
Abstract
Thus far, there are more than known 150 modifications to RNA, in which common internal modifications of mRNA include N6-methyladenosine (m6A), N1-methyladenosine, and 5-methylcytosine. Among them, m6A RNA modification is one of the highest abundance modifications in eukaryotes, regulating mechanisms controlling gene expression at the post-transcription level. As an invertible and dynamic epigenetic marker, m6A base modification influences almost all vital biological processes, cellular components, and molecular functions. Once the m6A modification process is abnormal, a series of diseases—including cancer, neurological diseases, and growth disorders—will be caused. Besides, several base modification activities also have been created by noncoding RNAs (ncRNAs), for instance, microRNAs, and circular RNAs, long ncRNAs, which were dynamically regulated during bone and cartilage pathophysiology processes. Therefore, it has now been clear that dynamic modification on coding RNAs and ncRNAs represents a completely new way to modulate genetic information. In this review, we highlight up-to-date progress and applications of m6A RNA modification in bone and cartilage pathophysiology, and we discuss the pathological roles and underlying molecular mechanism of m6A modifications in osteoarthritis and osteoporosis and osteosarcoma pathogenesis.
Introduction
RNA N6-methyladenosine methylation
Epigenetics refers to the reversible and heritable change in gene expression without changing the genetic sequence of DNA [1]. According to the central dogma, genetic information flows from DNA to proteins via RNA, and it regulates various biological processes. N6-methyladenosine (m6A) is a dynamic methylation at the N6 site of adenosine. Although the existence of m6A was discovered in RNA as early as the 1970s [2 –4], the exact mechanism of this field is still unknown for several decades. However, with the development of high-throughput sequencing technology and epigenomics, accurate mapping of transcriptome m6A distribution map had come true [5,6].
M6A has entered the stage of rapid research in recent years. As the most abundant internal modification of mRNA in eukaryotic cells, the per-mRNA molecule has 3.0–5.0 m6A residues on average, whose sites are enriched in the coding sequence and 3′untranslated region (3′UTR), couple with a particularly high enrichment near the stop codon area [5,7]. Such distinctive m6A distribution demonstrates that it is involved in downstream molecule functions, regulating various stages of mRNA metabolism. In 2011, the fat mass and obesity-associated protein (FTO), a member of the Alkb protein family, has been discovered as the first m6A demethylase [8,9]. The identification of FTO demonstrated that m6A is not merely a passive RNA base modification, but also a modification that can be actively regulated by the cell.
“Writers,” “erasers,” and “readers” proteins of m6A methylation
As the first known reversible mRNA modification, m6A is respectively, dynamically regulated by dedicated methyltransferase (writers), demethylase (erasers), and binding protein (reader) [10]. The m6A deposition on nuclear mRNA inside eukaryotic cells is executed co- or post-transcriptionally by a multicomponent m6A methyltransferase complex known as writers, which is composed of the catalytic subunit methyltransferase-like 3 (METTL3); cofactors such as METTL14 methyltransferase-like 14 (METTL14), WTAP, RBM15 [11], METTL16 [12], KIAA1429 [13], and ZC3H13 [14]. The core of m6A writers are Mettl3 and Mettl14, forming a stable heterodimer complex in a 1:1 ratio combined with CCCH motifs that constitute the minimally required regions for creating m6A modifications [15]. Mettl3 is a sole catalytic enzyme that binds to the methyl donor S-adenosyl-methionine or S-adenosylhomocysteine in the catalytic site, whereas Mettl14 plays a structural role that is critical for recognizing substrate RNAs and stabilizing METTL3 conformation [16]. Though WTAP is incapable of methylation activity, it interacts with the METTL3–14 heterodimer and significantly affects the deposition of m6A in cells [17]. Besides, RBM15/RBM15B is reported to recruit this complex to certain mRNA and long noncoding RNA (lncRNA) XIST to promote m6A formation [11]; KIAAI1429 and ZC3H13 are identified to regulate m6A methylation and play distinct functions in different cells [13,14].
The m6A deposition could also be removed by α-ketoglutarate (and)-dependent and Fe(II)-dependent demethylases, typically the FTO and alkB homolog 5 (ALKBH5) termed erasers, sequentially suggesting the m6A methylation that regulates the flow of genetic information is dynamically reversible [9,18]. Unlike FTO, ALKBH5 appears to be an m6A-specific demethylase, which could directly abrogate m6A modification to adenosine [18].
Diverse post-transcriptional fates of m6A-methylation rely on its specific binding proteins. The RNA m6A-binding proteins mainly include YT521-B homology (YTH) domain family proteins (YTHDF1–3), YTH domain-containing proteins (YTHDC1–2), IGF2BP1–3, HNRNPC/G/A2B1, and eIF3, regulating pre-mRNA splicing, mRNA transcription, mature, nuclear export, localization, translation, and stability. YTHDC1 regulates mRNA nuclear export and alternative splicing [19,20]. YTHDF1 selectively recognizes the 3′UTRs and stop codon of m6A-containing RNA and promotes translation initiation and protein synthesis by interacting with eIF3 [21]. YTHDF2 could recognize m6A mRNA within the GACU/A and it recruits the CCR4-NOT deadenylase complex to induce deadenylation and degradation of the transcripts [22,23]. YTHDF3 seems to serve as a co-factor to enhance the translation effects of YTHDF1 and accelerate the degradation of YHDF2 mediated on M6A-containing mRNAs [24,25]. Moreover, HNRNPC could change mRNA local structure, or HNRNPA2/B1 involves precursor miRNA (pre-miRNA) transcription to affect the abundance as well as alternative splicing of target mRNAs [26,27]. IGF2BPs enhance mRNA stability and storage (Fig. 1) [28].
