
Abstract
Directed differentiation of human pluripotent stem cells (hPSCs) uses a growing number of small molecules and growth factors required for in vitro generation of renal lineage cells. Although current protocols are relatively inefficient or expensive. The first objective of the present work was to establish a new differentiation protocol for generating renal precursors. We sought to determine if inducer of definitive endoderm 1 (IDE1), a cost-effective small molecule, can be used to replace activin A. Gene expression data showed significantly increased expressions of nephrogenic markers in cells differentiated with 20 nM IDE1 compared with cells differentiated with activin A. Thus, renal lineage cells could be generated by this alternative approach. Afterward, we determined whether coculture of endothelial and mesenchymal cells could increase the maturation of three-dimensional (3D) renal structures. For this purpose, we employed a 3D coculture system in which hPSC-derived kidney precursors were cocultured with endothelial cells (ECs) and mesenchymal stem cells (MSCs), hereafter named RMEM (renal microtissue derived from coculture of renal precursors with endothelial and mesenchymal stem cells). hPSC-derived kidney precursors were cultured either alone [renal microtissue (RM)] or in coculture with human umbilical vein endothelial cells and human bone marrow-derived mesenchymal stem cells at an approximate ratio of 10:7:2, respectively. Immunofluorescent staining showed expressions of kidney-specific markers synaptopodin, LTL, and E-cadherin, as well as CD31+ ECs that were distributed throughout the RMEMs. Quantitative real-time polymerase chain reaction analysis confirmed a significant increase in gene expressions of the renal-specific markers in RMEMs compared with RMs. These findings demonstrated that renal precursors cocultured with endothelial and MSCs showed greater maturity compared with RMs. Moreover, ex ovo transplantation induced further maturation in the RMEM constructs. Our novel approach enabled the generation of RMEM that could potentially be used in high-throughput drug screening and nephrotoxicology studies.
Introduction
Renal diseases are a major worldwide health problem. Failure of kidney function is caused by irreversible loss of the structural and filtration units of the kidneys (nephrons) due to acute and chronic insults. Human nephrogenesis (de novo nephron formation) only occurs until the 36th week of prenatal development. After birth, the human kidney has limited regenerative potential by intrarenal stem cells. Thus, replacement of the damaged renal cells remains a challenge. Recent studies have assessed the use of stem cells as a therapeutic approach for acute and chronic kidney injuries to offset the increase of donor shortages and end-stage renal failure [1]. Furthermore, in vitro-generated human pluripotent stem cell (hPSC)-derived kidney cells provide a suitable platform for studying kidney development, physiology, disease modeling, and drug screening. Although hPSCs-derived kidney organoids express some markers of fully differentiated kidney tissues, they have relatively immature characteristics [2]. Several strategies have been reported to improve the maturity of in vitro-differentiated kidney structures. These strategies include chemical compounds [3], three-dimensional (3D) cell culture, cell transplantation into a highly vascularized site [4], fluid flow [5], extracellular microenvironment [6], and coculture systems [7,8].
Embryological studies demonstrated that spatiotemporal cell–cell communication and signals from the complex microenvironment during development have important roles in the proper maintenance and differentiation of renal cells. PSC-derived renal cells can be generated following the same developmental events that occur during the embryonic period [2]. In recent years, multistep protocols have been used for differentiation of hPSCs into the renal lineage by recapitulating stages of in vivo kidney development. Stepwise addition of small molecules and growth factors is essential for in vitro kidney differentiation, and includes intermediate cell populations such as the primitive streak, intermediate mesoderm (IM), renal progenitors, pretubular aggregate, renal vesicle, and the mature kidney [3]. The results of several studies indicate that long-term application at a high concentration of CHIR99021, as a canonical Wnt agonist, could induce T-positive posterior primitive streak (PPS) cells from hPSCs [9]. The highly conserved activin/Nodal-like signaling pathway promotes the specification of IM cells during early development [10 –12]. This finding has led us to hypothesize that inducer of definitive endoderm 1 (IDE1), a cost-effective small molecule that activates the Nodal signaling pathway, could be an alternative to activin A to induce expression of IM markers when added after T-positive PPS generation. Small molecules are stable, cell permeable, easy to use, cost-effective, and have low batch-to-batch variation compared with other molecules that are used in differentiation protocols [13].
Nephrogenic IM includes four major progenitor cell populations: ureteric-bud progenitor cells, nephron progenitor cells (NPCs), stromal progenitors, and endothelial progenitor cells (EPCs). The mature kidney consists of various types of specialized cells such as different nephron segments, collecting duct, interstitial stromal and endothelial cells (ECs) [2]. Kidney development depends on the migration of endothelial and stromal cells from other embryonic tissues into the developing kidney [12,14,15]. ECs play a principal role in delivery of oxygen and micronutrients to the developing kidney, and paracrine factors secreted from ECs have a key role in proper differentiation and maturation of renal progenitors [2,16]. On the contrary, several renal cell types have been shown to promote survival, migration, and tube formation of ECs, as well as the proper spatial distribution of renal vessels during kidney development [2]. ECs generation in the majority of current hPSC protocols was very low (≤0.3% of total cells) [17], and many of these ECs have immature features [18]. Direct cell–cell interactions and angiogenic growth factors secreted by mesenchymal stem cells (MSCs) have a critical role in the development of functional blood vessels [19,20]. Like pericytes, MSCs are required for the stabilization and maturation of the ECs [21]. In this study, we first developed a new method for generating renal precursors, and then determined whether coculture of supportive cells (MSCs and ECs) could increase the maturation of hPSC-derived renal precursors in a 3D culture. We observed that renal microtissue derived from coculture of renal precursors with endothelial and mesenchymal stem cells (RMEMs) derived from coculture of supportive cells and hPSC-derived renal precursor cells in ultralow attachment (ULA) 96-well plates enhanced gene expressions of renal-specific markers. RMEMs could potentially be used at a relatively low cost for high-throughput drug screening and nephrotoxicology studies.
