
Abstract
The primitive state (stemness) of mesenchymal stromal cells (MSCs) is responsible for supporting the function of tissue-specific stem cells to regenerate damaged tissues. However, molecular mechanisms regulating the stemness of MSCs remain unknown. In this study, we found that the primitive state of MSCs is hierarchically regulated by the expression levels of the chromatin remodeling complex, CHD1, with CHD1 expression levels higher in the undifferentiated state, and decreasing upon MSC differentiation. Consistently, CHD1 expression levels decrease during progressive loss of clonogenic progenitors (CFU-F) induced by passage cultures. Moreover, knockdown (KD) of CHD1 decreased CFU-F frequency, whereas CHD1 overexpression increased it. In addition, the expression of stem cell-specific genes was down- or upregulated upon KD or overexpression of CHD1, respectively, accompanied by associated changes in chromatin condensation. Importantly, altering CHD1 expression levels affected the ability of MSCs to support the self-renewing expansion of hematopoietic stem cells (HSCs). Furthermore, CHD1 levels were significantly decreased in MSCs from acute myeloid leukemia or aplastic anemia patients, where CFU-F and HSC-supporting activities are lost. Altogether, these findings show that chromatin remodeling by CHD1 is a molecular parameter that influences the primitive state of MSCs and their stem cell-supporting activity, which controls tissue regeneration.
Introduction
Mesenchymal stromal cells (MSCs) are a non-hematopoietic adherent cell population. MSCs are derived from bone marrow (BM), adipose tissue, or placental tissue. MSCs exhibit multilineage differentiation potential toward diverse types of tissues, including bone, cartilage, and adipose tissues [1 –3]. Accumulating studies have shown that the primary mode of action for MSCs is the paracrine support of tissue regeneration both by inhibiting apoptosis and fibrosis [4], and by stimulating the regeneration of endogenous stem cells such as hematopoietic stem cells (HSCs), neuronal stem cells, and other tissue-specific endogenous stem cells [5,6]. As such, MSCs have been widely utilized for many clinical cell therapy trials. So far, more than 425 clinical trials have been performed using ex-vivo expanded MSCs to facilitate the regeneration of injured tissues [7].
However, MSCs are highly heterogeneous, and only specific subsets of primitive MSCs have the ability to form colonies (CFU-F), or to support endogenous stem cells. For example, in BM, only primitive MSC subsets that retain colony-forming potential could contribute to the stem cell niches [8,9]. Subsequent studies revealed that only the subsets of MSCs expressing nestin [10], leptin-receptor [11], or prx-1 [12], which are enriched with CFU-F, serve as major HSC-supporting niche cells. Moreover, in ex-vivo cultured MSCs, there was a positive correlation between the frequency of CFU-F and their ability to support HSC self-renewal, despite the functional discrepancies between cultured MSCs and in-vivo isolated MSCs [13]. Accordingly, interests are highlighted on the mechanisms that maintains the primitive state of MSCs, but the key molecular regulators have yet to be identified.
Recently, studies have shown that maintenance of the undifferentiated state in stem cells is characterized by a dynamic epigenetic plasticity. Specifically, pluripotent embryonic stem cells (ESCs) contain less-condensed chromatin structures [14,15] and there is poised (primed but held-in-check) expression of lineage-associated regulatory genes through a bivalent mode of histone modifications by polycomb (PcG) group proteins [14,16]. Moreover, a hierarchy of stemness was correlated to the degree of open, “loose” chromatin with higher rates of histone modification exchange, which became “condensed” heterochromatin upon differentiation [17 –19], thus illustrating the dynamic nature of chromatin in the undifferentiated state of stem cells.
The dynamic nature of chromatin and its remodeling are mediated by multiple family members of chromatin helicase DNA-binding domain (CHD) proteins through binding to methylated histones [20]. The CHD family of chromatin remodeling enzymes comprises two chromo domains and two Sucrose NonFementable2 (SNF2)-like ATP-dependent helicase domains [21], while each subfamily harbors additional specific domains [20]. Accordingly, CHD1 was shown to be necessary for the maintenance of open chromatin and the pluripotency of ESCs, as well as for reprogramming fibroblasts into a pluripotent state [22]. However, little is known about whether these chromatin remodeling activities could be mechanisms for regulating the primitive state of MSCs to control their clonogenic potential and ability to support stem cell regeneration. In this study, we investigated the possibility that CHD1 controls the cell fate of MSCs to influence their stem cell niche function in supporting HSCs.
Materials and Methods
Human cells and culture
Human MSCs were separated from BMs of acute myeloid leukemia (AML) and aplastic anemia (AA) patients under informed consent. Human cord bloods and MSCs were obtained from normal donors under informed consent MC19TNSI0012. This study was approved by the Institutional Review Boards of St. Mary's Hospital and Catholic University of Korea. Human MSCs were cultured in low-glucose Dulbecco's modified Eagle's medium (DMEM; HyClone, Waltham, MA) supplemented with 10% fetal bovine serum (FBS; HyClone) as described [23].
Coculture
Human CD34+ cells from umbilical cord blood were isolated by immunomagnetic column using Dynabeads (Invitrogen, Carlsbad, CA) according to the instructions. For coculture assays, MSCs were irradiated with 15Gy of 137Cs γ-rays (Gammacell 1000; MDS Nordion, Ottawa, ON, Canada) 24 h before coculture with human CD34+ cells for 4 days in DMEM supplemented with FBS in the presence of a cytokine mixture (20 ng/mL human SCF; 20 ng/mL human Flt3L (ProSpec-Tany TechnoGene Ltd.), 4 ng/mL human IL3, IL6 (R&D Systems, Minneapolis, MN), and 4 ng/mL human G-CSF (ProSpec) supplemented with 10−6M hydrocortisone (Sigma-Aldrich, St Louis, MO).