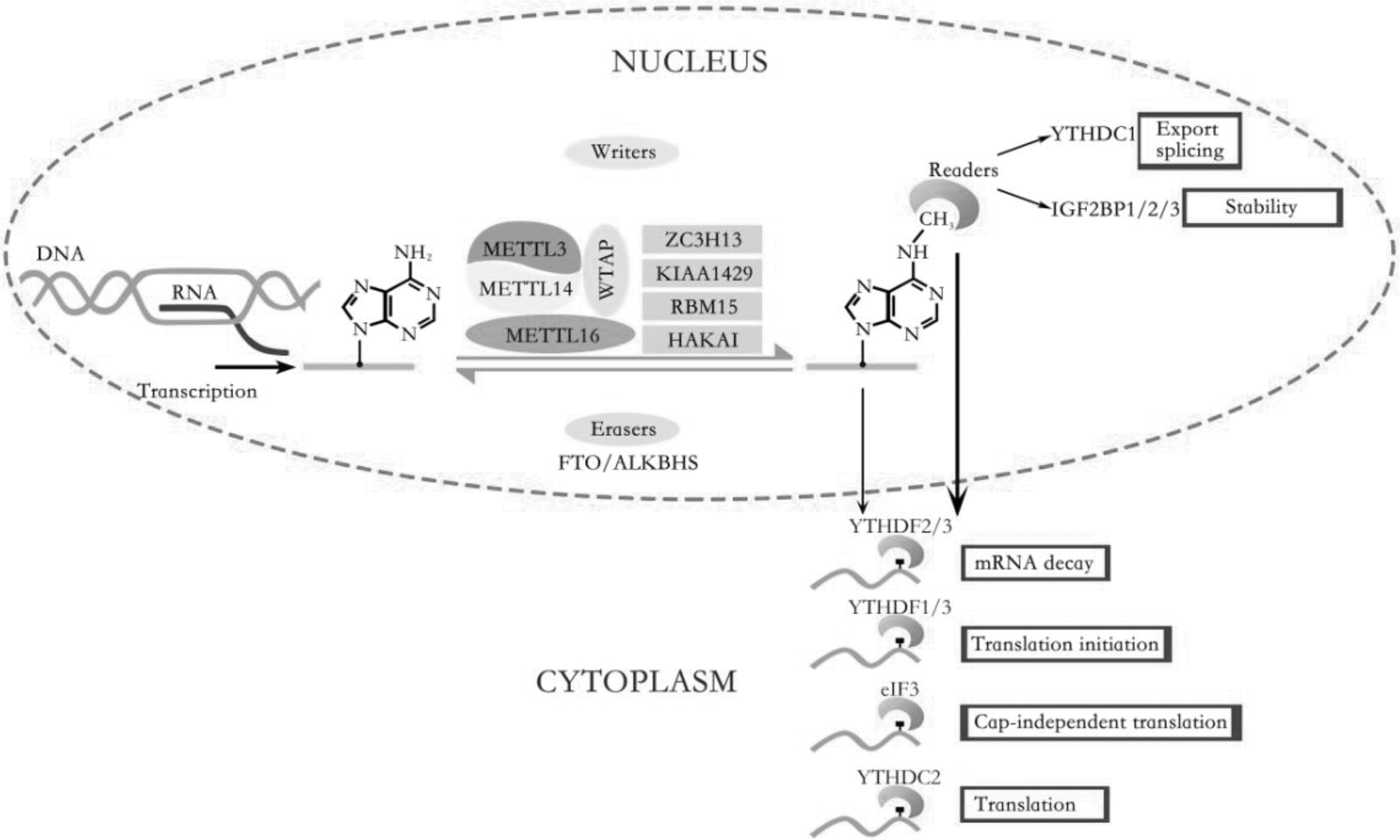
The dynamic regulation and function of RNA m6A modification: The m6A RNA methylation is composed of installation by writers, removal by erasers, and identification performed by readers. The adenosine (A) bases located in mRNA could be methylated to form m6A by the “writer.” The eraser is the demethylases FTO and ALKBH5, which catalyzed the oxidative demethylation from methylated adenosine. m6A could be recognized by m6A binding proteins (readers), including YTHDF1–3, YTHDC1–2, IGF2BP1–3, and eIF3. m6A modification regulates the life cycle of RNA metabolism, including pre-mRNA splicing, mRNA transcription, mature, nuclear export, localization, translation, and stability. ALKBH5, alkB homolog 5; FTO, fat mass and obesity-associated protein; m6A, N6-methyladenosine; YTHDF, YT521-B homology (YTH) domain family proteins.
Maintaining the dynamic balance of writers and erasers in cells and tissues is essential for normal biological processes and molecular functions, such as the proliferation and differentiation of bone. The development of certain diseases, for instance, osteoporosis, is usually associated with abnormal changes of m6A in vital biological functions.
m6A methylation and noncoding RNAs
Recent studies have shown that several noncoding RNAs (ncRNAs) also accept m6A modification [29]. ncRNAs is a kind of endogenous RNA molecules that are transcribed from the genome with no open reading frame and protein translation ability, but they have crucial biological functions in gene expression regulation. Based on the size of the transcript, ncRNAs consist roughly of lncRNAs with length exceeding 200 nt and small ncRNAs <200 nt, such as microRNAs (miRNAs) and circular RNAs (circRNAs) [30].
For LncRNAs, on the one hand, m6A modifications might interact with m6A regulators to facilitate their function. On the other hand, m6A modifications also regulate RNA–protein interactions by RNA structural switches. By enriching lncRNA MALAT1, a highly conservative lncRNA located in chromosome 11q13.1, from the total poly-A+RNA, Liu et al. have identified m6A sites on their hairpin-stem [26,29]. The m6A modification could alter the MALAT1 local structure and enhance the accessibility of its base-paired residues or nearby regions to modulate protein binding. Such a mechanism that regulates RNA–protein interactions via m6A-dependent RNA structural switches is known as “m6A-switch” [26]. X chromosome inactivation in mammals is regulated by the lncRNA XIST, which inhibits the chromosome from which it is transcribed. A latest study reported that RBM15 binding to the Xist A-repeat recruits the m6A complex METTL3/14 to the 5′end Xist m6A region and that this region is closely associated with Xist-mediated chromosome silencing [11]. miRNA is mainly involved in the regulation of post-transcriptional gene expression. The first step of miRNAs after DNA transcription is the forming of primary RNA (pri-miRNAs), in which the recognition of DGCR8 plays a significant role, and then is a series of cleavages to generate hairpin precursor, pre-miRNAs, and mature miRNAs [31]. Although pre-miRNA and mature miRNAs have little GGAC, the motif GGAC is highly abundant for pri-miRNA. Further, the pri-miRNAs with METTL3-mediated m6A modification could be bound by hnRNPA2B1, which interacts with DGCR8 and promotes the initiation of miRNA biogenesis [32]. Compared with m6A of linear RNAs that are enriched near 3′UTRs and near stop codons, m6A is enriched around the translation start site in circRNAs [33,34]. Although m6A reader YTHDF2 seems to fail to promote the degradation of circRNAs, it plays a role in regulating the stability of mRNA and the immunity of circRNA [35].