Materials and Methods
Expansion culture of hPSCs
The study protocols and informed consents for human cells were approved by Medical Ethics Committee of Royan Institute (Tehran, Iran;
Differentiation of hPSCs toward renal lineages
The hPSCs were enzymatically dissociated into single cells with 0.05% trypsin/EDTA (Cat. No. 25300-062; Gibco) at 37°C for 2 min and then plated at 5 × 104 cells/cm2 on Matrigel-coated multiwell plates in the same hPSC medium with 10 mM Rho-associated protein kinase (ROCK) inhibitor (Cat. No. Y0503; Sigma-Aldrich). After 24–48 h, the first step of differentiation was performed by treating hPSCs with 8 μM of the small molecule CHIR99021 (CHIR, Cat. No. 04-0004; Stemgent, Cambridge, MA) in differentiation medium for a total of 4 days. CHIR99021 is an activator of the canonical Wnt/catenin pathway. In this study, differentiation medium consisted of advanced RPMI 1640 basal medium (Cat. No. 12633-012; Gibco) supplemented with 2% B-27 (Cat. No. A18956-01; Gibco), 2 mM GlutaMAX, 0.1 mM NEAAs, 1% penicillin/streptomycin, and 0.1 mM β-mercaptoethanol. To achieve IM, we changed the medium to the step 2 differentiation medium that contained 10 ng/mL activin A (Cat. No. A4941; Sigma-Aldrich). For some of the experiments, we used varying concentrations (20, 50, and 100 nM) of IDE1 (Cat. No. 04-0026; Stemgent) as a Nodal agonist molecule for 3 days. Then, the differentiated cells were harvested on day 7 and analyzed by quantitative real-time polymerase chain reaction (qRT-PCR). The differentiation medium was supplemented with 10 ng/mL human fibroblast growth factor 9 (FGF9, Cat. No. 272-F9-025; R&D Systems) for 2 days. On day 9 of directed differentiation, the medium was switched to step 4 medium that contained 10 ng/mL human FGF9 and 3 μM CHIR for 2 days. On day 11, the medium was changed to step 5 differentiation medium that contained 10 ng/mL human FGF9 for 3 days. On day 14 of differentiation, the medium was changed to differentiation medium (without growth factor) for a total of 4 days.
Isolation and culture of human umbilical vein endothelial cells from human umbilical cords
The study protocols and informed consents for human cells were approved by Medical Ethics Committee of Royan Institute (
Human bone marrow-derived mesenchymal stem cells culture
The characterized human bone marrow-derived mesenchymal stem cells (hBMMSCs) were obtained from Royan Stem Cell Bank. hBMMSCs were cultured in low-glucose DMEM (Cat. No. 1885-084; Gibco) supplemented with 10% fetal bovine serum (Cat. No. 10270; Gibco), 2 mM GlutaMAX, 0.1 mM NEAAs, and 1% penicillin/streptomycin. The hBMMSCs were used for up to four passages.
Generation of renal microtissues (and RMEM) on a presolidified Matrigel bed
RMEMs were generated from hPSC-derived renal precursors and supportive cells [hBMMSCs and HUVECs]. First, we evaluated previously described method for aggregate formation [7]. We examined several different cell ratios at a total cell number of 200,000 in 96-well growth factor-reduced (GFR) Matrigel (Cat. No. 354230; Corning) -coated plates. All cell types were incubated at 37°C in a humidified 5% CO2 environment. Each cell type was dissociated by incubation in 0.05% trypsin/EDTA at 37°C for 2–5 min. The cell suspension was centrifuged for 5 min at 300g and resuspended in RMEM culture media. The resultant cell suspension was transferred onto a presolidified Matrigel bed [50% dilution of GFR Matrigel (Cat. No. 354230; Corning), solidified at 37°C for 20 min] with the same RMEM culture medium that was composed of a 1:1 EGM-2/kidney differentiation medium supplemented with 10 ng/mL human FGF9 and 10 mM ROCK inhibitor. On day 2 after coculture, the medium was switched to step 4 RMEM medium that contained 10 ng/mL human FGF9 and 3 μM CHIR for 2 days. Then, the medium was changed to step 5 RMEM medium that contained 10 ng/mL human FGF9 for 3 days. On day 14 of differentiation, the medium was changed to RMEM medium without growth factor for 4 days.
Generation of renal microtissues (and RMEM) in ULA plates
Then, we examined several different cell ratios (10:5:5, 10:2:1, and 10:7:2) at a total cell number of 200,000 in 96-well U-bottom ULA plates. All cell types were incubated at 37°C in a humidified 5% CO2 environment. Each cell type was dissociated by incubation in 0.05% trypsin/EDTA at 37°C for 2–5 min. The cell suspension was centrifuged for 5 min at 1,500 rpm. The resultant supernatant was replaced by the RMEM medium that was composed of 1:1 EGM-2/kidney differentiation medium supplemented with 10 ng/mL human FGF9 and 10 mM ROCK inhibitor. The cell suspension was seeded into a 96-well ULA U-bottom plate (Cat. No. CLS7007; Corning® Costar). After 12 h of culture, the cells were aggregated at ∼200 000 cells/aggregate in each well of the 96-well U-bottom ULA plates. The RMEM medium was changed according to the same conditions described for the 3D coculture system onto a presolidified Matrigel bed.
LIVE/DEAD assay
Cell viability of the RMEMs was evaluated using LIVE/DEAD Viability/Cytotoxicity Kit (Cat. No. L3224; Invitrogen) on day 18 according to the manufacturer's instructions. In brief, the RMEMs were washed with phosphate buffered saline (PBS) and stained using 0.2 mM calcein-AM and 0.1 mM ethidium homodimer for 30 min at room temperature (RT). Then, the samples were washed and investigated by a fluorescence microscope (IX71; Olympus, Tokyo, Japan).
Nephrotoxicity assays
Next, we conducted an in vitro analysis of nephrotoxicity. The RMEMs, 11 days after coculture, were exposed to 5 and 50 μM cisplatin (Cat. No. P4394; Sigma-Aldrich), a known anticancer drug for 24 h, and 5 mg/mL gentamicin (Cat. No. G1264; Sigma) for 48 h. The RMEMs were subsequently collected for qRT-PCR analysis. Furthermore, RMEMs were lysed, and kidney injury molecule-1 (KIM-1) protein concentration was assessed by enzyme-linked immunosorbent assay (ELISA; MesoScale Discovery) according to the manufacturer's instructions.
Ex ovo chorioallantoic membrane transplantation of RMEMs
Fertilized Hy-line W-36 eggs from laying hens were supplied by a commercial farm (Iran). The eggs were incubated horizontally at ∼37°C in a 60% humidified incubator. The experimental steps were carried out inside a biological laminar-flow hood. First, the eggs were cracked, and their contents were transferred to a large sterile petri dish. The yolk-embedded embryo was then transferred to the first surrogate shell, which was slightly (3–4 g) heavier than the egg shell. Then, the shell was sealed with a conventional plastic tape for incubation in forced air circulation for 60 h at 37°C and 60% humidity. This was considered to be embryonic day 0 (ED 0). At ED 2.5, the yolk-embedded embryo was transferred to another surrogate shell, which was 35–40 g heavier than the egg shell, sealed with a conventional plastic tape, and incubated for 7 days. Then, day 18 RMEMs were implanted onto the surface of the chorioallantoic membrane (CAM) at ED 7.5 distal from the embryo and proximal to major blood vessels, and maintained in an incubator for 7 additional days (until ED 12.5). Under a dissecting microscope, fluorescein isothiocyanate–dextran (Cat. No. FD-2000S; Sigma-Aldrich) was injected into one of the CAM vessels to evaluate vascularization. The RMEMs were collected for hematoxylin and eosin (H&E) staining and gene expression analysis.