Flow cytometry and cell cycle analysis
Human CD34+ cells were stained with CD45-PE, CD34-APC, and CD90-FITC antibodies (BD PharMingen). Cell cycles of MSCs were analyzed by staining the MSCs with 10 μmol/L Hoechst33342 at 37°C for 45 min, and Pyronin Y was then added to give a final concentration of 2.5 μg/mL. Data were analyzed with Diva software using a LSRII.
Transduction and transfection of MSC
A negative scrambled control shRNA sequence and a CHD1/shRNA sequence were cloned into the pMirzip lentiviral vector (System Biosciences, Palo Alto, CA). For overexpression of CHD1, wild-type CHD1 cDNA was cloned into the pMIRNA1 lentiviral vector (SBI). MSCs were transduced with each lentiviral vector and sorted for transduced (GFP+) cells. For transfection of siRNA into MSCs, CHD1-specific siRNA and the control siRNA were purchased from GenePharma (Shanghai, China) and each siRNAs (50 nM) were transfected in the presence of oligofectamine (Invitrogen).
CFU-F assay and differentiation of MSCs
For CFU-F assays, MSCs were plated in 100 mm tissue culture dish. Fourteen days after plating, the cells were fixed with methanol and then stained with Crystal Violet (Sigma) for visualization. For osteogenic or adipogenic differentiation of MSCs, MSCs under growth medium were switched to differentiation-inducing medium for 14 days for osteogenic and 14 days for adipogenic differentiation. The extent of terminal differentiation was quantified by mineralization or lipid droplets in the differentiated cells using Alizarin Red S or Oil Red O staining, followed by spectrophotometry at 550 and 520 nm, respectively.
Real-time polymerase chain reaction
Total RNA was extracted from MSCs using TRIzol (Invitrogen). cDNA was synthesized from 1 μg of total RNA with reverse transcriptase (Invitrogen). Real-time quantitative polymerase chain reaction (RT-qPCR) was performed with SYBR Premix Ex Taq (TaKaRa, Japan). The threshold cycle (Ct) value for each gene was normalized to the Ct value of 18sRNA. The relative mRNA expression was calculated by using the formula; 2−ΔΔCt, where ΔCt = Ctsample− Ct18sRNA and ΔΔCt = ΔCtsample − ΔCtreference group.
Western blotting
For western blot analysis, MSC lysates were subjected to gel electrophoresis [sodium dodecyl sulfate/polyacrylamide gel electrophoresis (SDS-PAGE)], transferred to membranes, and incubated with primary antibodies against each specific antibody. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies, and proteins were visualized using a chemiluminescence substrate (Thermo Scientific, Abbott Park, IL). Primary antibodies against CHD1 were purchased from Santa Cruz Biotechnology (Dallas, TX).
Senescence-associated β-galactosidase assay
Senescence-associated β-galactosidase (SA-β-gal) activity was examined by the Senescence β-Galactosidase Staining Kit (Cell Signaling Technology, Danvers, MA) according to the instruction. Stained cells were observed under a light microscope.
Chromatin immunoprecipitation asssay
Chromatin immunoprecipitation (ChIP) was performed with the EZ-ChIP™ Kit (Millipore/Upstate) according to the manufacturer's instruction. Briefly, MSCs were fixed in 1% formaldehyde, washed and lysed, then sonicated to shear DNA. Subsequently, anti-tri-MeH3K9 (Cell Signaling Technology) was added for immunoprecipitation along with protein A/G sepharose beads. After washes, protein/DNA complexes were reverse crosslinked for 4 h at 65°C, and analyzed by quantitative PCR for each gene promoter-specific sequences.
Chromatin accessibility
Chromatin accessibility of the Oct4 gene promoter in hMSCs was assessed by nuclease protection assay using the EpiQ Chromatin Analysis Kit (Bio-Rad, Munich, Germany). Briefly, cells were incubated in EpiQ chromatin buffer with or without nuclease at 37°C for 1 h, then genomic DNA was extracted. Chromatin accessibility was assessed by quantitative PCR and calculated using the EpiQ Chromatin Kit data analysis tool. The β hemoglobin (HBB) gene was used as the inaccessible reference for measuring relative chromatin accessibility at Oct4 gene locus. PCR primers: Oct4, 5′-CACTGCACTCCAGTCTGGGCAACAA and 5′-TGCCTAATGGTGGTGGCAATGGTGT; HBB, 5′-AGCCAGTGCCAGAAGAGCCAAGGA and 5′-CCCACAGGGCAGTAACGGCAGACTT.
Results
To determine whether CHD1 is involved in regulating the primitive state of MSCs, we first investigated whether CHD1 expression in MSCs changes with respect to the frequency of mesenchymal progenitors. For this, we examined the expression levels of CHD1 in MSCs during serial passage culture that causes a loss in the number of colony-forming unit fibroblasts (CFU-F). As shown, serial culture of MSCs caused a progressive loss of CFU-F with increased passage numbers (Fig. 1A). This decrease in the frequency of CFU-F was accompanied by a concomitant decrease of CHD1 expression levels in MSCs (Fig. 1B).