The Role of m6A in Bone Development
The development of bone is regulated by transcription factors, cytokines, multiple signaling pathways, and epigenetics [36,37]. Bone marrow mesenchymal stem cells (BMSCs) are multipotent stem cells that are extensively applied in tissue engineering stem cell transplantation and gene therapy, which could differentiate into and form different types of tissues, including osteoblasts, chondrocytes, and adipocytes both in vitro and in vivo [38,39].
In recent years, how to regulate the differentiation of BMSCs into desired cells at the post-transcriptional level is a promising perspective. Therefore, numerous researches have reported the function of m6A methylation in the MSCs differentiation. For example, in 2018, Mo et al. found that m6A-single nucleotide polymorphisms (SNPs) have a potential relationship and mechanism with osteogenic differentiation through genome-wide identification [40]. Likewise, Yan et al. found that METTL3-based m6A modification enhances osteogenic differentiation of BMSCs through m6A-based direct and indirect regulation of RUNX2, and aberrant downregulation of METTL3 may be one of the mechanisms of osteoporosis phenotype in patients and mice [41]. On the other hand, Shen et al. found that the expression of FTO increased in the process of adipose differentiation of BMSCs, and it decreased during osteogenesis differentiation [42]. The absence of METTL3 in porcine BMSCs could promote adipogenesis and JAK1 protein expression in an m6A-dependent way [43]. In addition, m6A could also modify the ncRNA, which changes the state of the methylation of the gene of the human bone or cartilage. These would be discussed in more detail in later sections (Table 1, Figs. 2 and 3).
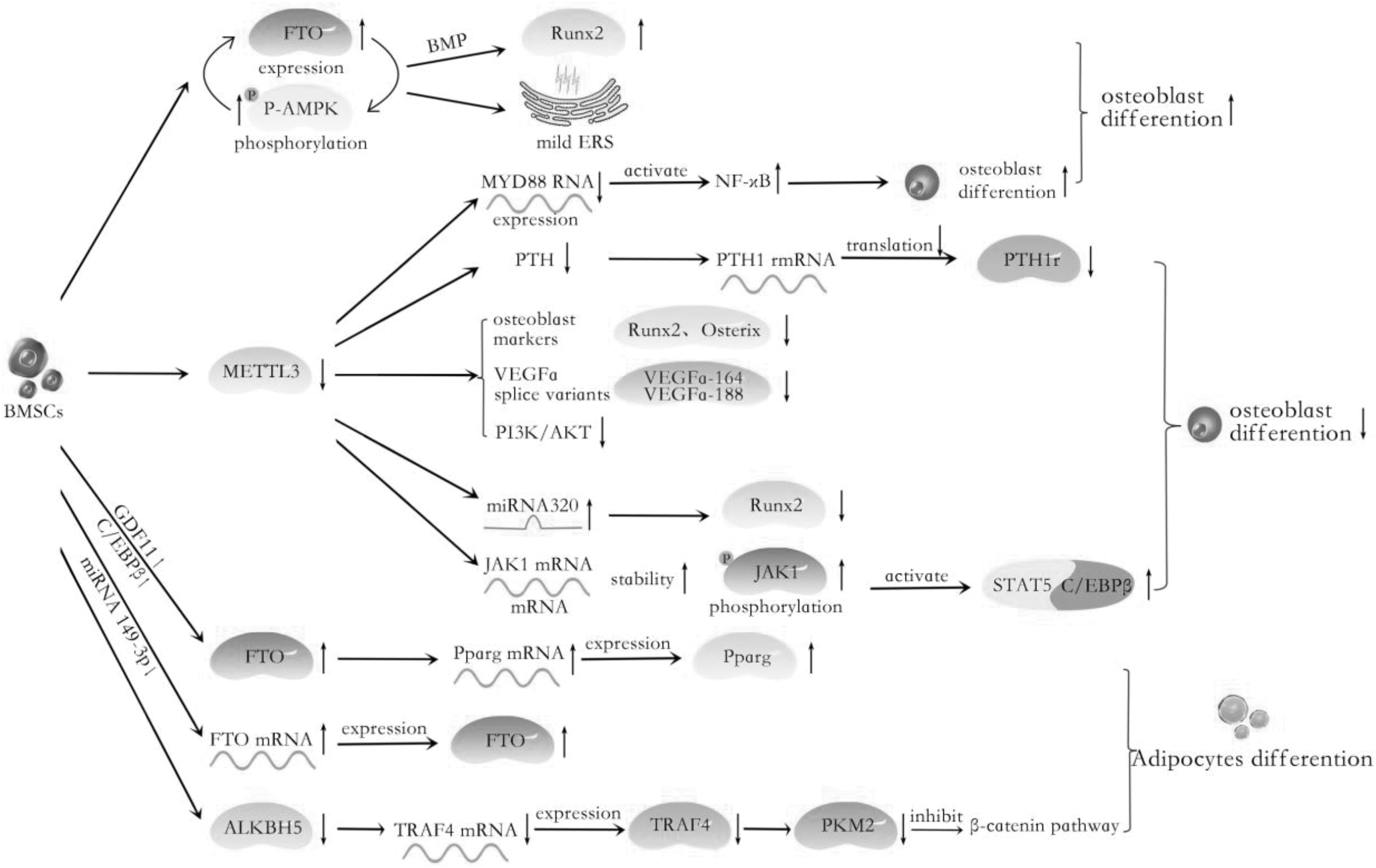
The role of m6A regulators in BMSCs adipogenesis and osteogenesis: The model in which METTL3 is decreased activates the JAK1/STAT5/C/EBPβ pathway and MiRNA320 level; inhibits the PTH/Pth1r signaling axis and the expression of RUNX2, Osterix, and Vegfa. GDF11-C/EBP and MiRNA149-3P target FTO, resulting in an increase in its expression and thereby promoting adipogenic differentiation. ALKBH5 is involved in adipogenic differentiation by regulating the mRNA expression of TRAF4. The FTO activates osteogenic differentiation by inducing mild ER stress and FTO and p-AMPK positive feedback loop. In MenSCs, METTL3 facilitates MYD88-RNA m6A methylation modification, inducing the activation of NF-κB, suppressing osteogenesis. BMSC, bone marrow mesenchymal stem cell; METTL3, methyltransferase-like 3.
