
Abstract
Autosomal recessive hypercholesterolemia (ARH) is a rare monogenic disorder caused by pathogenic variants in the low-density lipoprotein receptor (LDLR) adaptor protein 1 (LDLRAP1) gene, encoding for the LDLRAP1 protein, which impairs internalization of hepatic LDLR. There are variable responses of ARH patients to treatment and the pathophysiological mechanism(s) for this variability remains unclear. This is in part caused by absence of reliable cellular models to evaluate the effect of LDLRAP1 mutations on the LDLRAP1 protein function and its role in LDLR internalization. Here, we aimed to validate patient-specific induced pluripotent stem cell (iPSC)-derived hepatocyte-like cells (HLCs) as an appropriate tool to model ARH disease. Fibroblasts from an ARH patient carrying the recently reported nonsense mutation, c.649G>T, were reprogrammed into hiPSCs using Sendai viral vectors. In addition, we used clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) to create an LDLRAP1 gene (also known as ARH) knockout in two different human iPSC lines. ARH patient-derived iPSCs, ARH-knockout iPSC lines, and control iPSCs were efficiently differentiated into HLCs. Western blot analysis demonstrated the absence of LDLRAP1 in HLCs derived from patient and knockout iPSCs, and this was associated with a decreased low-density lipoprotein cholesterol (LDL-C) uptake in ARH-mutant/knockout HLCs compared to control HLCs. In conclusion, we determined that the recently described c.649G>T point mutation in LDLRAP1 induces absence of the LDLRAP1 protein, similar to what is seen following LDLRAP1 knockout. This causes a decreased, although not fully absent, LDL-uptake in ARH-mutant/knockout HLCs. As knockout of LDLRAP1 or presence of the c.649G>T point mutation results in absence of LDLRAP1 protein, residual LDL uptake might be regulated by LDLRAP1-independent internalization mechanisms. Patient-specific iPSC-derived HLCs can therefore be a powerful tool to further decipher LDLRAP1 mutations and function of the protein.
Introduction
The liver is the major site for the synthesis and clearance of cholesteryl ester–rich lipoproteins. More than 70% of low-density lipoprotein cholesterol (LDL-C) is cleared from the blood circulation through hepatic low-density lipoprotein receptor (LDLR)-mediated endocytosis [1]. However, when the rate of LDL-C clearance from the blood is decreased due to a reduction in activity or membrane expression of LDLR this may result in hypercholesterolemia [2].
Familial hypercholesterolemia (FH) is one of the most frequent monogenic metabolic disorders that results in abnormally elevated levels of LDL-C. Clinical manifestations of this disorder comprise large xanthomas in different parts of the body and premature coronary heart disease [3].
Mutations in several genes, including the LDLR (MIM 606945), apolipoprotein B-100 (APO-B, MIM 107730), and proprotein convertase subtilisin/kexin type 9 (PCSK9, MIM 607786) have been linked to autosomal dominant hypercholesterolemia. Most mutations are generally located in the LDLR gene and result in defective LDLR protein. This prohibits LDL-C internalization and subsequent degradation within the cells [4]. In addition, several mutations, located in the APO-B gene alter the affinity of LDLR for LDL-Cs [5]. Gain-of-function mutations of PCSK9 can increase PCSK9-mediated lysosomal degradation of LDLR, causing reduced LDLR levels and consequently a decrease in plasma clearance of LDL-C [6,7]. On the other hand, LDLR adaptor protein 1 (LDLRAP1) protein (GenBank Accession no. NP_056442) is required for receptor-mediated endocytosis of LDL-C and mutations in LDLRAP1 (MIM 605747) result in an autosomal recessive form of FH [autosomal recessive hypercholesterolemia (ARH); Online Mendelian Inheritance in Man (OMIM) catalog number 603813], which has low prevalence (1–9/1,000,000) [1,8].