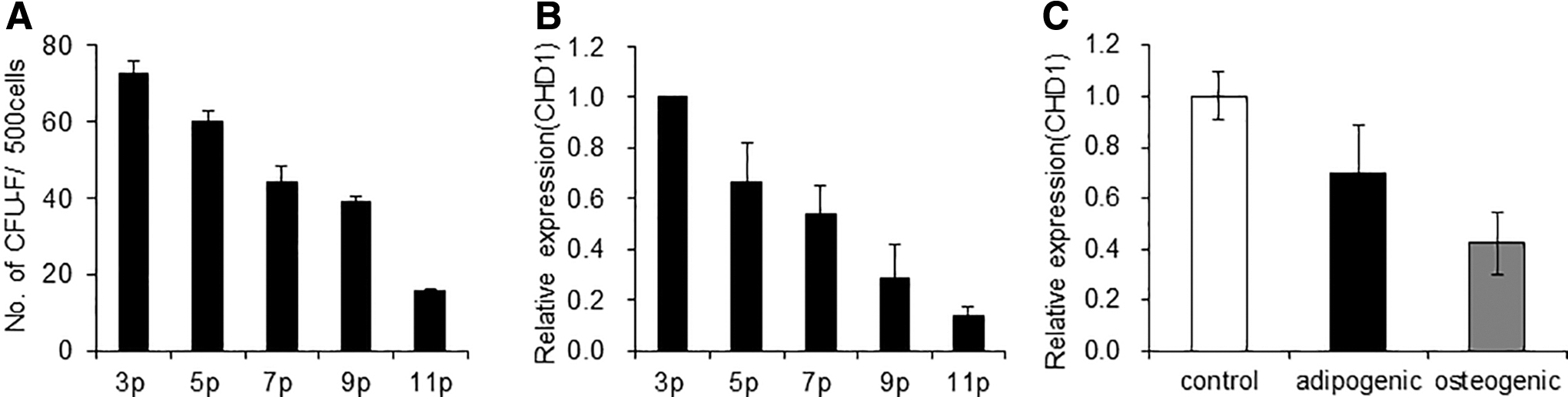
Changes in CHD1 expression levels in MSCs during passage cultures and differentiation.
We also compared the expression of CHD1 during osteogenic or adipogenic differentiation of cultured MSCs. The expression level was highest in the undifferentiated state of MSCs, but decreased during their terminal differentiation into osteogenic or adipogenic lineages (Fig. 1C and Supplementary Fig. S1). These results indicate that CHD1 expression levels of MSCs are regulated along with changes in cell fate such that higher levels of expression of CHD1 are associated with the primitive, undifferentiated state of MSCs.
To further examine the functional significance of CHD1 expression levels in MSCs, we next examined the effect of CHD1 knockdown (KD) on the clonogenic potential and multilineage differentiation of MSCs. Lentiviral vector expressing shRNA against CHD1 was transduced into MSCs, which caused a significant decrease in CHD1 expression levels (Fig. 2A–C and Supplementary Fig. S2).
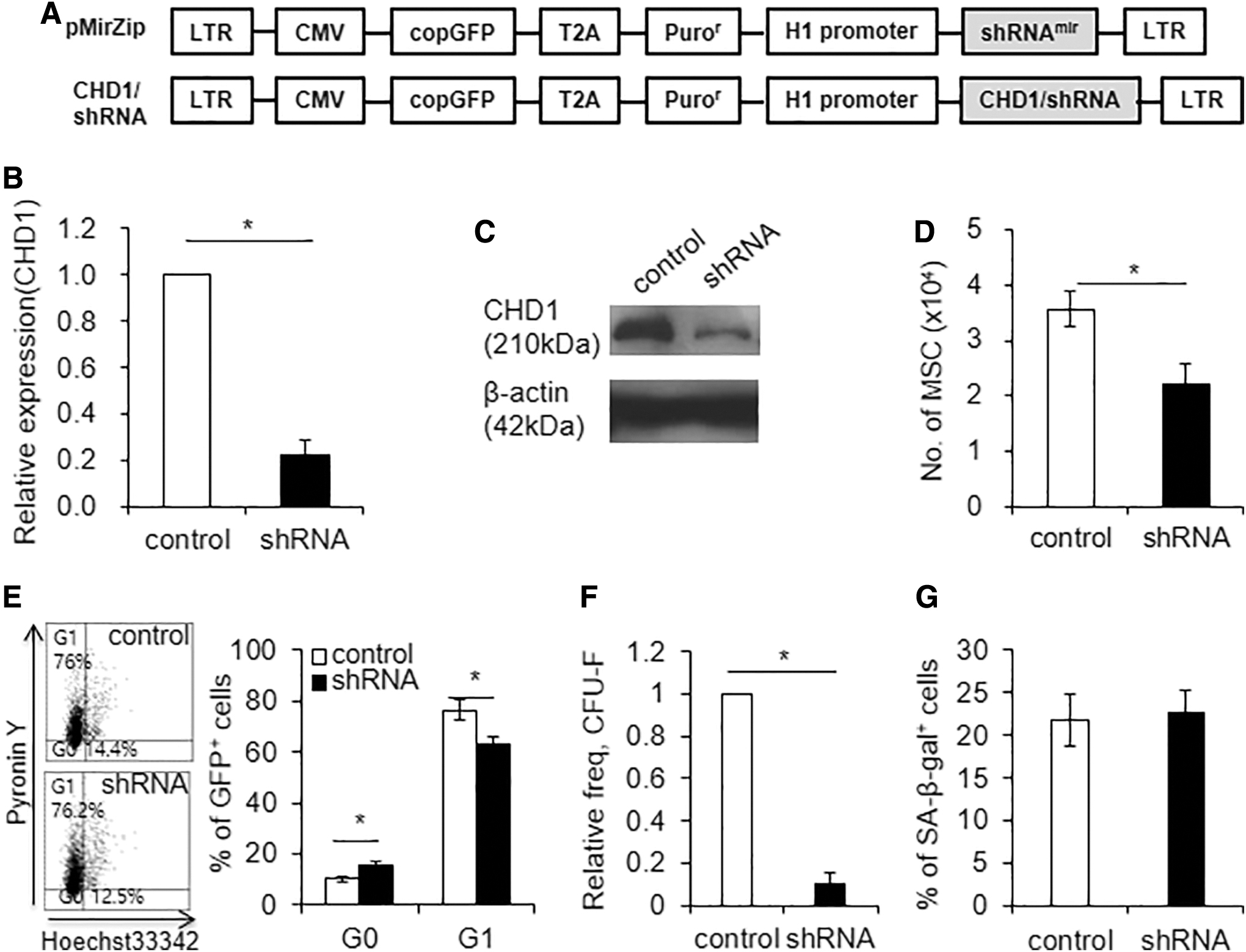
Effects of CHD1-KD on proliferation and frequency of CFU-F.