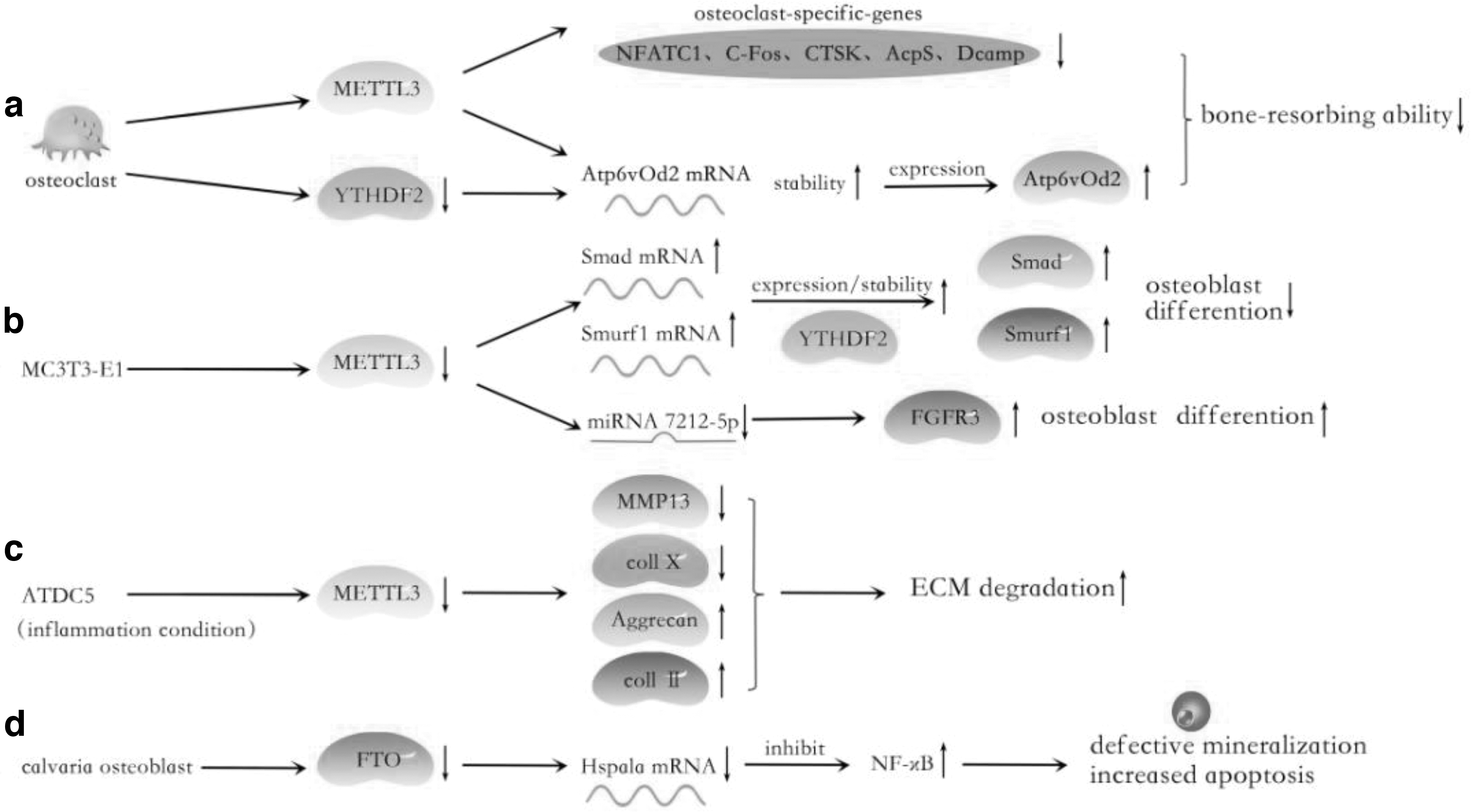
The role of m6A regulators in osteoclast, MC3T3-E1, ATDC5, and calvaria osteoblast:
Roles of N6-Methyladenosine Key Regulator in Bone Development and Disease
ALKBH5, alkB homolog 5; BMMS, bone marrow derived macrophage; BMSC, bone marrow mesenchymal stem cell; FTO, fat mass and obesity-associated protein; m6A, N6-methyladenosine; METTL3, methyltransferase-like 3; OA, osteoarthritis; pBMSC, pigs bone marrow mesenchymal stem cells; YTHDF2, YT521-B homology (YTH) domain family protein 2.
The role of m6A in osteogenic differentiation
The positive regulation of m6A in osteogenesis
In 2018, Wu et al. confirmed that Mettl3-mediated m6A RNA methylation regulates the osteogenesis differentiation fate of BMSCs through regulating the translation efficiency of MSCs lineage allocator Pth1r [44]. When treated with knockout Mettl3, the PTH-induced osteogenic effects were blunted and the translation efficiency of Pth1r mRNA-Pth1r, a critical regulator of lineage allocation in MSCs and osteoblast precursors, was decreased [44]. Moreover, PTH also depresses adipocytic differentiation to increase bone formation. Similarly, another research confirmed that through RNA sequencing that after the knockout of the METTL3 gene, a large number of affected genes were related to osteogenic differentiation and bone mineralization, such as Runx2, Osterix, and ALP. METTL3 affects the osteogenic differentiation of BMSCs by regulating the PI3K-Akt signaling pathway and the expression level of VEGF. Mettl3 inhibition decreased the expression level of Akt phosphorylation, Vegfa, and its selective shear during the osteogenic differentiation of BMSCs [45].