The human LDLRAP1 gene, located on the short arm of chromosome 1 (1p36.11), is comprised of nine exons and encodes the LDLRAP1 protein with 308 amino acids. LDLRAP1 mediates LDLR internalization through clathrin-coat machinery. LDLRAP1-based LDLR internalization depends on simultaneous binding of the N-terminal phosphotyrosine-binding (PTB) domain of LDLRAP1 to the FDNPxY motif within the cytoplasmic tail of LDLR, Clathrin Box module (C) to clathrin proteins, and the LDLRAP1 C-terminus to the β2-adaptin subunit of the adaptor protein 2 (AP2) [9,10].
In comparison with homozygous familial hypercholesterolemia (HoFH; OMIM number 143890) patients carrying mutations in the LDLR gene, which completely abolish receptor function (receptor-negative), ARH patients exhibit lower serum LDL-C levels, and the later onset of cardiovascular diseases [11]. Skin fibroblasts from ARH patients represent a normal activity of receptor-mediated endocytosis of LDL-C uptake, as Disabled 2 (Dab-2) has been shown to compensate for the absence of LDLRAP1 in fibroblasts [12]. ARH patients, however, show a significant reduction of hepatic LDL-C uptake [13]. Generally, ARH is more responsive to statins or a combination of statins and ezetimibe in comparison with HoFH patients [14,15]. Nevertheless, some ARH patients, despite receiving maximal tolerated doses of statins and an apheresis treatment, fail to attain normal LDL-C levels [16]. The pathophysiological mechanism for this variability remains unclear.
Different LDLRAP1 gene mutations have been reported in ARH patients [17]. Here, we investigated the effect of a homozygous mutation, c.649G>T, on LDLRAP1 protein expression and LDLR activity in patient-specific hepatocyte-like cells (HLCs) derived from induced pluripotent stem cells (iPSCs). This c.649G>T mutation was recently reported by our team [18]. Existing cellular models available to decipher LDLRAP1 function are restricted to hepatoma cells [19,20], fibroblasts [10,21], and lymphocytes [22]. However, these are not appropriate to study the function of LDLRAP1, as lipid metabolism in fibroblasts and lymphocytes serves as a limited surrogate for hepatocytes [23,24]. Therefore, the current study, establishing ARH patient-specific iPSCs and subsequently ARH patient-derived HLCs, represents a novel tool for modeling ARH and investigating of the role of LDLRAP1 in the internalization of LDLR/LDL-C complexes.
Materials and Methods
Cell cultures
BJ1 iPSCs were made in-house from BJ1 fibroblasts [25] and Sigma iPSCs (Sigma0028) were purchased from Sigma-Aldrich (Saint Louis, MO). Human iPSCs were maintained in feeder-free conditions on human matrigel (BD Biosciences, San Jose, CA) coated plates in Essential 8 Flex medium (A2858501; Gibco) with 0.1% U/mL penicillin–streptomycin in a humidified 5% CO2 incubator at 37°C. Colonies were split twice a week with 0.5 mM EDTA (Gibco) in phosphate-buffered saline (PBS). Cell cultures were regularly analyzed for potential mycoplasma contamination by quantitative PCR (qPCR). HepG2 hepatoma cells were maintained in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin–streptomycin in a humidified 5% CO2 incubator at 37°C. Brightfield pictures of the iPSC colonies were taken with a Leica MC120 HD microscope.
Generation of patient-specific iPSCs from patient-derived fibroblasts
Fibroblasts were harvested from a 17-year-old Iranian female ARH patient with the approval of the Ethics Committee of the Royan institute (
Quality control assays included single nucleotide polymorphism (SNP) quantitative polymerase chain reaction (PCR) to demonstrate cell identity using the TaqMan® SNP Genotyping Assay (Applied Biosystems), Array Comparative Genomic Hybridization (aCGH) to demonstrate genome integrity, pluripotency maintenance by embryoid body (EB) formation and TaqMan hPSC scorecard® analysis (Applied Biosystems), RT-qPCR for OCT3/4, SOX2, and NANOG mRNA (primer sequences are listed in Supplementary Table S1), and immunostaining for pluripotency proteins, including OCT4, SOX2, TRA-1-60, and SSEA4 (primary antibodies are listed in Supplementary Table S2). In addition, PCR-sequencing was used to identify the c.649G>T mutation in the LDLRAP1 gene in the ARH-iPSC line (primers used were FW:5′-CAGTCTAAGCCATTAGTCAGGTTCT and RV: 5′-CCTTTGGCTGGTCTTGTGGT).