The KD of CHD1 caused a moderate decrease in cell proliferation, with an increase in the number of cells in the G0 phase of the cell cycle (Fig. 2D, E). Strikingly, MSCs with CHD1-KD exhibited a profound decrease in the number of CFU-F compared with control MSCs (Fig. 2F and Supplementary Fig. S3A). This decrease of CFU-F was not associated with an increase in cell senescence, as evidenced by comparable levels of SA-β-gal activity (Fig. 2G and Supplementary Fig. S3B). These results show that high expression levels of CHD1 are critical for the maintenance of mesenchymal progenitor cells, and decreased expression of CHD1 causes the loss of clonogenic potential.
To determine whether the effects of CHD1-KD on the clonogenic potential of MSCs is related to changes in stemness secondary to its known chromatin remodeling activity, we examined the expression changes of pluripotency-related genes. As shown, the expression levels of OCT4, NANOG, and SOX2 were significantly decreased in CHD1-KD MSCs compared with the control MSCs (Fig. 3A). The decrease in expression levels of these pluripotent genes were similarly reproduced in MSCs with siRNA-induced KD of CHD1 (Supplementary Fig. S4). The decrease in pluripotent gene expression was associated with condensation of chromatin, as evidenced by an increase in the repressive histone mark, H3K9-me3 [24], in the OCT4 promoter (Fig. 3B) as well as a decrease in chromatin accessibility to nuclease at the promoter (Fig. 3C, D). Of note, the ChIP assay for MSCs with CHD1-KD exhibited increase of H3K9-me3 in the promoters of genes for primitive state of MSCs, including SOX2, KLF4, and NESTIN [10], but not tissue-specific genes such as RUNX2 or OSTEOCALCIN (Supplementary Fig. S5), suggestive of preferential silencing of genes involved in the primitive state of MSCs. These results indicate that CHD1 regulates the stemness of MSCs through its chromatin remodeling activities.
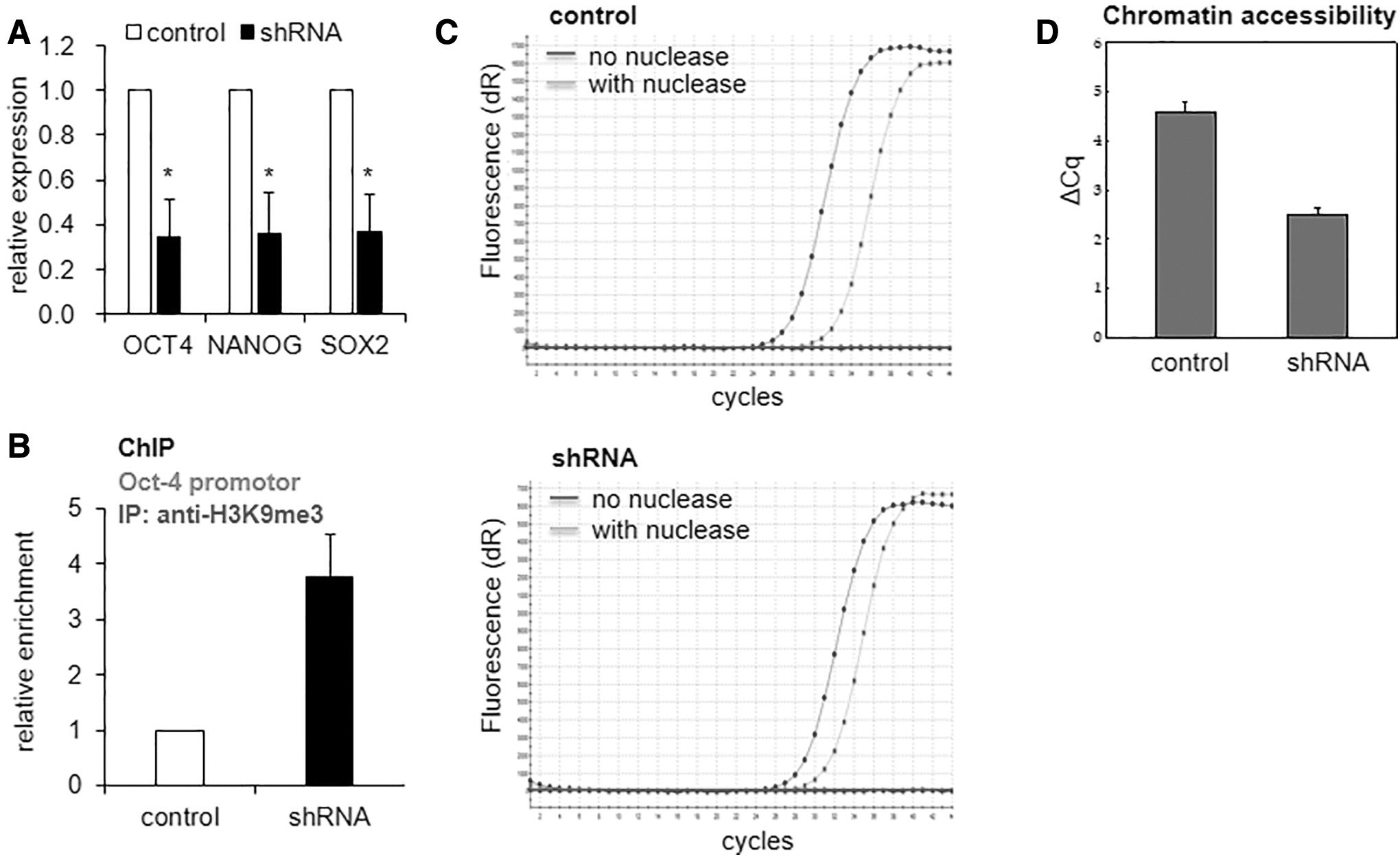
Effects of CHD1-KD on the transcription of pluripotent genes.