Yan et al. found that among the METTL3 target genes in BMSCs used an m6A RNA immunoprecipitation microarray, the m6A methylation level of pre-Mir-320 significantly changed, and its methylation was significantly reduced after METTL3 silencing. Moreover, they further demonstrated that RUNX2 is a target gene for miR-320, and METTL3/m6A implements the osteogenesis through upregulating expression levels of RUNX2 through dual mechanisms: (1) METTL3/m6A upregulates the RUNX2 level by inhibiting pre-miR-320 and miR-320; (2) METTL3/m6A promotes direct methylation of RUNX2 mRNA and enhances its expression level and cellular stability. Intriguingly, pre-miRNAs could be methylated in the nucleus, causing reduced biogenesis of mature miRNAs [41]. Notably, miRNAs have been reported to play crucial roles in the fine regulation balance of biological processes. For example, the expression changes of MiR-188, MiR-23a/B, and MiR-320 could regulate the expression of osteogenic genes in BMSCs, in which how m6A plays a role in this change might also be an interesting topic in further studies. A bold guess here is that the same gene may have multiple m6A-mediated pathways for regulation in many cases. Besides, METTL3-YTHDF2-mediated m6A methylation is involved in osteogenic differentiation not only under physiological conditions but also under inflammatory conditions. When METTL3 of MC3T3-E1 is knocked out in inflammatory conditions, YTHDF2 participates in stabilizing Smad7 and Smurf1 mRNA transcripts, resulting in Smad-dependent signaling blockage and osteoblast differentiation decrease [46].
The negative regulation of m6A in osteogenesis
There is, however, a different sound. Yu et al. found that METTL3 positively regulates expression of MYD88, a diffusely accepted regulator of NF-κB, subsequently activating the NF-κB signaling, which is regarded as an inhibitor of osteogenesis and therefore negatively regulates osteogenesis. Merely, this tendency could be reversed by the demethylase ALKBH5 [47]. In a separate study, Mi et al. also demonstrated that METTL3 suppresses osteogenic processes both in vitro and in vivo, and this effect is achieved through METTL3/miR-7212-5p/FGFR3 axis. Specifically, the increased level of miR-7212-5p was mediated by METTL3, and miR-7212-5p suppressed osteoblast differentiation in MC3T3-E1 cells by targeting FGFR3 [48]. Moreover, Son et al. also confirmed for the first time that an underlying mechanism FTO activates osteogenic differentiation by inducing mild ER stress. In C3H10T1/2 cells, p-AMPK upregulated the expression of FTO, FTO induced phosphorylation of AMPK, and a positive feedback loop existed between FTO and p-AMPK. Mechanistically, m6A demethylase FTO regulates demethylation in promoters of ER stress genes to stimulate ER stress and then promotes osteogenic differentiation [49].
The dual mechanism of m6A modification on osteogenic differentiation
Just as METTL3 overexpression promotes gastric cancer progression; down the expression of METTL3 induces endometrial cancer, so differences in m6A methylation levels may lead to opposite results [50,51]. In the studies just cited, mettL3-mediated methylation of m6A has markedly different effects on the regulation of osteogenic differentiation. This difference may be caused by the high or low m6A modification abundance. Mettl3-meditated m6A methylation of different genes or different abundance of methylation of the same gene may lead to different results. Perhaps, we might be tempted to assume that m6A promotes or inhibits osteogenic gene expression by regulating different signaling pathways and ncRNAs. Such a dual mechanism of m6A level on osteogenic differentiation remained to be further investigated.
The role of m6A in chondrogenic differentiation
In the monolayer culture of primary chondrocytes, chondrocytes undergo dedifferentiation and lose their phenotype and the capacity to form cartilage. Based on the microarray analysis, Ma et al. found that in vitro expanded human chondrocytes showed an overall decrease in total gene expression. Partly, DNA methylation results in the expression of downregulation of many genes [52]. Chondrocyte hypertrophic differentiation is also accompanied by changes in DNA methylation in vitro by the integration of RNA-Seq and ERRBS datasets. In addition, in the inflammatory condition, the levels of METTL3 mRNA and m6A methylated were upregulated in IL-1b-treated ATDC5 cells in a dose-dependent manner. Interestingly, only MELLT3 expression was increased, but the expression of other regulators was not significantly affected. When METTL3 was silenced, ECM accelerated degradation by decreasing the expression of MMP-13 and Coll X, upregulating the expression of Aggrecan and Coll II. This suggests the significance of METLL3 in mediating secretion of inflammatory factors, collagen synthesis and degradation in chondrocytes, and the possibility of METTL3 as a therapeutic target of osteoarthritis (OA) (Fig. 3) [53].
The role of m6A in adipogenic differentiation
Just like the idea that “bone loss is fat gain,” the opposite of osteogenesis is adipogenesis [54]. It has been reported that FTO, a gene contributed to human obesity, regulated adipogenesis via controlling m6A levels and mRNA splicing. Zhao et al. demonstrated that m6A sites show a spatial overlap with SRSF1- and SRSF2-binding clusters, and then the m6A modification may be regarded as a novel exonic cis-regulatory signal for RNA splicing. The FTO via regulating m6A levels and hence SRSF2 binding at splice sites controls alternative splicing of RUNX1T1, which modulates preadipocyte differentiation [9]. Likewise, a study by Merkestein et al. revealed that FTO promotes adipogenesis by regulating mitotic clonal expansion through RUNX1T1 in a demethylation-dependent manner [55]. MiRNAs have had a great effect on regulating the differentiation of BMSCs into a particular lineage. MiR-149-3p via directly targeting FTO regulated the alternative lineages of BMSCs into adipocytes and osteoblasts, whose expression in BMSCs tended to be decreased during adipogenic differentiation, whereas it increased during osteogenic differentiation [56]. Besides FTO, ALKBH5-mediated RNA demethylation is also involved in fine-tuning the balance between adipogenic and osteogenic differentiation by regulating the mRNA expression of TRAF4. Mechanistically, TRAF4, as a classical ubiquitin enzyme binds, binds with PKM2 through the RING domain and N- and C-terminals on PKM2 to activate the kinase activity of PKM2, and it subsequently inhibits adipogenesis by activating the β-catenin signaling [57].