RNA isolation and qRT-PCR
Total RNA was extracted with an RNeasy Micro Kit (Cat. No. 740902.10; Nucleospin RNA XS) according to the manufacturer's instructions from the following groups of cells: hPSCs; hPSC-derived cells at 4, 7, 9, and 18 days of differentiation in a two-dimensional (2D) culture; hPSC-derived renal microtissues (RMs); RMEMs; and kidneys from a 21-year-old male and a 21-week-old fetus. Complementary DNA (cDNA) was synthesized from 1 μg of resultant RNA using a Revert Aid First Strand cDNA Synthesis Kit (Cat. No. 4368813; Thermo Fisher Scientific). qRT-PCR was performed to quantify gene expression from cDNAs using SYBR Green Master Mix (Cat. No. RR820L; Takara Bio, Kusatsu, Japan) by the Step One Plus™ Real-Time PCR System. The data were normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and calibrated with pluripotent status. Values were calculated by the CT method (2−ΔΔCt). Table 1 shows the primer sequences used in this research. Samples were obtained from three to five independent biological replicates, and all reactions were performed in technical duplicates.
List of Primers Used for Quantitative Real-Time Quantitative Polymerase Chain Reaction
Immunostaining assays
The cultured cells were washed twice with DPBS and fixed in 4% paraformaldehyde (Cat. No. P6148; Sigma-Aldrich) for 20 min. The fixed cells were washed three times in DPBS and incubated in blocking buffer that consisted of 0.1% Triton X-100, 10% secondary antibody host serum, and 0.5% bovine serum albumin (BSA) for 1 h at RT. Then, the cells were allowed to incubate overnight at 4°C or for 2 h at RT with primary antibodies (Table 2). The cells were then washed three times in DPBS and incubated with secondary antibodies (Table 2) for 2 h. When the samples were immunostained with biotinylated LTL (Cat. No. B-1325; Vector Labs), a blocking step using the Streptavidin/Biotin Blocking Kit (Cat. No. SP-2002; Vector Labs) was performed, and fluorescent conjugates of streptavidin (Alexa Fluor 488) were used to detect biotinylated LTL. Cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, Cat. No. D8417; Sigma-Aldrich) for 1 min. Immunofluorescence staining was visualized under a microscope (IX71; Olympus) and captured by an Olympus DP72 digital camera. Quantification was performed using ImageJ (RRID: SCR_003070) by counting at least five random images per experiment in three independent repeats.
List of Antibodies Used for Immunostaining
The day 18 microtissues were collected for immunohistofluorescence, washed with DPBS, and fixed overnight in 4% formalin. The samples were then incorporated into pre-embedding agar gel (2%) and embedded in paraffin wax. The paraffin-embedded samples were sectioned into 5 μm sections using a microtome (MicromHM325; Thermo Fisher Scientific). The microtissue slides were then subjected to H&E staining. For immunohistofluorescence, the sections were deparaffinized, and a heat-mediated antigen retrieval step using Dako target retrieval solution (S2368; Glostrup, Denmark) was performed according to the manufacturer's instructions. Permeabilization was performed using 0.5% Triton X-100 in DPBS for 20 min at room temperature. Then, the microtissue sections were blocked for 45 min at RT with 10% BSA in DPBS. The samples were incubated overnight at 4°C with primary antibodies (Table 2). The slides were then washed three times with DPBS that contained 0.5% Tween-20 (PBS/T) for 15 min each, and incubated with secondary antibodies (Table 2) for 45 min at 37°C. The microtissue sections were washed three times with PBS/T for 15 min each and then stained with DAPI for 1 min. Images were acquired with a fluorescence microscope (IX71; Olympus) equipped with an Olympus DP72 digital camera.
Flow cytometry analysis
hPSCs-derived cells were dissociated into single cells using trypsin/EDTA (for 5 min at 37°C) and fixed in 2% paraformaldehyde for 15 min at 4°C. Samples were permeabilized in 0.1% Triton X-100 for 15 min at 4°C and blocked with 5% donkey serum in PBS for 15 min at RT. Cells were incubated with primary antibody for 30 min, then washed with 1% BSA in PBS two times. The secondary antibody was applied for 30 min at 4°C, followed by twice washing with 1% BSA in PBS. Cells were then analyzed using a flow cytometer (FACS Calibur; BD Biosciences, San Jose, CA) and flowing software, version 2.5.1 (RRID: SCR_015781). All experiments were repeated three times.
Statistical analysis
All data are presented as the mean ± standard deviation from at least three biological and two technical replicates. Statistical analysis was done using the unpaired two-tailed Student's t-test for pairwise comparisons or one-way and two-way analyses of variance, followed by the appropriate post hoc test in SPSS software (SPSS 16.0) for assessment of differences between more than two groups. P < 0.05 was considered to be statistically significant.
Results
Differentiation and characterization of hPSC-derived renal lineages
We used a two-step differentiation protocol that included primitive streak and IM to produce nephrogenic IM (Fig. 1A). Before starting the differentiation, the pluripotency of hPSCs was confirmed by immunostaining for NANOG and OCT4 (Fig. 1B). The hPSCs were treated with a high dose of CHIR (8 μM) for 4 days in a 2D monolayer culture. qRT-PCR analysis revealed the upregulation of the PPS marker gene, T (Fig. 1C and Supplementary Fig. S6A). Furthermore, immunofluorescent staining showed a large population of brachyury-positive cells (90.06% ± 4.4%) (Fig. 1D, E). To determine whether IDE1 could replace activin A in the differentiation method, we added varying concentrations of IDE1 (20, 50, 100 nM) to T-positive cells for an additional 3 days as a replacement for activin A. Then, the differentiated cells were harvested on day 7 and analyzed by qRT-PCR. We observed significantly increased expressions of nephrogenic markers WT1, HOXD11, EYA1, and FOXD1 in cells differentiated with 20 nM IDE1 compared with cells differentiated using activin A (Fig. 1F). qRT-PCR analysis confirmed increased expressions of LHX1, OSR1, PAX2, and HOXD11 (nephrogenic IM markers) in IDE1-treated IM (day 7) relative to undifferentiated cells (day 0). In contrast, the PPS marker (T) was downregulated (Fig. 1C and Supplementary Fig. S6A). Furthermore, flow cytometry analysis revealed that 88.32% of differentiated cells were WT1+, 77.18% PAX2+, and 49.78% LHX1+ (Fig. 1G). Treatment of nephrogenic IM cells (day 7) with FGF9 (10 ng/mL) for 2 days resulted in the formation of NPCs on day 9. Immunofluorescence staining showed that 89.33% ± 6.6% of the hPSCs-derived cells expressed WT1, and 88.45% ± 3.6% of cells expressed SIX2 (Fig. 1D, E). Using qRT-PCR, we documented the expressions of renal-specific transcripts that characterized nephron progenitors (EYA1, SALL1, WT1, and SIX2) and stromal progenitors (FOXD1) on day 9, as well as mesangial cells (PDGFR-β), podocytes (WT1, PODXL, NPHS1), proximal tubules (AQP1), loops of Henle (UMOD), distal tubules (CDH1), and collecting duct (GATA3) on day 18 in the monolayer culture (Fig. 1C, H and Supplementary Fig. S6A). Figure 1 provides details of the differentiation methods and analysis of gene expressions at each stage of differentiation during 18 days in the monolayer culture. Furthermore, Supplementary Fig. S1 shows the morphology of the cells during the differentiation process. These data indicated that renal lineage cells could be generated from this approach.