Creation of ARH knockout iPSC lines
To test the guide RNAs (gRNAs) targeting LDLRAP1, we used the hepatoma cell line, HepG2. Cells were transduced with lentiviral particles containing the LentiCrispr v2 construct (Addgene Plasmid #91977) [26] and a single guide (sg) RNA against LDLRAP1. The sgRNAs used were as follows: CGCCGCCATCAAGAGGATCG for sgRNA 1, ATGGACGCGCTCAAGTCGGC for sgRNA2, GTACCTGGGCATGACGCTAG for sgRNA3, and TCAAGAGGATCGTGGCTACA for sgRNA4. To assess the knockout efficiency, protein knockout was determined by means of western blot analysis for LDLRAP1 following hygromycin (Sigma-Aldrich) selection.
To create ARH knockout iPSC, two normal donor-derived iPSC lines were used. The Sigma iPSCs were purchased from Sigma (0028 iPSC line). The BJ1 iPSCs were generated in-house from BJ1 fibroblasts (CRL-2522; ATCC) as previously described [27]. The Sigma and BJ1 iPSCs were transduced with lentiviral particles containing the LentiCrispr v2 construct and sgRNA1 or sgRNA2. After hygromycin selection, ARH-knockout (KO) iPSCs underwent the following quality control assays. Quality control assays included SNP-qPCR, EB formation and TaqMan hPSC scorecard analysis (Applied Biosystems), and immunostaining for pluripotency proteins. To demonstrate ARH knockout, PCR-sequencing for indels in the LDLRAP1 gene was done (primers are listed in Supplementary Table S3); and iPSCs were differentiated into HLCs and western blot for LDLRAP1 was performed. In addition, we assessed the cells for off-target genomic cuts. The top 3 off-target regions of sgRNA 1 and 2 were determined using the clustered regularly interspaced short palindromic repeats (CRISPR) genome editing tool from IDT. Primers were designed 200 to 300 bp up-and downstream from the target sites (Supplementary Table S3). A PCR reaction was performed using Phusion DNA polymerase (NEB) following the PCR program: 95°C for 5 min, 30 × (95°C for 30 s, 58°C −63°C (depending on Tm of the primer set) for 30 s, 70°C for 30 s), 70 degrees for 5 min. A part of the PCR product was loaded on a 1% agarose gel and gel electrophoresis was performed. The rest of the PCR product was PCR purified using the PureLink PCR Purification Kit (Invitrogen). Purified PCR product was sent for sequencing (Eurofins).
hiPSC differentiation to the hepatocyte lineage
Differentiation of iPSCs toward the hepatocyte lineage was performed as previously described by Boon et al. [28]. Briefly, iPSCs were split at 8.75 × 104 cells/cm2 using accutase (Sigma-Aldrich) and plated on matrigel-coated plates in mTeSR medium (Stem cell technologies). When the cell confluency reached 70%–80%, iPSCs were differentiated in liver differentiation medium (LDM; the composition is listed in Supplementary Table S4), supplemented with 0.6% dimethyl sulfoxide (DMSO; Sigma-Aldrich) until day 12 of differentiation. From day 12 onward, LDM was supplemented with MEM Amino Acids Solution (50 × , 8 mL/100 mL LDM), MEM Non-Essential Amino Acids Solution (100 × ; 16 mL/100 mL LDM, Thermo Fisher Scientific), and 2% DMSO; and from day 14 onward, 2% glycine (Sigma), combined with MEM Amino Acids Solution and MEM Non-Essential Amino Acids Solution, was used, but DMSO was omitted. Growth factors used included Activin-A (50 ng/mL; day 0–day 4), Wnt3a (50 ng/mL; day 0–day 2), BMP4 (50 ng/mL; day 4–day 8), FGF1 (20 ng/mL; day 8–day 12), and HGF (20 ng/mL; day 12 till the end of the hepatocyte differentiation). All cytokines were purchased from Peprotech (NJ).