We next examined the effect of CHD1-KD on the multilineage differentiation potential of MSCs. When subjected to osteogenic or adipogenic differentiation, MSCs with CHD1-KD exhibited a significant decrease in their terminal differentiation into both lineages (Fig. 4A, B). These results show that CHD1, while contributing to the maintenance of the undifferentiated state, also plays a role in guidance of multilineage differentiation process.
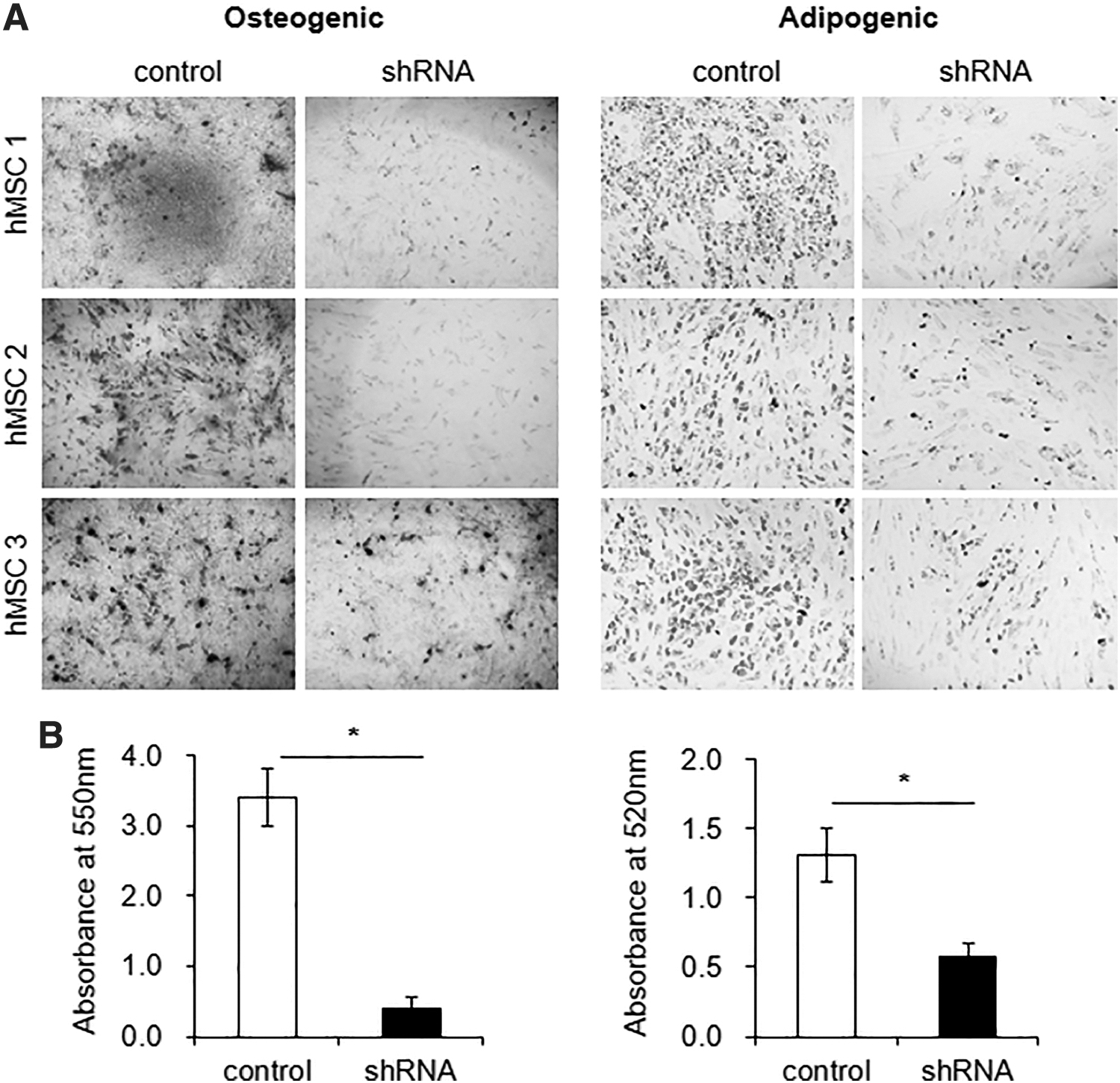
Effects of CHD1-KD on multilineage differentiation of MSCs. CHD1-KD or control MSCs were subjected to osteogenic or adipogenic differentiation, and stained with Alizarin Red S or Oil Red, respectively. Shown are the representative profiles for osteogenic and adipogenic differentiation
Based on the effects of CHD1-KD in MSCs, we next questioned whether increasing CHD1 expression would cause the opposite effects on the characteristics of MSCs. Thus, MSCs overexpressing CHD1 were generated by transduction with lentiviral vector encoding wild-type CHD1 (Fig. 5A–C). As shown, the CHD1-overexpressing MSCs exhibited an increase in the frequency of CFU-F and increased expression of pluripotent genes without changes in SA-β-gal activity (Fig. 5D–F). The increased expressions of pluripotent genes were associated with decondensation of chromatin as evidenced by increased accessibility to nuclease (Fig. 5G, H). Similarly, MSCs overexpressing CHD1 exhibited an increase in osteogenic differentiation potential while maintaining adipogenic differentiation potential (Fig. 5I). Altogether, these results show that the expression levels of CHD1 is a factor controlling the stemness and multilineage differentiation potential of MSCs.