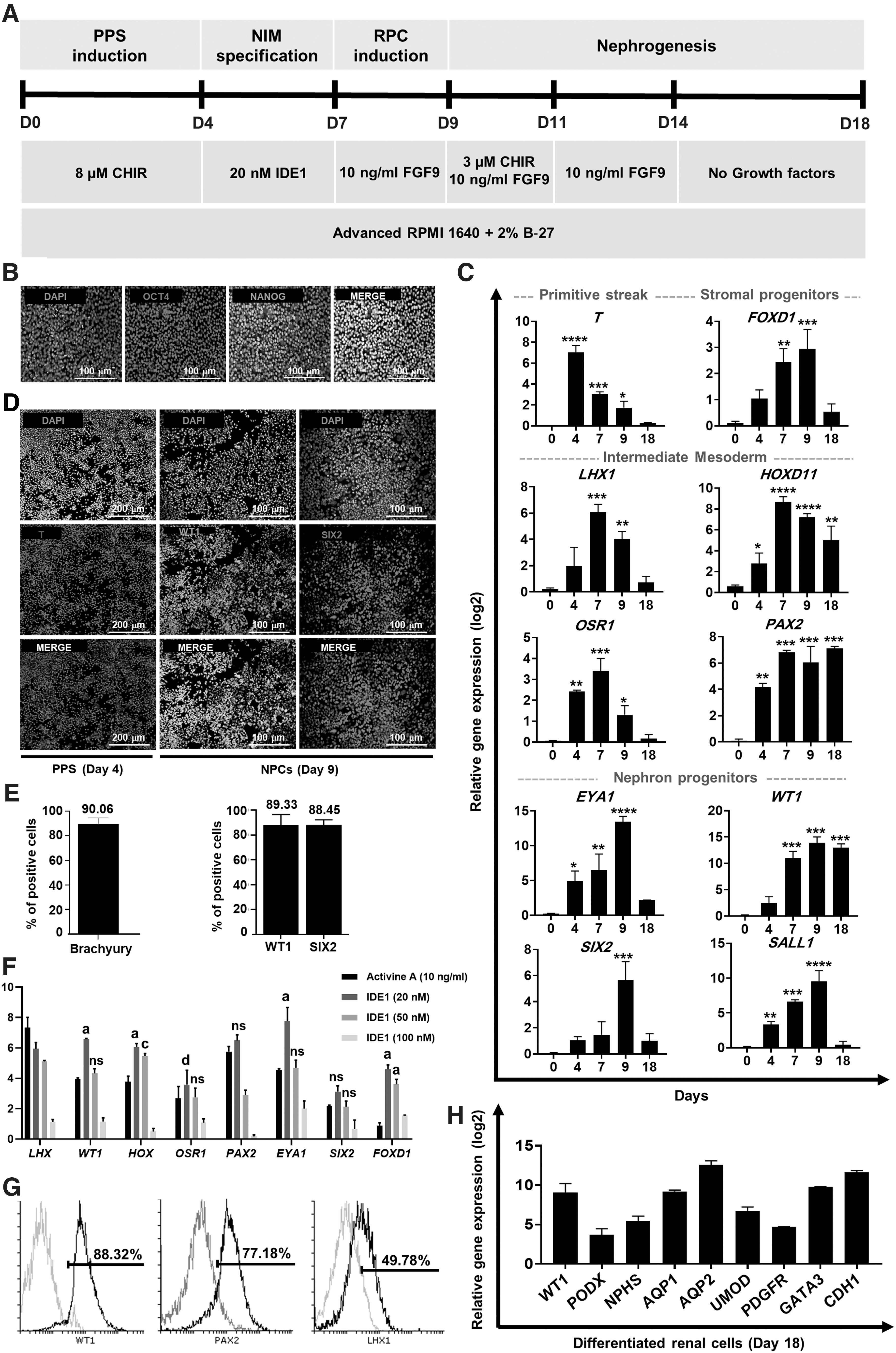
Differentiation of hPSCs into renal lineages in a 2D culture.
Generation and characterization of 3D RMEM in a coculture system
Initially, we evaluated the Takebe method for generation of aggregates [7]. hPSCs-derived nephrogenic IM cells were cocultivated with HUVECs and hBMMSCs at several different cell ratios on a presolidified Matrigel bed. At 24 h after the coculture, we observed that the self-assembled aggregates were formed in groups with supportive cells (Supplementary Fig. S2). We did not observe any obvious condensed structures in groups without supportive cells. At 6 days after coculture on the Matrigel bed, the MSCs dispersed outward from the aggregates and the organoid structures were disrupted. Therefore, the aggregates could not be maintained for an extended period of time in the in vitro culture with this method (Supplementary Fig. S2).
Next, day 7 inducible nephrogenic IM were cocultivated with HUVECs and MSCs at several different cell ratios (10:5:5, 10:2:1, and 10:7:2) in ULA U-bottom plates (Fig. 2A). Condensed multicellular aggregates were formed 12 h after the coculture (Fig. 2B). These aggregates were cultured for 11 days in vitro. To determine the optimal cell ratios in forming RMEMs, we used qRT-PCR analysis of renal-specific markers on day 18. We observed that several renal-specific genes were expressed at remarkably higher ratio of 10:7:2 compared with other groups (P < 0.01) (Supplementary Fig. S3). Thus, the qRT-PCR results showed that an optimal cell ratio was 10:7:2. Immunohistofluorescence analysis for endothelial marker CD31 and renal-specific markers LTL, SYNPO, and E-cadherin was performed on day 18 (Fig. 2C). Immunohistofluorescence for CD31 showed that the endothelial structures were distributed throughout the RMEMs. Immunohistofluorescence of RMEM showed kidney-like structures. Expressions of proximal tubule marker (LTL), distal tubular marker (E-cadherin), and podocyte marker (synaptopodin) were observed (Fig. 2C). Furthermore, live/dead staining showed that most cells inside the RMEMs were viable on day 18, with only a small number of dead cells (Supplementary Fig. S4). The data suggest that the differentiation protocol and coculture system had no adverse effect on the cell viability of RMEMs.