Functional assessments
The rate of albumin secretion was evaluated using an enzyme-linked immunosorbent assay (ELISA) following the manufacturer's protocol (Bethyl, Montgomery, TX). The supernatant of iPSC-HLCs was collected at day 35 and incubated with a primary albumin antibody at room temperature (RT) for 1 h. Subsequently, through adding HRP-detection antibody at RT for 1 h and incubation with TMB-peroxidase solution in the dark for 15 min, the reaction was stopped by adding stop solution. The absorbance was determined on a plate reader at a wavelength of 450 nm. Data were normalized for cell number and presented as albumin secretion during 1 h per 10,000 differentiated cells. A fluorescence-based CYP3A4/CYP1A2-dependent metabolization assay was performed by incubation of iPSC-HLCs at day 35 with fluorometric probe 7-benzyloxy-4-trifluoromethylcoumarin (BFC) specific for the CYP3A4/CYP1A2 metabolization, as described previously [29]. Metabolization was determined after 4 h of incubation. Data were normalized for cell number.
Western blot analysis
Cell pellets were collected and lysed in RIPA buffer (ThermoFisher Scientific), supplemented with phosphatase and protease inhibitors (4906845001; Sigma). Lysates were cleared by centrifugation at 12,000 g for 15 min at 4°C and supernatant was recovered for protein quantification. The amount of protein was measured using a pierce BCA protein assay kit (ThermoFisher Scientific). Seventy micrograms of protein were loaded on a 4%–20% ExpressPlus PAGE Gel (Genscript), and separated via electrophoresis. Next, the proteins were transferred onto a nitrocellulose membrane using the iBlot Dry Blotting System (Invitrogen). The membrane was blocked with 5% skim milk in TBST for 1 h and then incubated overnight at 4°C with LDLRAP1 primary antibody (1:1,000; ab181043) and beta-tubulin (1:1,000; ab179513). After overnight incubation, the membrane was washed three times with TBST for 5 min and subsequently, incubated with an anti-rabbit HRP-conjugated secondary antibody (1:2,000, 61-9520; ThermoFisher Scientific) for 1 h at RT. Bands were revealed using the Super Signal West Femto Chemiluminescent Substrate (ThermoFisher Scientific) and detected with GelDoc system (Bio-Rad).
RNA extraction, cDNA synthesis. and RT-qPCR
Total RNA was isolated using the GenElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich). After concentration determination and integrity validation using the Nano-Drop 1000 (ThermoFisher Scientific), reverse transcription was performed using the SuperScript® III First-Strand Synthesis kit (Invitrogen). After cDNA synthesis, qPCR was performed in technical triplicates using the Platinum SYBR® Green qPCR supermix-UDG kit (Invitrogen) and the ViiA7 Real-Time PCR instrument with 384-well plate (ThermoFischer Scientific). Relative quantification was done using the ΔCt and 2−ΔCt method, and GAPDH was used for normalization. The sequences of all used RT-qPCR primers can be found in Supplementary Table S1.