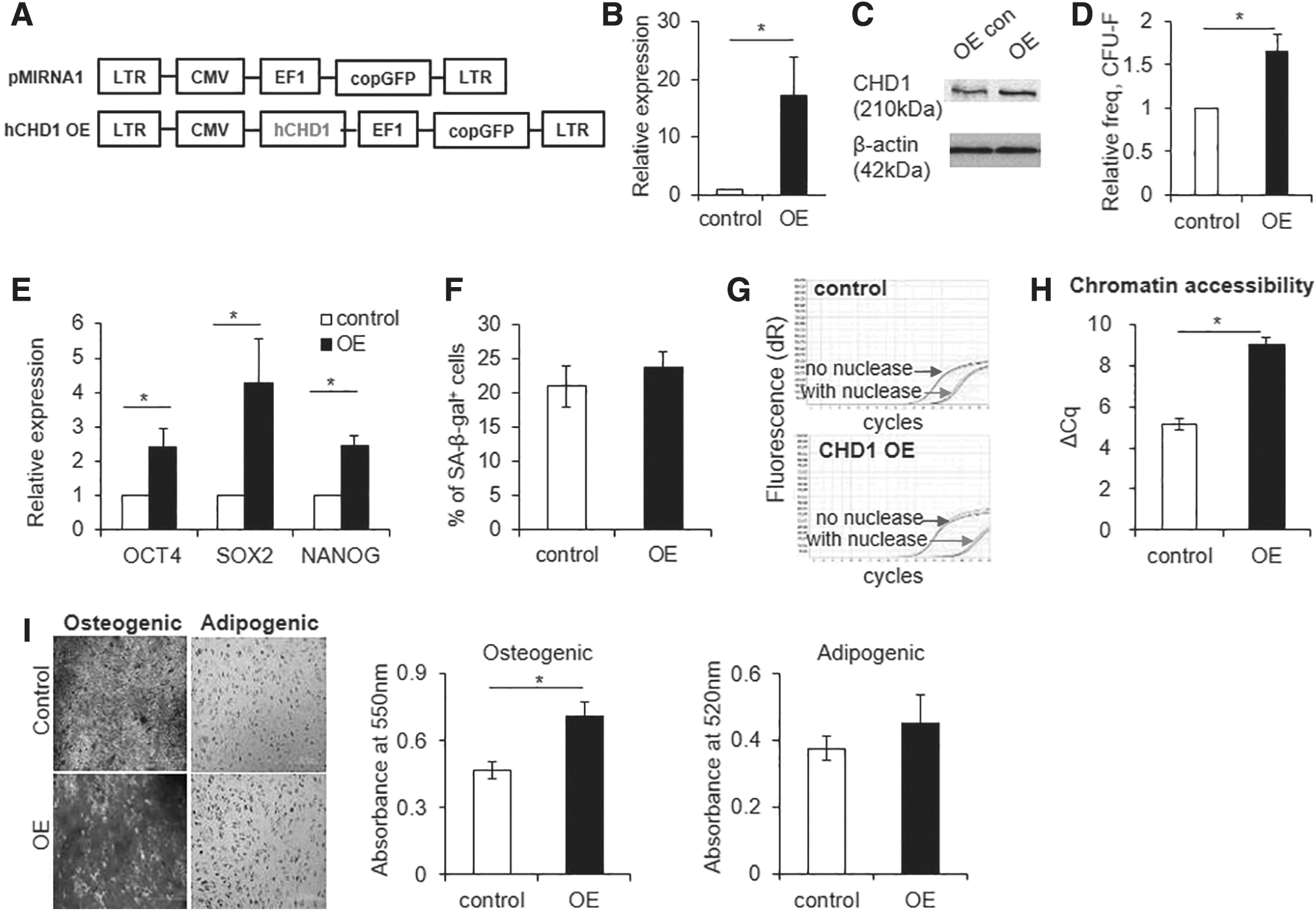
Effect of CHD1 overexpression in MSCs.
Since the primitive stemness of MSCs is related to their niche function supporting endogenous stem cells [10,23,25 –27], we next questioned whether CHD1 in MSCs was also involved in the control of niche activity to support HSCs.
The HSC-supporting activity of MSCs is reflected by a shift toward expressing epithelial/mesenchymal transition (EMT) genes [28,29]. Thus, we first examined the influence of CHD1 expression levels on expression changes of EMT genes. MSCs with CHD1-KD exhibited significantly lower expression levels of EMT-regulating genes, including ZEB1, ZEB2, TWIST1, SNAI1, and SLUG (Fig. 6A). Moreover, KD of CHD1 caused a significant decrease in NESTIN and CD146 levels, which are markers for mesenchymal niche cells displaying HSC-supporting activities [9,10,25,30] (Fig. 6B). In contrast, overexpression of CHD1 in MSCs upregulated the EMT-related genes (Fig. 6C) and the ratio of E-cadherin to N-cadherin reflecting EMT was concomitantly changed with KD or overexpression of CHD1 (Fig. 6D), indicating that EMT genes are controlled by CHD1 expression levels.

Effects of CHD1 expression changes on the HSC-supporting activity of MSCs. MSCs were transfected with control or siRNA against CHD1.
Consistent with the effects on MSCs, umbilical cord blood-derived CD34+ cells cocultured with CHD1-KD MSCs also exhibited a significant decrease in the self-renewing expansion of primitive hematopoietic subsets (CD34+CD90+), the subset previously characterized as long-term repopulating HSCs [31]. In contrast, coculture of CD34+ hematopoietic cells with CHD1-overexpressing MSCs significantly enhanced the self-renewing expansion of CD34+90+ hematopoietic cells (Fig. 6E). Altogether, these results show that the changes in expression levels of CHD1 in MSCs is a molecular parameter that can cause a dynamic change in the HSC-supporting activity of MSCs.