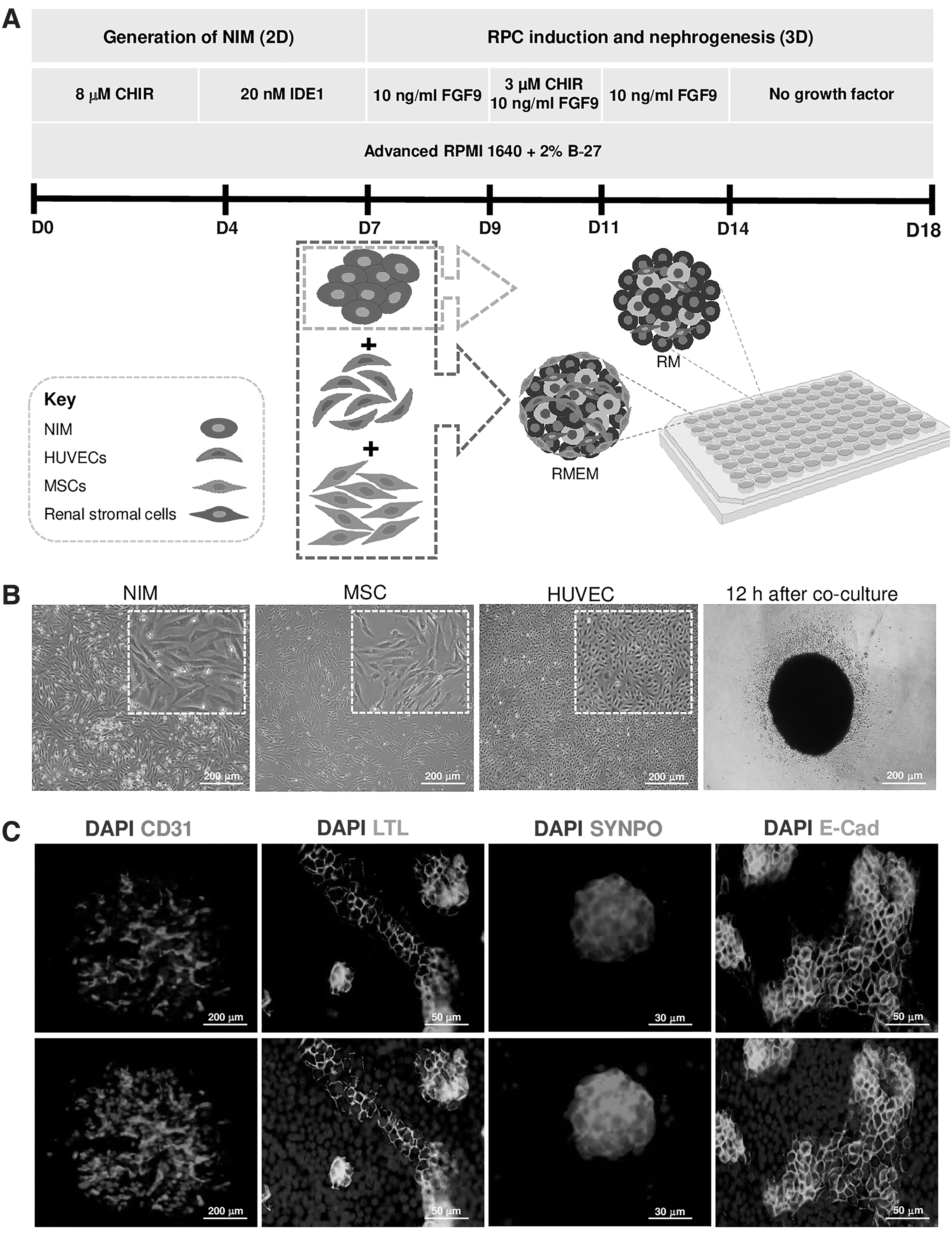
Generation of RMEMs.
Gene expression of renal-specific markers in 3D RMEMs
We used qRT-PCR to assess the impact of supportive cells on differentiation and maturation of RMEMs by measuring the expressions of AQP1, AQP2, CDH1, WT1, NPHS1, PODXL, PDGFR-β, UMOD, and GATA3. There was a significant increase in gene expressions of the renal-specific markers AQP1, AQP2, CDH1, PODXL, PDGFR-β, UMOD, and GATA3 in RMEMs compared with RMs without supportive cells. We observed that the expression levels of the renal signature genes AQP1, AQP2, GATA3, WT1, and PODXL reached levels similar to those detected in fetal kidney tissue. Expressions of all of the renal-specific genes were lower than those of the adult kidney tissue (Fig. 3 and Supplementary Fig. S6B).
