors [8 –10,15,20,51,52]. Identifying methods to consistently maximize MSC osteogenic differentiation will likely facilitate development of cell-based orthopedic treatment strategies. One strategy to address MSC variability is the use of iPS-derived MSCs, as these cells and their expressed proteins represent a theoretically limitless supply of MSCs.
Although human and canine MSCs share more similarities than differences [11 –13], one important difference between the two species appears to be the poor response of cMSCs to traditional in vitro osteogenic induction media. A growing body of work has demonstrated that additional agonists are necessary for robust osteogenic differentiation of cMSCs [8,17 –19]. While a number of agonists have been evaluated, rhBMP-2 has received the most attention [8,17 –19,53]. For example, Volk et al. reported that the combination of ascorbate-2-phosphate and BMP-2 exhibited a synergistic effect to significantly increase ALP activity of marrow-derived cMSCs [17]. This finding was subsequently confirmed by Kamishina et al. and Bearden et al. Kamishina et al. demonstrated that canine marrow-derived MSCs failed to form mineralized nodules when cultured in osteogenic media lacking BMP-2 [53]. In an extensive characterization study, Bearden et al. evaluated 15 cMSC preparations from canine marrow, synovium, and adipose tissue. While there were differences between tissues of origin, all 15 cMSC preparations required BMP-2 for late-stage osteogenesis [8]. Thus, it appears that autologous cMSCs require additional agonists, such as BMP-2, to consistently undergo in vitro differentiation. The in vitro osteogenic ability of canine iPS-derived MSCs, as well as their need for BMP-2 or other agonists, is not well described.
Conditions that minimize reliance on BMP-2 while maintaining in vitro osteogenic abilities are of the utmost importance, as the use of rhBMP-2 for canine bone healing studies presents significant regulatory and translational challenges. Good manufacturing practice-quality BMP-2 is not widely available, carries considerable expense, and has been associated with adverse effects in vivo such as ectopic bone formation, osteolysis, and local inflammation [54 –56]. Moreover, use of BMP-2 in vivo in tandem with MSCs or MSC-derived proteins introduces substantial confounding variables that must be addressed by more complex experimental design and data analysis. In contrast to BMP-2, dexamethasone is widely available, inexpensive, and commonly used in clinical setting. Dexamethasone is known to promote osteogenic differentiation, although the exact mechanism of action remains unclear [35,38 –40]. For these reasons, the objective of this study was to assess the effect of both dexamethasone and BMP-2 on early- and late-stage osteogenesis in cMSCs. In addition, our goal was to comprehensively assess the in vitro differentiation of canine iPS-derived MSCs [21] and compare the osteogenic properties of iPS-derived cMSCs to autologous cMSCs.
In our early-stage osteogenic differentiation assays, increasing concentrations of dexamethasone reduced ALP activity at all BMP-2 concentrations in both autologous and iPS-derived cMSCs (Fig. 2). These results suggest that dexamethasone is inhibitory to ALP activity within cMSCs. Investigators using the ALP activity assay and either canine bone marrow MSCs or iPS-derived MSCs should avoid inclusion of dexamethasone in osteogenic induction media during early-stage osteogenesis. In sharp contrast to the early-stage assays, inclusion of higher concentrations of dexamethasone (as early as Day 1) in late-stage osteogenic differentiation assays led to increased monolayer mineralization for the autologous bone marrow-derived cMSCs (Figs. 3–5). Moreover, dexamethasone was required for successful late-stage monolayer mineralization of the two representative autologous cMSCs used in this study. Higher concentrations of dexamethasone supplemented at Day 1 dramatically increased monolayer mineralization. Importantly, the concentration of BMP-2 and the concentration and timing of delivery of dexamethasone could be modulated such that a marginally osteogenic preparation of cMSCs (H012) could be stimulated to mineralize at a level equivalent to an excellent preparation of cMSCs (IC018) (Figs. 3 and 4). The ability to improve the monolayer mineralization of poorly performing autologous cMSCs by modulating the concentration and temporal delivery of a widely available and inexpensive drug such as dexamethasone represents an important contribution to the cMSC field. The techniques described in the present study provide investigators with straightforward inexpensive methods to maximize late-stage osteogenic differentiation in marginally performing canine MSCs while simultaneously reducing the concentration of rhBMP-2 in canine osteogenic cultures. Importantly, dexamethasone was not required for early- or late-stage osteogenesis of iPS-derived cMSCs. In fact, dexamethasone was inhibitory to late-stage osteogenic differentiation when assessed by the monolayer mineralization assay and ALZ staining (Fig. 5). Investigators using iPS-derived cMSCs should avoid use of dexamethasone in early or late-stage osteogenic assays.