In 3T3L1 cells, Zfp217, a classical transcription factor for stem cell differentiation, binds directly to the promoter of FTO, and it interacts with YTHDF2, enhances the accessibility of FTO to m6A target RNAs and demethylase activity [58]. This finding indicated that Zfp217 could coordinate transcriptional and post-transcriptional regulation in an m6A-YTHDF2-dependent manner and ultimately promote adipogenic differentiation. In pig preadipocytes, FAM134B promotes fat deposition by upregulating PPARγ and C/EBPβ level function through the whole adipogenic differentiation progress, whose expression was distinctly regulated by its m6A abundance [59]. On the other hand, FTO promotes adipogenic differentiation via regulating the downstream signaling pathway m6A level. METTL3-YTHDF2 mediated m6A methylation, and it suppressed the porcine BMSC adipogenesis process by targeting the JAK1/STAT5/C/EBPβ pathway [43]. FTO regulates the fate of BMSCs through the GDF11-FTO-PPAR axis, inhibits the osteogenic process, and promotes the transfer of MSC lineage to adipocytes [42]. Zhang et al. showed that FTO is essential for osteoblast survival and differentiation and it protects osteoblasts from genotoxic damage through the Hspa1a–NF-κB signaling pathway by building both global and osteoblast-specific FTO KO models [60].
The role of m6A in osteoclast differentiation
Osteoclasts are large multinucleated cells that are derived from hematopoietic precursor cells [61]. As the sole bone-resorbing cells, osteoclast plays a crucial role in maintaining bone homeostasis and preserving skeletal integrity. de la Rica et al. revealed that during osteoclast differentiation and fusion, hypomethylation and hypermethylation occur in key functional pathways and genes through transcription factor binding motif analysis [62]. The total m6A levels and METTL3 but not FTO or ALKBH5 expression increased on osteoclast induction. When Mettl3 was knocked out, the size was increased but the bone-resorbing ability of osteoclasts was decreased. Mechanistically, Mettl3 knockdown inhibited the expression of osteoclast genes (Nfatc1, c-Fos, Ctsk, Acp5, and Dcstamp) and inactivated the phosphorylation levels of RANKL-induced MAPK, NF-κB, and PI3K-AKT signaling pathways. On the contrary, the expression of Atp6v0d2, which is a fusion-associated gene, accelerated cell–cell fusion and the multinucleation process, due to the depletion of Mettl3 and YTHDF2, elevated the Atp6v0d2 mRNA stability [63]. These findings elucidate the molecular basis of RNA epigenetic regulation in osteoclast development (Fig. 3).
The role of m6A in BMSCs
Gene expression is closely controlled at different layers, including transcription, post-transcription, translation, and post-translation. The m6A methylation has been found on some transcripts in BMSCs, particularly those encoding developmental regulators, which regulated the differentiation of BMSCs into a particular lineage. There are several cross-talks between m6A modification and downstream signal pathways regulating adipo-osteogenic differentiation, such as the PTH/PTH1r pathway [45], PI3K/AKT pathway [44], and wnt/β-catenin pathway [57], which broadens the multilayer regulation of stem cell differentiation. On the other hand, m6A regulators (“writers,” “erasers,” and “reader”) are also regulated by ncRNAs, for instance, miRNA320, miRNA149-3p, and miRNA7212-5p; such upstream miRNAs might also be interesting candidates in subsequent studies. This epigenetic mechanism could determine the fate of BMSCs and it provides new insights into the post-transcriptional level gene regulation network in stem cell biology. However, the clinical application efficiency of BMSCs is limited because of the possibility of differentiation into undesired tissues. Therefore, to successfully utilize MSCs to generate mesodermal lineages for therapies of bone disease via m6A regulation, it is crucial to understand the molecular mechanisms of their differentiation into specific terminal lineages (Fig. 2).
Bone and Cartilage Disease
m6A in OA
OA is the most prevalent degenerative joint disorder, and it is characterized by progressive degeneration of articular cartilage, subchondral bone sclerosis, joint pain stiffness, synovial inflammation, and osteophyte formation [66]. For epigenetics, the expression levels of several epigenetic factors are remarkably altered in OA, thus suggesting a potential regulatory relationship between OA and epigenetics. In 2017 Steinberg et al. reported that there is remarkably differential methylation among transcription factors in normal and OA knee articular cartilage. Concretely, compared with normal articular cartilage, DNA methylation increases and the expression of transcription factors reduces in human OA cartilage. Another study confirmed by integration that the epigenomic, transcriptome, and proteomic levels of OA chondrocytes, AQP1, COL1A1, and CLEC3B were significantly differentially expressed at the omics level [67]. Moreover, more than 90% of differentially expressed genes and differentially methylated genes changed in the same direction. A study by Zhao et al. revealed that TRAF1, CTGF, and CX3CL1 genes were hypomethylated in OA chondrocytes and expressed in line with mRNA through genome-wide methylation analysis of articular chondrocytes [68]. The alterations in methylation in key transcription factors may represent a mechanism that can explain changes in the transcriptome and function of OA chondrocytes.
In the lipopolysaccharide (LPS)-induced pathological inflammation condition, pre-osteoblast MC3T3-E1 cell differentiation and mineralization were inhibited when METTL3 was knocked out, verifying that METTL3 acted as a positive regulator in osteoblast mineralization [46]. METTL3 silence could protect ATDC5 cells treated by IL-1b. In addition, METTL3 regulates the occurrence and development of OA by regulating the expression levels of inflammatory reaction-related genes (IL-8, IL-6, IL-12, TNF-α) and the ECM degrading enzyme MMP-13 [53]. Recent evidence has demonstrated that epigenetic dysregulation of multiple molecular pathways underlies OA pathogenesis, providing a new mechanistic and therapeutic axis. Due to MELLT3, but not METTL4, FTO, and ALKBH5 were involved in the development of OA; thus, METTL3 may have the potential to be a target for the treatment of OA.
m6A and osteoporosis
Osteoporosis is a systematically metabolic skeletal disorder in which bone mineral density (BMD) decreases, resulting in deterioration in bone microarchitecture, excessive accumulation of adipose tissue in the bone; the resulting bone becomes fragile and prone to fractures [69,70]. Under the pathological stimulation of various risk factors, BMSCs preferentially differentiate into lipoblast cells rather than osteoblasts, leading to bone marrow fat increase and progressive bone loss [71].