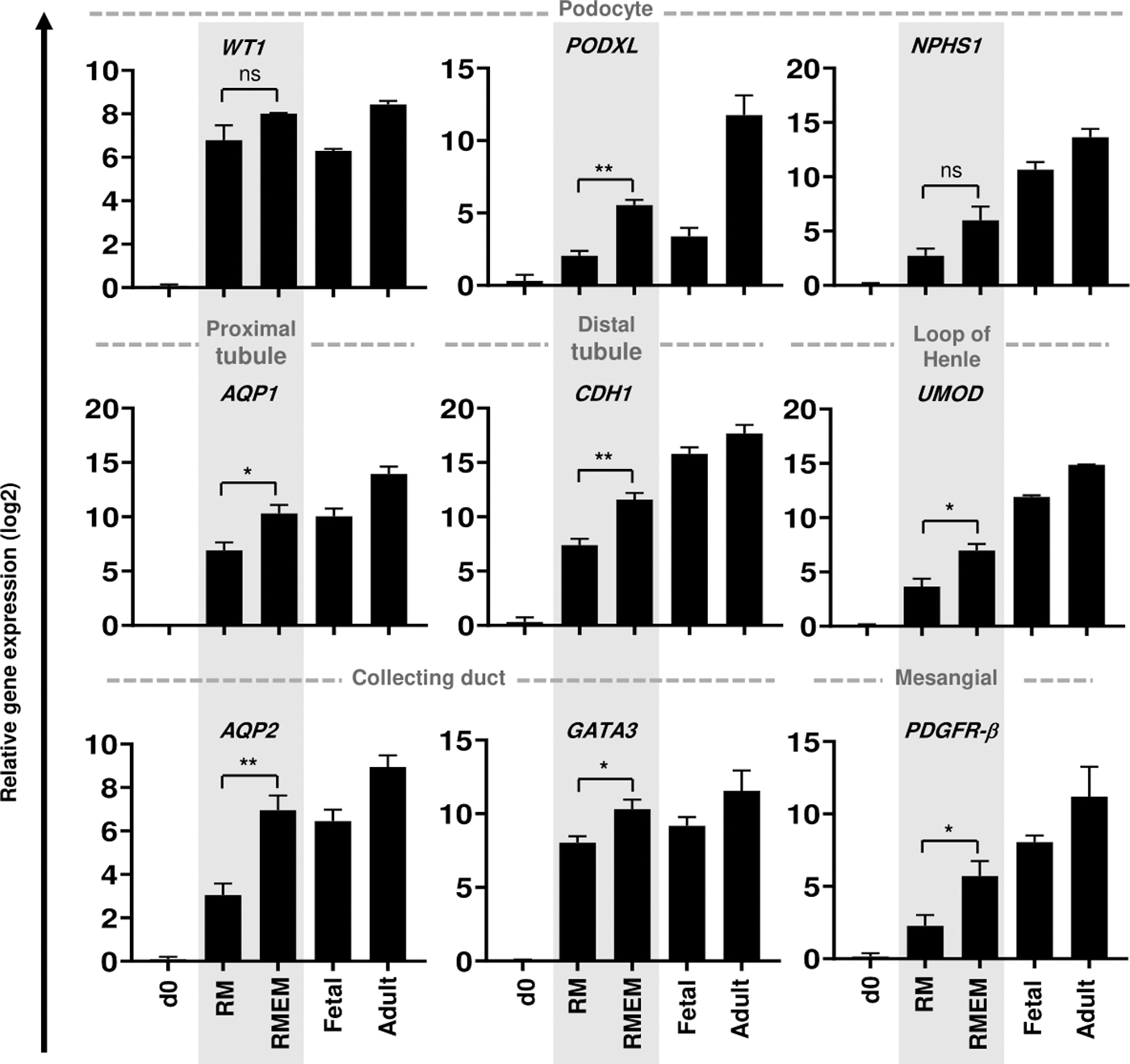
Gene expression analysis of RMEM and RM derived from RH5 hESCs. Transcriptional expressions of renal-specific genes that include podocyte markers (WT1, PODX1, and NPHS1), a proximal tubule gene (AQP1), a distal tubule gene (CDH1), the loop of Henle marker (UMOD), a mesangial gene (PDGFR-β), and collecting duct genes (AQP2, GATA3) in RM, RMEM, human fetal kidney tissues at 21 weeks of gestation, and an adult kidney sample from a 21-year-old male. Fetal and adult kidney tissues were the control groups. Each sample is a pool of four organoids. Data were normalized against GAPDH and compared with day 0 RH5 hESCs as the calibrator. Data are shown as mean ± SD (n = 3). Statistical analysis was performed using one-way ANOVA. *P < 0.05 and **P < 0.01.
Ex ovo transplantation of RMEMs enhances maturation of renal lineage cells
The d18 RMEMs were transplanted on the CAM with the hypothesis that the proper microenvironmental niche would improve maturation of the RMEMs. H&E staining and fluorescent dextran injection visualized chick embryo vessels in the vicinity of the RMEMs (Fig. 4A, B). We further explored the expressions of renal-specific markers 7 days after the ex ovo transplantation. qRT-PCR analysis showed significant increases in the expressions of the renal-specific genes that included podocytes (WT1, PODXL, NPHS1), proximal tubules (AQP1), loops of Henle (UMOD), distal tubules (CDH1), and collecting duct (AQP2, GATA3) in the transplanted RMEMs relative to d18 RMEMs (Fig. 4C). These findings suggested that various cellular communications and a suitable microenvironmental niche supported further differentiation and maturation of RMEM cells.
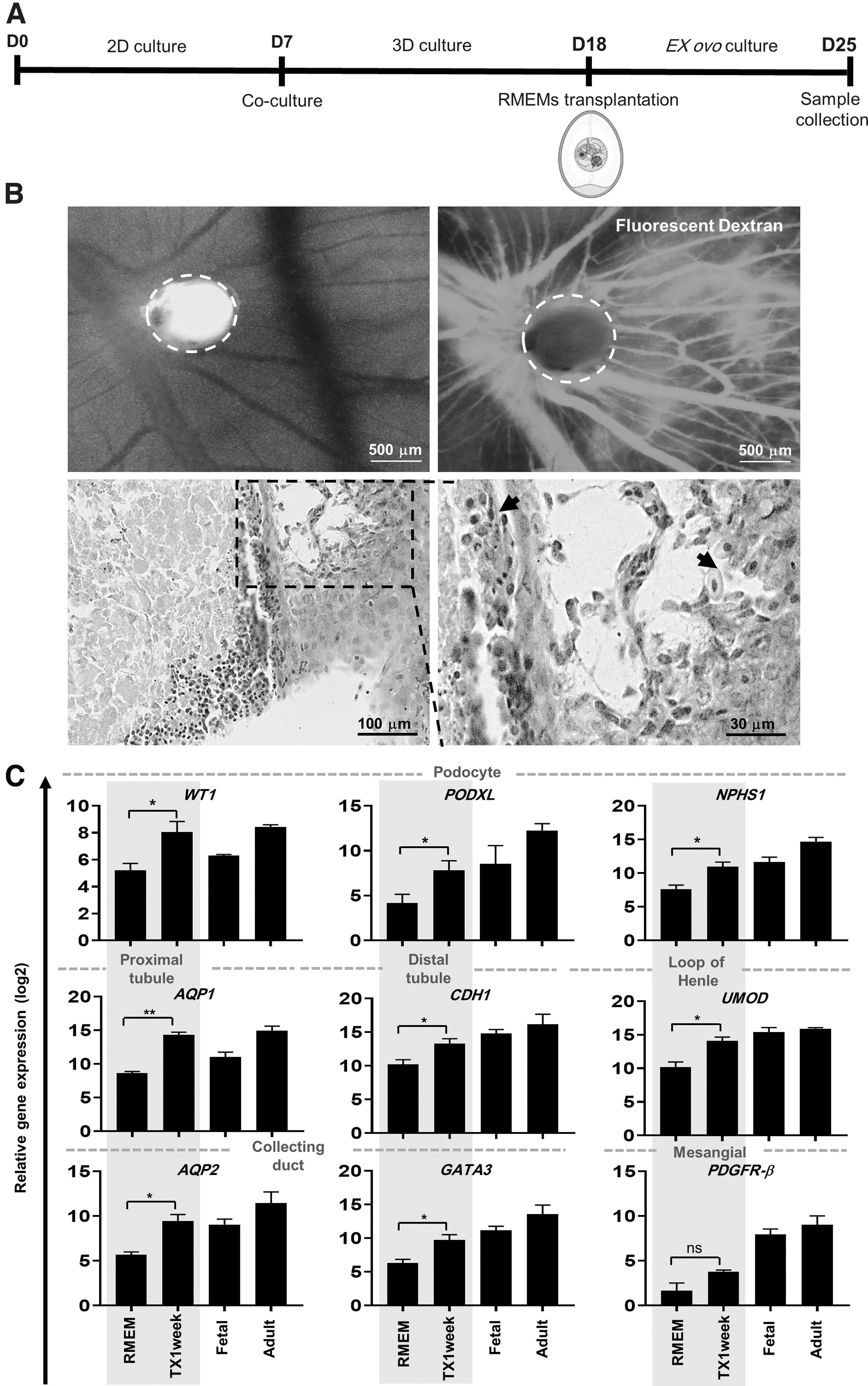
Ex ovo implantation of RM derived from coculture of endothelial and mesenchymal cells with hPSC-derived renal precursor cells (RMEM).