While the ALP activity and ALZ staining assays are classic assays for in vitro osteogenesis, they do not allow insight into transcriptional activity. For this reason, we performed qPCR over several time points (Days 3, 7, 14, 21) using a small panel of qPCR primers developed for cMSCs. Our qPCR results indicate that inclusion of BMP-2 in osteogenic media results in significantly increased osteocalcin and osterix expression in autologous cells (Fig. 6a,b) and that upregulation of these genes increases with time. These findings provide additional evidence to support the concept that cMSCs require additional agonists such as BMP-2 to undergo in vitro osteogenesis. Importantly, in the absence of BMP-2, there was minimal change in expression of osteocalcin, osterix, or Runx2. The qPCR results for iPS-derived cMSCs in the early-stage osteogenesis assay were much more unimpressive. Osteogenic genes exhibited modest changes in response to both dexamethasone and BMP-2 compared to the autologous cMSCs. One possible explanation for these results is altered transcriptional activity arising from (1) mutations present in the reprogrammed somatic cells, (2) reprogramming mutations during induction of the iPS state, or (3) passage-induced mutations secondary to prolonged in vitro culture and passage [57 –59].
From a mechanistic standpoint, it is reasonable to propose that experimental conditions maximizing early-stage osteogenesis would lead to maximal late-stage osteogenesis. Although our results suggest that this may be true for iPS-derived cMSCs, it appears that different BMP-2 containing osteogenic media are necessary to achieve robust early- versus late-stage osteogenesis of autologous marrow-derived cMSCs. It remains unclear why dexamethasone should be avoided for optimal short-term osteogenesis but included at high concentrations for optimal late-stage mineralization assays within these cell preparations. However, these results are consistent with previous studies that have demonstrated differential effects of dexamethasone on osteogenesis based on concentration, timing, and duration of supplementation [39,41 –43]. Rimando et al. reported that low concentrations of dexamethasone provided early in the assay promoted osteogenic differentiation of hMSCs, while prolonged exposure to high concentration of dexamethasone reduced monolayer mineralization and expression of late-stage osteogenesis markers (e.g., osteocalcin and osteopontin) [39]. Based on these data, Rimando et al. proposed a two-step protocol in which dexamethasone is supplemented early to enhance lineage commitment, followed by withdrawal at later stages for enhanced mineralization. In contrast, others have suggested that omitting dexamethasone in osteogenic medium during the first week but including dexamethasone during subsequent weeks of culture results in superior late-stage mineralization [30]. These findings are in contrast to what we have observed in the present study and highlight another prominent interspecies difference between the osteogenic performance of canine and hMSCs. We propose that this finding may be explained by differences in the interplay of the BMP-2 signaling cascade and a hormonal/glucocorticoid axis within each species. However, further work is necessary to fully elucidate these key differences.
While there is considerable variability in the endogenous osteogenic activity of autologous MSCs [8 –10,15,20,51,52], the results of the present study demonstrate that osteogenic performance of both poorly and strongly mineralizing cMSCs can be optimized by modulating the concentration and temporal delivery of dexamethasone and BMP-2 (Fig. 3 and 4). Previous work by Mendes et al. has demonstrated both BMP-2 and dexamethasone to be important bioactive factors for in vivo bone formation using hMSCs [60]. However, Mendes et al. evaluated a single concentration and timepoint of administration for these osteogenic induction agents. Additional canine studies are warranted to determine if the findings of the present study translate to the in vivo setting.
Although the present study demonstrates the ability to enhance osteogenic performance of autologous cMSCs, a notable limitation is that these cells provide a finite source for bone regenerative studies. As such, once autologous sources of cMSCs have been exhausted, new preparations must be isolated, characterized, and optimized. iPS-derived MSCs have emerged as a promising alternative due to their prolonged proliferative capacity, immunomodulatory properties, and ability to eliminate interdonor variability [21,26 –28]. Furthermore, as demonstrated in the present study, iPS-derived cMSCs are able to undergo early- and late-stage osteogenesis making them a potentially ideal cell source for bone regenerative studies using cells or cell-derived matrices. The results of the present study demonstrate that canine iPS-derived cMSCs are capable of undergoing consistent early- and late-stage mineralization in a BMP-2 dependent but dexamethasone independent manner. Additional in vitro and in vivo work is warranted to evaluate other preparations of iPS-cMSCs in both in vitro and in vivo settings.