Immunostaining
Cells were washed with PBS and fixed with 4% paraformaldehyde for 15 min at RT and again thoroughly washed with PBS. Preparations were permeabilized in 0.2% Triton-X100 in PBS (0.2% PBST; Sigma-Aldrich) and blocked in 5% goat serum (Dako) or donkey serum (Dako) in 0.2% PBST for 30 min at RT. Cells were then incubated overnight at 4°C with primary antibodies (Supplementary Table S2) followed by incubation for 30 min at RT with the appropriate secondary antibodies. Samples were incubated for 10 min at RT with Hoechst (1:2,000 dilution; Sigma-Aldrich) for nuclear staining and the slides were mounted with Prolong Gold antifade reagent (P36931; Life Technologies). Images were taken with an Axio imager Z1 fluorescence microscope (Carl Zeiss, Inc.) and the Operetta High Content Imaging system (PerkinElmer) using extended focus computation from z-stacks. In all immunofluorescence staining experiments, applicable isotype control antibodies were used.
LDL uptake assay
An immunofluorescence LDL uptake assay was performed according to the modified protocol of Karim Si-Tayeb et al. [30]. iPSC-HLCs were exposed to 10 μg/mL DiI-LDL (Invitrogen) at 37°C for 3.5 h in the cell culture medium. For uptake measurement, DiI-LDL-treated cells were rinsed with DPBS (Gibco) and the LDL uptake ability of the cells was analyzed by fluorescence microscopy (Nikon Eclipse Ti2).
Statistical analysis
Data are expressed as mean ± SEM and comparisons between two data groups analyzed using an unpaired or paired two-tailed Student's t-test. P values <0.05 (*), P < 0.01 (**), P < 0.001 (***), and P < 0.0001 (****) were measured statistically significant.
Results
Generation and characterization of an ARH-hiPSC line and ARH-KO iPSC lines
To generate a disease model and investigate the effect of the c.649 G > T LDLRAP1 mutation on LDLRAP1 protein expression and function, we generated iPSC from fibroblasts of an ARH patient with the mutation c.649 G > T and who developed premature atherosclerosis. Reprogramming was done using nonintegrating Sendai viral vectors, encoding for OCT3/4, KLF4, c-MYC, and SOX2. Reprogrammed clones were picked of which three clones (clone 8, 19, and 36) were subjected to an extensive quality control. Absence of Sendai viral vectors was confirmed by RT-qPCR analysis (Supplementary Table S5). Immunostaining of iPSC demonstrated homogenous expression of the pluripotency markers OCT3/4, SOX2, TRA1-60, and SSEA4 (Fig. 1A and Supplementary Fig. S1A), which was also confirmed by RT-qPCR for OCT3/4, SOX2, and NANOG mRNA (Fig. 1B and Supplementary Fig. S1B). Pluripotency was further confirmed by EB formation and TaqMan hPSC Scorecard@ analysis (Fig. 1C and Supplementary Fig. S1C). Genome integrity was demonstrated by aCGH (Supplementary Fig. S2A) and cell identity by SNP-PCR (Supplementary Table S6A). Furthermore, brightfield pictures, taken at high and low magnification, demonstrated similar colony and cell morphology (Supplementary Fig. S2B). As slight but significant lower pluripotency gene expression levels for OCT3/4 and SOX2 were seen in ARH patient-derived iPSC clone 8 and 19, respectively, we proceeded the rest of our studies with ARH patient-derived iPSC clone 36 (Supplementary Fig. S1B). Sequencing of the LDLRAP1 gene demonstrated that the c.649 G > T LDLRAP1 mutation was also present in this ARH-iPSC line (Supplementary Fig. S2C).

ARH patient-derived hiPSC characterization.
We also created ARH knockout iPSC lines, starting from two control iPSC lines. Four different sgRNAs against ARH were designed: sgRNA 2 designed to cut in exon 1 and sgRNA 1, 3, and 4 designed to cut in exon 2 of ARH. Their efficiency for gene knockout was tested in the HepG2 hepatoma cell line by transduction of a CRISPR/CRISPR-associated protein 9 (Cas9) containing lentiviral plasmid and the four different sgRNAs. Western blot analysis demonstrated that a complete knockout (absence of the LDLRAP1 protein) was induced by sgRNAs 1-3 (Supplementary Fig. S3). Subsequently, a lentiviral vector containing Cas9 and either sgRNA 1 or sgRNA 2 were transduced in wild-type BJ1 and Sigma iPSCs (termed BJ1-ARH-KO sg1, BJ1-ARH-KO sg2, and Sigma-ARH-KO sg1). Following selection with hygromycin, presence of Cas9 was confirmed by immunostaining (Supplementary Fig. S4).