METTL3 in osteoporosis
Mettl3 gain-of-function prevents estrogen deficiency-induced postmenopausal osteoporosis, establishing the indispensability of Mettl3 in defining bone marrow MSCs fate and thereby ensuring skeletal health [44]. METTL3 expression and m6A methylation are substantially decreased in patients with osteoporosis and ovariectomy, a classic model to induce postmenopausal osteoporosis [41]. Moreover, Del Real et al. found patients with fragility fractures due to insufficient activity of the bone-forming osteoblasts, osteogenic drivers RUNX2/OSX in hMSCs occur some signs of accelerating methylation aging [72]. Wu et al. have shown that the absence of Mettl3 in bone marrow MSCs results in osteoporosis pathological phenotypes such as impaired bone formation [44]. METTL3 in BMSCs promoted adipogenesis and inhibited the osteogenic differentiation process through regulating the JAK1/STAT5/C/EBPβ signal pathway via an m6A-YTHDF2–dependent regulatory pattern [43]. Conditional knockdown of the METTL3 in BMSCs could suppress PI3K-Akt signaling and PTH/Pth1r signaling, downregulate the expression of bone formation-related genes (such as Runx2 and Osterix) and VEGF, and ultimately inhibit bone formation [44,45].
FTO in osteoporosis
Further, FTO also affects the osteoporosis phenotypes [64]. FTO-mediated m6A demethylation regulates mRNA splicing and is involved in adipogenesis. Gregor Sachse a et al. found that FTO demethylase activity is essential for normal bone growth and bone mineralization in mice [65]. The FTO expression in the bone is upregulated during aging and osteoporosis. The latest research reported that by regulating the transformation axis of GDF11-FTO-PPAR, FTO determines the fate of BMSCs during osteoporosis, promotes the transfer of BMSCs to adipose cells, and inhibits bone formation [42]. McMurray et al. conducted an examination to assess the effect of FTO demethylase function both in vitro and in vivo through pharmacologically inhibiting FTO [73]. They found that IOX3, which is a known inhibitor of the HIF prolyl hydroxylases, decreased protein expression of FTO in C2C12 cells; mice were treated with IOX3; FTO protein levels were not significantly altered; BMD and content were significantly reduced; and adipose tissue distribution was altered [73].
Nucleotide polymorphisms in the FTO gene were associated with elevated body mass index, which is a predictor of hip fracture risk. Based on the candidate gene association study, Tran et al. have shown that genetic variation within the FTO gene is associated with hip fracture risk [74]. In Sachse's study, it was demonstrated that although FTO catalytic activity is not essential for normal body composition, it is needed for normal bone growth, mineralization, body size, and viability. When the FTO gene was knocked out, the mice developed postnatal growth retardation, low body length, body weight, and lower BMD as well as bone mineral content, which was similar to that seen in osteoporosis [73]. However, FTO enzymatic activity retained roughly 50% and 20% could rescue the bone phenotype adequately.
Currently, m6A-SNPs may influence BMD by altering the local gene expression. For 1,354 M6A-SNPs theoretically related to FN-BMD (femoral neck BMD) or LS-BMD (lumbar spine BMD), it was found that 10 m6A-SNPs were related with FN-BMD and 8 were associated with LS-BMD (P < 0.05). Forty-seven BMDS detected by m6A-SNPs (P < 0.0001 for FN-BMD, LS-BMD, or BMD) were associated with the expression of 46 corresponding local genes, among which rs1110720iNespl1 (P = 2.05 × 10−10) had remarkable genome-wide significance. Twenty-six of the 47 in which the m6A-SNPs showed cis-eQTL signals were differentially expressed in at least one study (P < 0.05), and m6A-SNPS were always very close to the methylation site, in particular, which might be the methylation site [40]. These 26 m6A-SNPs may affect BMD through local epigenetic modification, suggesting which might be important candidates for further genetic association and functional studies.
The FTO, as a novel regulator of bone growth and mineralization, may not determine the composition of the body, but it could affect the biological process and molecular function of osteoblasts to varying degrees. Disruption of the bidirectional balance between osteogenesis and adipocyte differentiation may lead to osteoporosis. This provides a new therapeutic target for studying the mechanism of osteoporosis.
m6A and osteosarcoma
Emerging evidence has demonstrated the m6A modification implicated in the pathogenesis and progression of cancer, including osteosarcoma (OS). The OS is the most frequent primary malignant bone tumor that originates from primitive transformed cells of mesenchymal-producing immature bone and osteoid [75,76]. It has been reported that dysregulated m6A-related regulators are correlated with metastasis and unfavorable prognosis in OS (Table 2, Fig. 4) [77]. Besides, Wang et al. analyzed the transcriptome-wide m6A methylome of OS stem cells enriched by chemotherapy, revealing that differentially methylated genes were enriched in some pathways regulating the pluripotency of stem cells.