Nephrotoxicity assay of RMEMs
To demonstrate the functionality of these RMEMs, the day 18 samples were administered 5 and 50 μM cisplatin, a known anticancer drug for 24 h, and gentamicin, an antibiotic agent (5 mg/mL) for 48 h. We analyzed the expression of KIM-1, a specific biomarker for tubular injury in RMEMs. qRT-PCR results indicated that KIM-1 expression markedly increased in RMEMs treated with cisplatin and gentamicin. Upregulation of KIM-1 in the cisplatin-treated RMEMs was dose dependent (Fig. 5A). ELISA confirmed an increase in protein concentration of Kim-1 in RMEMs treated with cisplatin and gentamicin (Fig. 5B).
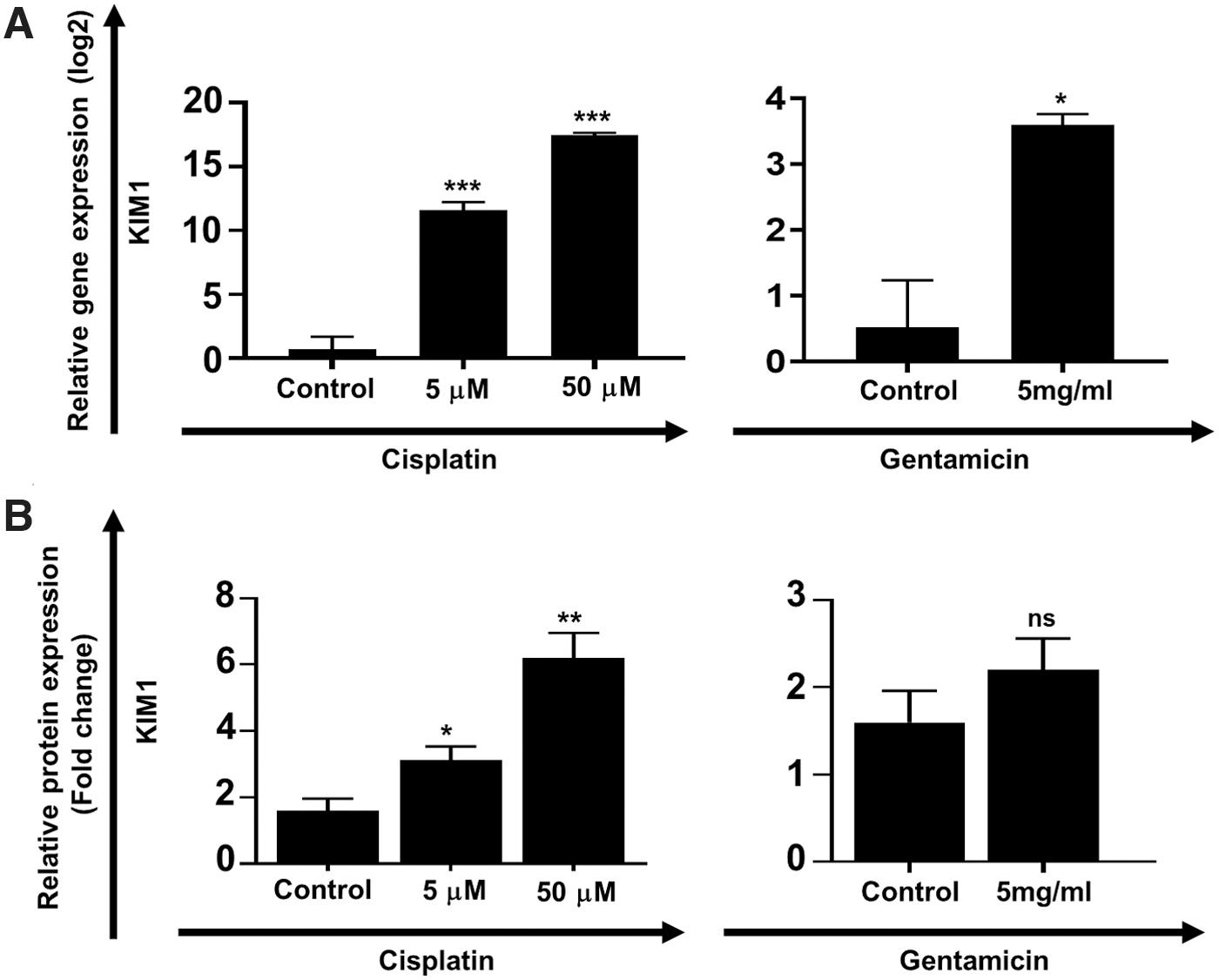
RMEMs as a model for toxicological studies.
Discussion
hPSC-derived differentiated cells have been introduced as a novel cell source for the study of kidney development, physiology, drug screening, and regenerative medicine [1]. The stepwise processes of directed differentiation that uses a growing number of small molecules and growth factors (eg, CHIR99021, Activin A, FGF9, and BMP7) drive forward differentiation of hPSCs to renal lineage cells [3,8]. Unfortunately, these protocols have numerous challenges, including extended time periods and expensive differentiation procedures, as well as the immaturity of differentiated cells [2]. These cells are not suitable for clinical applications due to graft immaturity and safety concerns [22]. Therefore, tremendous efforts have been taken to develop safe, efficient, inexpensive, and rapid approaches.
In this study, we first established a new protocol for generating renal precursors and then developed an organotypic coculture model using ECs and MSCs as supportive cells that favored further differentiation of renal precursors. The findings of a positive effect of the activin/Nodal-like signaling pathway on the specification of the posterior intermediate stage led us to use a cost-effective small molecule, IDE1, as a replacement for activin A. Gene expression analysis of multiple renal lineage markers in cells differentiated with IDE1 shows that this small molecule has higher efficiency than activin A. IDE1-induced SMAD2/3 phosphorylation has a lower toxicity and higher efficiency than activin A [23]. The results of studies show that this small molecule can be used for directed differentiation of definitive endoderm [24,25] and microglial cells [26]. To the best of our knowledge, this is the first study that has demonstrated the effect of IDE1 in differentiation of hPSCs into nephrogenic IM. Our data show that renal lineage cells can indeed be generated using this alternative method. Although the underlying mechanisms for the IDE1 effects in nephrogenic IM stage are beyond the goal of the present research, future studies might aim to answer questions that pertain to the mechanisms by which IDE1 regulates downstream targets.