The genome-edited iPSC lines underwent the same quality control studies as described for the ARH-iPSC line above (Fig. 2 and Supplementary Table S6B). In addition, PCR-amplification and sequencing analysis to determine on-target indels in the LDLRAP1 gene were performed: sgRNA 1 caused a deletion of an AT in the BJ1 ARH-KO sg1 iPSC line, while an addition of an A was found in the sigma ARH-KO sg1 iPSCs (Supplementary Fig. S5 and Table 3). We were not able to determine the indel caused by sgRNA 2 in the BJ1 ARH-KO sg2 iPSC line, as PCR-amplification proved difficult in that region. Finally, we also screened for off-target effects, potentially caused by the sgRNAs. sgRNA 1 showed no off-target effects, and also sgRNA 2 did not cause off-target effects. For the apparent off-target base replacement on chromosome 20, we found an identical SNP in both the control (BJ1 iPSCs) and ARH-KO iPSC line (BJ1 ARH-KO sg2) (Supplementary Fig. S6).
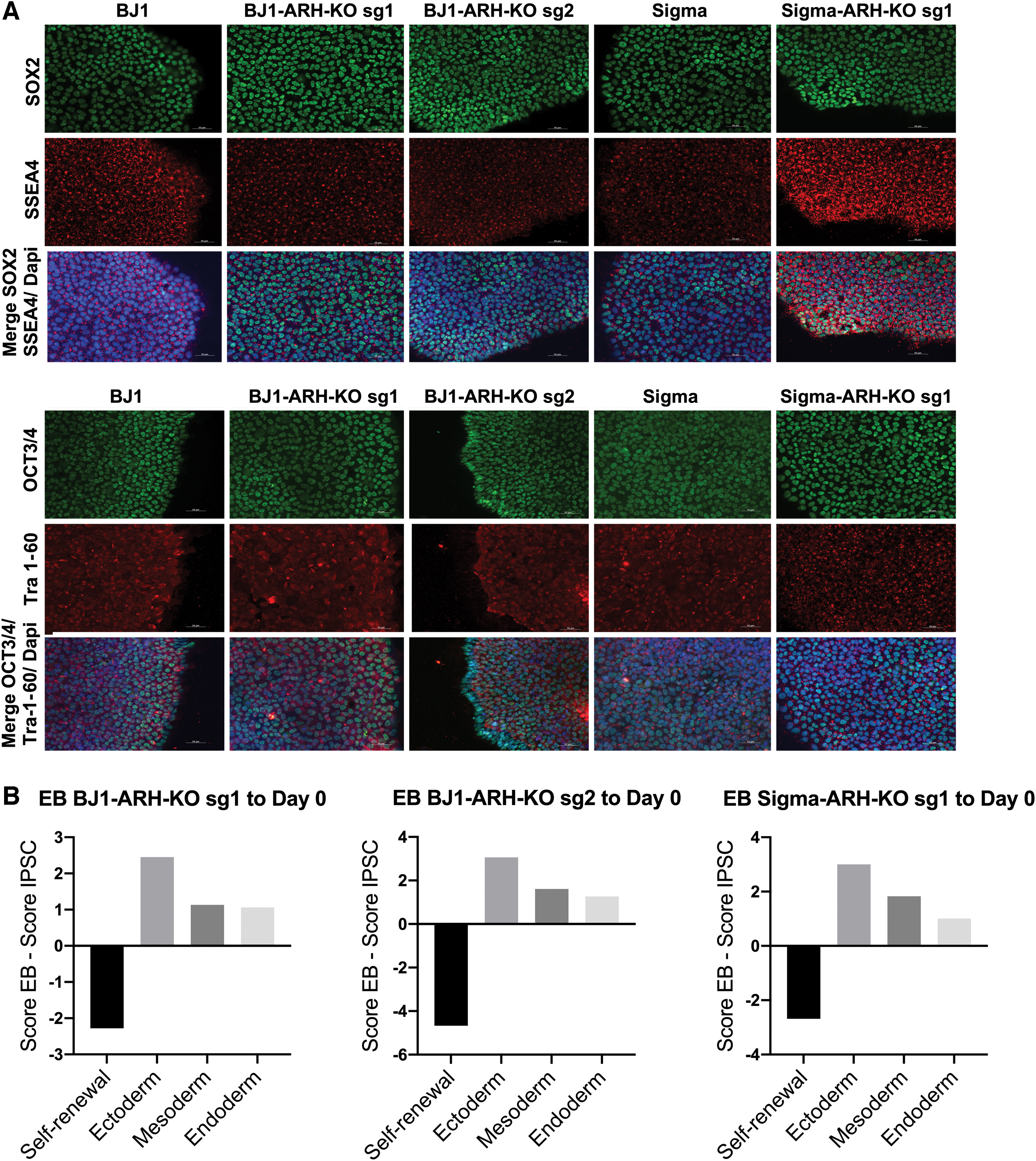
ARH-KO hiPSC characterization.
ARH-iPSC and ARH-KO iPSCs differentiate toward HLCs
To study the effect of the c.649 G > T LDLRAP1 mutation/knockout on LDLRAP1 and its function in hepatocyte-like progeny, we differentiated the ARH patient-derived and ARH-KO iPSCs toward HLCs. Control (BJ1 and Sigma iPSCs), BJ1-ARH-KO sg1, BJ1-ARH-KO sg2, Sigma-ARH-KO sg1, and ARH-iPSC efficiently differentiated toward HLCs. The differentiated cells co-expressed the hepatic markers ALB and HNF4α, as seen by immunofluorescent staining (Fig. 3A). The