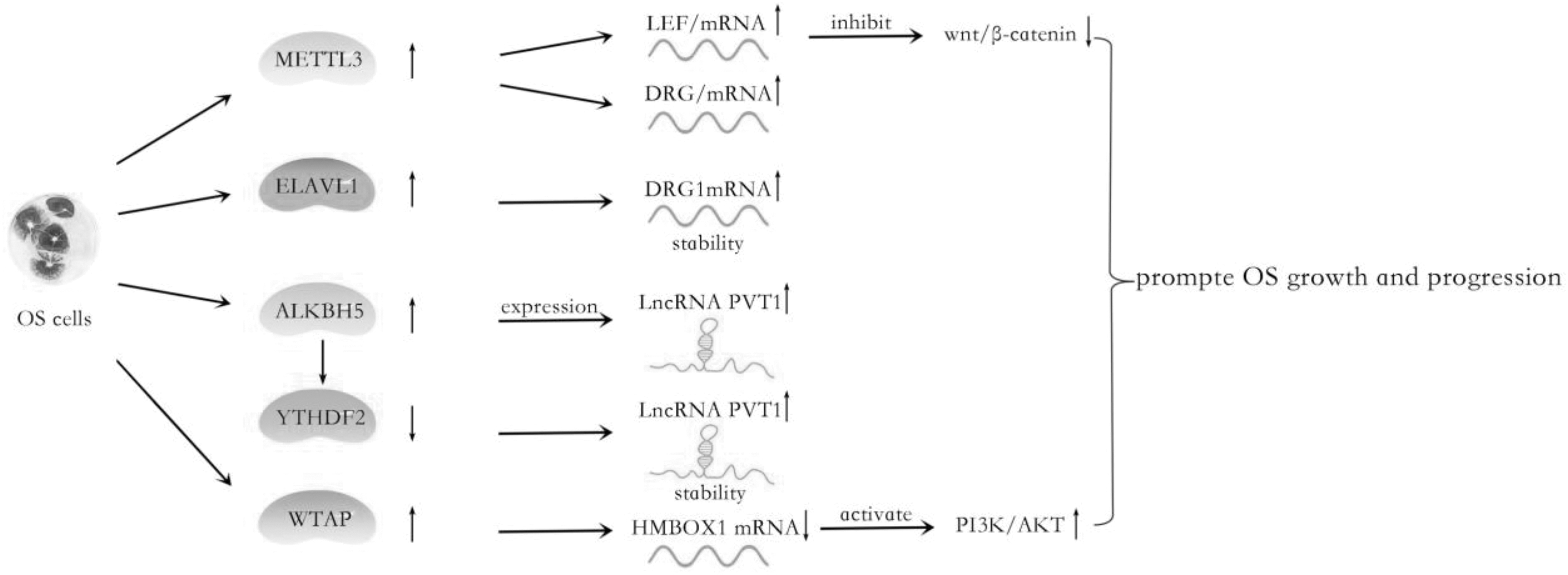
The role of m6A regulators in OS cells: The increase of m6A regulators METTL3, ELAVL1, ALKBH5, and WTAP modulates the expression of downstream genes LEF1, DRG1, PVT1, and HMBOX1, and it then promotes OS growth and progression. OS, osteosarcoma.
Roles of N6-Methyladenosine Key Regulator in Osteosarcoma
OS, osteosarcoma.
In 2019, Miao et al. identified that both the m6A level and METTL3 are elevated in human OS tissues and cell lines compared with the normal osteoblast cells. Their further research indicated that METTL3 promotes OS cell progression through regulating the m6A level of LEF1 and the activity of the wnt/β-catenin signaling pathway [78]. Simultaneously, DRG1 was abnormally upregulated in OS tissues compared with paracancerous tissues, which exerted cancer-promoting effects. Silencing METTL3 reduced m6A, mRNA, and protein levels of DRG1 in OS cells. When ELAVL1 was knocked out, the stability of DRG1 mRNA was decreased; thus, both the mRNA and protein levels of DRG1 were decreased. Upregulated DRG1 was elevated by METTL3 and ELAVL1 in an m6A-dependent manner; it promoted viability, migration, and colony formation abilities of OS cells [79]. TRIM7 was also involved in OS development and was an independent risk factor in predicting a bad prognosis. Zhou et al. detected that upregulated TRIM7 was regulated by the METTL3/14-YTHDF2-mRNA in a decay-dependent manner in OS cells [80].
In recent years, compelling evidence has revealed that methyltransferase WTAP was found as an oncogene in various cancers, including promoting the m6A modification and progression of OS [81,82]. Mechanistically, HMBOX1, a potential target gene of WTAP, was regulated by WTAP with m6A modification at the 3′UTR, inhibiting OS growth and metastasis in vivo. Further investigations demonstrated that WTAP/HMBOX1 regulated OS growth and metastasis via the PI3K/AKT pathway [83]. In 2020, Chen et al. reported that demethylase ALKBH5 expression was upregulated in OS tissues compared with normal tissues. Moreover, ALKBH5 mRNA level was positively associated with PVT1—a well-known oncogenic lncRNA—transcript level. Further research found that ALKBH5 could elevate PVT1 expression via associating with PVT1 and inhibiting its degradation. ALKBH5 reduced the m6A modification level of PVT1, subsequently suppressing the binding of reader protein YTHDF2 in PVT1 [84].
Evidence is emerging that m6A abundance and expression of m6A regulators are abnormal in OS, and their abnormality could be related to drug resistance and poor prognosis. Due to both m6A writers and erasers being overexpressed in OS, such as ALKBH5, METTL3, and WTAP, the total m6A abundance may not be adequately informative. Therefore, certain particular transcripts and m6A signatures could be better biomarkers for early cancer diagnosis, prediction, and risk assessment. This provides a future direction for OS therapy that warrants further studies.
Summary and Prospect
Growing research has demonstrated that m6A is deposited on native RNA transcripts that are tightly associated with the bone development process during transcription and it affects gene expression post-transcriptionally through altering RNA local structure or specific recognition by the m6A-binding protein. Therefore, the dynamic balance of m6A modification is essential for normal bioprocesses and development, disorders that could cause various diseases. In this review, the effects of m6A RNA modification in bone and cartilage pathophysiology as well as in pathogenesis have been revealed. Advances in the m6A field underscore the complex confluence of genetic and environmental factors as key factors in bone homeostasis and other common bone tissue diseases. As such, they also help explain the mechanisms by which various nongenetic factors (from nutrition to psychological stress) affect our bodies, such as the effect of both psychological stress and bad habits on temporomandibular joint (TMJ) OA.
However, gene-specific effects of methylation are not clear, the results of the differential abundance of methylation in the same gene remain to be explored, and the other components of m6A also need to be further discovered. The SNPs detection method, high resolution, and high-throughput sequencing technology of m6A methylation also need to be improved. Although the relevant research is little, predictably, research interests in this field will remarkably increase over the coming years and m6A methylation is expected to be an effective method to regulate bone homeostasis and treat bone diseases.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This article was supported by the National Natural Science Foundation of China (no. 31870929) and the Natural Science Foundation of Shandong Province (no. ZR2019MH007).