To further explore the physiological significance of CHD1 expression changes for controlling HSC-supporting activity of MSCs, we examined CHD1expression levels under pathological conditions of altered niche activity. For this, we first examined MSCs derived from AML patients. The mesenchymal niche cells from AML patients exhibited loss of CFU-F and multilineage differentiation potential (Fig. 7A, B) and accordingly, loss of HSC-supporting activities (Fig. 7C), consistent with previous findings [30,32,33]. When CHD1 expression levels were examined, we found significantly lower levels in AML-derived MSCs compared with MSCs derived from normal donors (Fig. 7D). Thus, functional changes of MSCs induced by CHD1-KD, including loss of CFU-F, multilineage differentiation and HSC-supporting activity, were similarly reproduced in MSCs of AML patients, where CHD1 expression level is decreased.
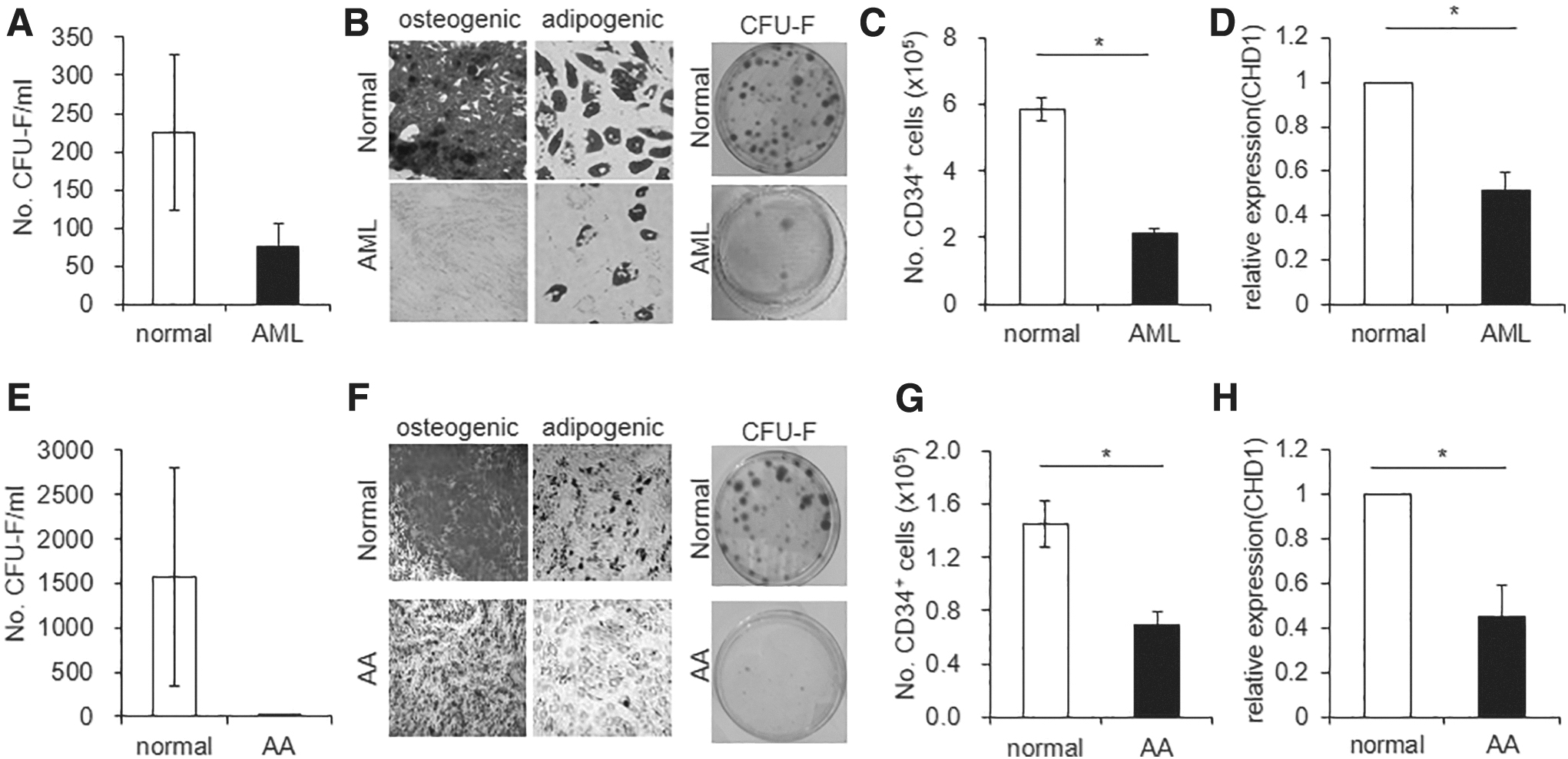
Changes of CHD1 expression in MSCs from hematological disorders with degeneration of the stem cell niche.
We also examined MSCs from patients with AA, as an independent model for loss of stemness in MSCs. MSCs from AA similarly exhibited significantly decreased levels of CFU-F and multilineage differentiation potential (Fig. 7E, F), decrease of HSC-supporting activities (Fig. 7G). Similar to the MSCs from AML, CHD1 levels in AA-derived MSCs were significantly lower than the levels in normal MSCs (Fig. 7H), similarly mirroring the MSCs with CHD1-KD.
Altogether, these results show that the changes of the CHD1 expression levels in MSCs are related to the molecular mechanism regulating the niche activity of MSCs in the context of diseases characterized by degenerative niche conditions and could comprise another pathogenic mechanism for alteration of HSC-supporting activities in patients with hematological insufficiency.