As with all studies, ours was not without limitations. First, although both high and low performing autologous cMSC preparations were selected, this study was limited to evaluation of two preparations of bone marrow-derived cMSCs. While we could have evaluated cMSCs from additional donors and tissues, we preferred to perform a more in-depth analysis of representative marginal and exceptional autologous bone-marrow cells. Additional work is necessary to determine if our findings are universally present in all bone marrow-derived cMSCs and/or cMSCs isolated from other tissue sources. To our knowledge, only a handful of laboratories have generated iPS-derived cMSCs [21,24,25,61], and as such, only a single preparation of iPS-derived cMSCs were available for evaluation in this study. Given the significant interdonor variability in osteogenic performance of autologous MSCs, it is possible that there is also variability in performance of iPS-derived MSCs. Although only a single preparation of iPS-derived cMSCs was evaluated, previous work has demonstrated that these cells exhibit similar biological and immunomodulatory properties to traditional tissue-derived cMSCs [21].
Second, while the qPCR results presented in the present study provide a preliminary transcriptional confirmation of early and late-stage osteogenesis, additional work is necessary to gain a deeper understanding of the mechanistic pathways explaining the differential response of autologous cMSCs to dexamethasone in early- and late-stage osteogenesis. This study evaluated several important genes involved in osteogenic differentiation and osteogenesis but was by no means exhaustive. Evaluation of additional genes (e.g., Dlx5 and Msx2) and/or signaling pathways (e.g., canonical Wnt, noncanonical Wnt, and mitogen-activated protein kinase) may be warranted. Furthermore, use of more powerful technologies such as RNA sequencing may be of benefit to evaluate transcriptional changes during early- and late-stage osteogenesis in canine MSCs. These areas will be the focus of future studies in our laboratory.
Finally, as mentioned above, it is unknown if these in vitro findings translate to an in vivo setting. The results of the present study suggest that dexamethasone and BMP-2 can be modulated to enhance cMSC in vitro osteogenesis. Although such findings may be relevant in the clinical setting, in vivo studies are necessary to evaluate this hypothesis. In addition, in vivo studies are necessary to evaluate the osteogenic capacity of autologous versus iPS-derived cMSCs. Our laboratory has recently confirmed that in the murine calvarial defect model, inclusion of subtherapeutic concentrations of BMP-2 with autologous bone marrow cMSCs leads to improved defect healing compared to defects treated with cMSCs alone (article in preparation).
In conclusion, we demonstrated that the in vitro osteogenic performance of autologous and iPS-derived cMSCs can be enhanced by modulating the concentration and time-of-delivery of dexamethasone and BMP-2. Based on these results, we suggest that not only should BMP-2 (or other agonists) be included in osteogenic media, but that osteogenic media be tailored to individual cell preparations to maximize osteogenic performance. When using early-stage assays such as ALP activity, dexamethasone should be omitted from culture media. However, if late-stage mineralization assays are required, inclusion of dexamethasone as early as Day 1 and at a concentration as high as 100 nM will result in robust mineralization of autologous marrow-derived cMSCs. If working with iPS-derived MSCs in late-stage mineralization assays, dexamethasone should be avoided. iPS-derived cMSCs hold much promise for bone tissue engineering, but their response to dexamethasone in late-stage osteogenesis is in sharp contrast to that of autologous cMSCs. Further work will be necessary to characterize inherent differences between autologous and iPS-derived cMSCs. This work is significant in that it provides insight into important similarities and differences between autologous and iPS-derived cMSCs and will facilitate in vitro osteogenic assays for investigators unfamiliar with the nuances of cMSCs. This work may prove useful for translational studies evaluating osteogenic differentiation of iPS-derived MSCs derived from other species.
Footnotes
Acknowledgment
The authors thank and acknowledge Dr. Gus Wright at the Texas A&M University Flow Cytometry Facility for his assistance with flow cytometry methods, data analysis, and figure creation.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Funding for this work was provided by the American Kennel Club-Canine Health Foundation (AKC-CHF) GCHP Hill Country's Let's Get Ready to Rumble Clinician-Scientist Fellowship, an NIH T32 Training grant (5T32OD011083-09 [S.B.G.]), and the Bone & Joint Fund, Texas A&M Foundation, College Station, TX, USA.
Supplementary Material
Supplementary Data
Supplementary Table S1
Supplementary Table S2
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.