In this study, we showed that the coculture of hPSCs-derived renal precursors with MSCs and ECs could improve maturation of renal cells in a 3D culture. Nephrogenic IM includes at least four major cell populations: NPCs, ureteric-bud progenitor cells, stromal progenitors, and ECs [2]. In vitro recapitulation of this nephrogenic niche is a novel approach to the generation of 3D renal structures from hPSCs. ECs generation in the majority of current hPSC protocols was very low [17], and many of these ECs have immature features [18]. Our results confirm previous evidence (Supplementary Fig. S5). Numerous researchers observed that when hPSC-derived progenitor cells were combined with stromal cells and ECs, the cell mixture self-organized into 3D structures included pancreatic [27,28], liver [29], and cardiac [30] organoids. Coculture methods have been used to generate higher order and vascularized 3D kidney structures. All of these studies were based on 3D coculture of murine primary renal cells with endothelial and stromal cells [7,8,31]. Takebe and colleagues used a triculture system that combined E13.5 kidney-derived cells with HUVECs and bone marrow-derived MSCs onto Matrigel. Implantation of organ buds inside the cranium further promoted differentiation and neovascularization in the kidney buds. However, organoids generated by this approach had some disadvantages, including limited maturation before transplantation, and a large size that contributed to cell death in the center regions of the aggregates and inhibited its potential to differentiate well in vitro [7,32]. This approach produced kidney structures with a 3D system based on Matrigel. The use of Matrigel has several disadvantages, including poorly defined compositions, interbatch variability, and xenobiotic product derived from mouse sarcoma [33]. Furthermore, long-term culture of aggregates on a Matrigel bed would lead to the dispersion of MSCs outward from the aggregates and disruption of the organoid structures (Supplementary Fig. S2).
In the current research, we established a novel coculture method to generate small diameter renal structures that can improve micronutrient permeation and absorption. For this purpose, RMEMs were generated under supportive 3D coculture conditions in ULA 96-well round-bottom plates that were suitable for high-throughput screening. We observed that seeded mixed cell populations endured condensation, and differentiated into the renal lineage and endothelial structures. Our results showed that RMEMs derived from ECs, MSCs, and hPSC-derived renal precursors had enhanced expressions of renal signature genes that included AQP1 (proximal tubule), CDH1 (distal tubule), UMOD (loop of Henle), WT1, PODXL, and NPHS1 (podocyte), and PDGFR-β (mesangial). Further maturation of RMEM was achieved by subsequent transplantation of these aggregates into the CAM. The CAM is a naturally immunodeficient host that maintains grafted microtissues without species-specific limitations [6,29]. Studies indicated that soft substrates with mechanical properties similar to early embryonic microenvironment such as CAM could improve the maturation of renal microstructures [6,7]. Our results showed an increase in the expression of the renal-specific genes in the transplanted RMEMs.
Paracrine factors secreted from ECs play a principal role in proper kidney organogenesis and regeneration [1,2,33]. Myosin IIA is expressed by MSCs; it directs mechanical movements of dispersed cell populations, and acts to ensure that cells are triggered to undergo self-condensation [7]. In addition to their role in microtissue formation, MSCs possess beneficial immunomodulatory and regenerative effects that might provide further advantages for clinical applications [1]. Previous research indicated that stromal cells produced various cytokines and growth factors (such as FGFs, HGF, and VEGF), which have key roles in kidney development and regeneration [1,2]. MSCs have been used to treat various kidney diseases, including acute [34], chronic [35], and polycystic [36,37].
Drug-induced nephrotoxicity is one of the most common causes of acute renal failure in hospitalized patients. In vitro renal cell-based models provide a platform for drug nephrotoxicity assay. The utility of these models will be dependent upon the functional maturation of renal cells. To demonstrate whether our RMEMs could be used to study drug-induced nephrotoxicity, day 18 samples were administered 5 and 50 μM cisplatin, a known chemotherapeutic agent, and gentamicin (5 mg/mL). Cisplatin-induced nephrotoxicity involves the proximal tubular cells and to a lesser extent the distal tubule and glomerular cells. Cisplatin causes DNA damage, apoptosis, necrosis, and inflammation in renal cells [38]. Gentamicin, an antibiotic agent, causes well-established tubular cytotoxicity through apoptosis and necrosis of cells. Gentamicin-induced cytotoxicity involves the proximal tubular cells and to a lesser degree the distal tubule [39]. We analyzed the expression of KIM-1, the most specific biomarker for tubular injury that is very low under normal circumstances and highly upregulated in the kidney after acute injuries [38]. Our results indicated that KIM-1 expression markedly increased in RMEMs treated with cisplatin and gentamicin. Thus, RMEMs could be used as an in vitro model for drug-induced nephrotoxicity assays.
Although the current method proves useful, it has some limitations. However, our RMEMs expressed some markers of terminal differentiation; they have relatively immature tubular and vascular structures. The expressions of renal-specific genes were lower than those of the adult kidney tissue. Furthermore, HUVECs have been used for RMEM formation, which exert a limited proliferation rate [40]. Coculture of EPCs derived from hPSCs with renal precursors may induce further renal-specific vascular specification. Despite the advantages of BMMSCs, hPSCs-derived MSCs (ES-MSC) have a higher proliferative rate during in vitro expansion and overcome the limitations of harvesting MSCs from bone marrow [41]. The generation of fully hiPSC-derived organoids may be advantageous as in vitro kidney models with a homogeneous genetic background, thereby opening up a new approach in regenerative medicine and drug screening applications.
Conclusion
In conclusion, we established a low-cost, rapid method to generate a 3D RMEM from hPSCs. We used a cost-effective small molecule, IDE1, to generate nephrogenic IM cells that had less toxicity and higher efficiency than cells generated with activin A. By combining these cells with BMMSCs and HUVECs in a 3D coculture system, we generated RMEMs. The presence of endothelial and stromal cells provided a microenvironmental niche that mimicked human kidney features in vitro. Our novel approaches have helped in the low-cost generation of 3D models that may potentially be used in high-throughput drug screening and nephrotoxicology studies.
Footnotes
Acknowledgments
We express our appreciation to Saeed Yakhkeshi for his technical assistance. All listed authors of this article are members of the Royan Institute. Royan Institute is a nongovernmental research and educational institute dedicated to translational and clinical research, stem cell research, and infertility treatment.
Author Disclosure Statement
The authors declare no conflicts of interest.
Funding Information
This work was supported by a grant from Royan Institute (Grant 96000089).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.