Discussion
Despite the widespread use of MSCs in clinical cell therapy trials, significant functional heterogeneity exists in their ability to stimulate tissue-specific stem cells, where primitive subset of MSCs with clonogenic potential play the major role as niche cells in-vivo [8 –12,34] and in-vitro [23]. However, factors that maintain the stemness of MSCs have not been well characterized.
In the current study, we found that the primitive state of MSCs is maintained by high expression levels of the chromatin remodeling complex, CHD1, which functions in the dynamic plasticity of chromatin structure.
The CHD1 expression levels are hierarchically regulated in a manner that higher expression levels are associated with the primitive and undifferentiated state of MSCs and KD or overexpression of CHD1 in MSCs could concomitantly influence their primitive state. Thus, dynamic remodeling of chromatin mediated by CHD1 may play a major role in the maintenance of the primitive state of MSCs, reminiscent of their role in maintenance of the pluripotent state and reprogramming into the stem cell state [19,20,22,35].
Supporting the possibility, KD of CHD1 caused selective downregulation in the transcription of pluripotent genes, but not tissue-specific genes. However, taking that the principal role of CHD1 has been implicated in the chromatin remodeling and hypertranscription phenomenon [35], it is not clear yet how the selectivity of target genes are determined in the transcription process.
Interestingly, the role of CHD1 in MSCs was not confined to the maintenance of primitive state, but was also involved in the guidance of multilineage differentiation process, that is, KD of CHD1 in MSCs inhibited their osteogenic or adipogenic differentiation.
Studies have shown that the stem cell state with multilineage differentiation potential is associated with higher epigenetic plasticity with open chromatin structure to permit the cell fate changes into various types of differentiated cells [36 –39]. In contrast, differentiated cells exhibit less epigenetic plasticity and epigenetic alterations leads to the apoptosis of the cells, stably maintaining the given cell identity [17].
Supporting the possibility, it was shown that CHD1 was also necessary for induction of osteoblast-specific genes [40] or for multilineage differentiation of ESCs [41]. Thus, dynamic remodeling of chromatin mediated by CHD1 is necessary for guidance of a differentiation program as well as for maintenance of primitive state for multiple spectrums of stem cells with different levels of stemness.
However, it is noteworthy that multiple members of the chromatin helicase domain (CHD) family have been shown to mediate chromatin remodeling [20] in a manner that is cell type specific. For example, chromatin remodeling by chd2 is required for determination of myogenic cell fates and induction of muscle-specific genes, including MyoD [42]. Similarly, chromatin remodeling by chd4 establishes a lymphopoiesis-specific gene expression profile [43], and chd9 is involved in the expression of osteocalcin for bone development [44,45]. In this regard, it is possible that CHD1, among multiple CHD family members, is primarily involved in the establishment of stemness signatures in multiple types of stem cells.
Of note, the primitive state of mesenchymal cells was directly related to their niche cell function to promote regeneration of tissue-specific stem cells, HSCs, as inferred from the influence of CHD1 expression on the self-renewing expansion of cocultured HSCs.
Previous studies have shown that subsets of MSCs that can serve as niche for HSCs in BM are characterized by subsets exhibiting higher frequencies of colony-forming cells and expression of specific markers such as Prx1, Nestin, Leptin, or CD51/PDGFR [11,12,46 –48]. Moreover, increase of colony-forming cells were correlated to higher HSC-supporting activities during in-vitro culture model [23], suggestive of the relationship between primitive state of MSCs and their niche function.
Moreover, given that HSC-supporting activities of MSCs were either upregulated or downregulated with respect to CHD1 expression levels, it is possible that CHD1 expression is a molecular parameter that can dictate the dynamic changes of MSC niche activities during their adaptive response to various extrinsic signals for regulating hematopoiesis [49]. Supporting this possibility, CHD1 expression levels were significantly downregulated in MSCs from patients with AML or AA, where MSCs exhibit a loss of stemness and decreased HSC-supporting activities [30,32,33]. The functional defect of AML is caused by extensive complexity of cellular and molecular alterations, but exhibit degenerative changes of MSCs with loss of HSC-supporting activities [30,50 –54]. Similarly, the cellular changes in the MSCs of AA is caused by multiple idiopathic etiologies, exhibiting similar degenerative changes [55 –57].
Taking that the degenerative changes of MSCs caused by CHD1-KD, including loss of CFU-F, multilineage differentiation or loss of HSC-supporting activity, were similarly reproduced in MSCs from AML or AA patients with a decrease of CHD1 expression, it is possible that changes in CHD1 expression comprise another target of molecule leading to the degenerative changes of MSCs in those patients exhibiting decrease of HSC-supporting activities.
At present, the upstream regulators that can modulate the expression changes of CHD1 remain unclear. Therefore, further studies are warranted to dissect the mechanism controlling dynamic changes of CHD1 expression levels in MSCs as well as signals that can control their expression under various physiological conditions. Nevertheless, our findings show another physiological role of chromatin remodeling factor, CHD1, as a new regulatory mechanism controlling the microenvironment of stem cells, in addition to role of the chromatin remodeling for maintenance of stem cell state, self-renewal or differentiation of normal and cancer stem cells [36 –39,58].
Conclusion
Our findings reveal that the chromatin remodeling activity mediated by CHD1 is a molecular parameter that can influence the primitive state of MSCs and their stem cell niche activity, pointing it as potential molecular target to control tissue regeneration.
Footnotes
Acknowledgment
The human MSCs from healthy donors (MASTER Cells) were supplied from the Catholic Institute of Cell Therapy (CIC, Seoul, Korea).
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study is supported by the NRF of Korea and funded by the Ministry of Science, ICT, & Future Planning (2017M3A9B3061947).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.