differentiation efficiency of the ARH-KO-derived and ARH patient-derived HLCs was determined by the quantification of HNF4α-positive cells. Similar percentages of HNF4α-positive cells were observed in all differentiated progeny (±70%–75%), except in Sigma-ARH-KO sg1 HLCs (±55%) (Supplementary Fig. S7C). However, as no differences in HNF4α-positive cells were observed in BJ1-HLCs and BJ1-ARH-KO sg1 HLCs, we believe that this difference is not caused by the KO of LDLRAP1 itself. The gene expression levels of several immature and mature hepatic markers, including HNF4α, ALB, AFP, AAT, HNF1α, HNF6, G6PC, and PEPCK in addition to genes involved in cholesterol metabolism SREBF2, HMGCR, LDLR, and AP2; metabolizing enzyme genes CYP2D6, CYP1A2, CYP2C9, and UGT1A1; and transporters genes OATPB1, OATPB3, MDR1, and NTCP were found to be similar between control HLCs and ARH-KO/ARH patient-derived HLCs (Fig. 3B, C and Supplementary Fig. S7A). In line with Boon et al. [28], the transcript levels of several hepatic markers, metabolizing enzymes, and transporters in HLCs differed from those found in cryopreserved primary human hepatocytes (PHHs), consistent with an immature phenotype of PSC-derived HLC progeny (Fig. 3B and Supplementary Fig. S7A). Nevertheless, a significant reduction in LDLRAP1 mRNA levels was observed in ARH patient-derived HLCs compared to control BJ1-HLCs (74.5%; P < 0.001) and Sigma-HLCs (76.9%; P < 0.0001) (Fig. 3C). Albumin secretion and the metabolization rate of BFC were comparable in ARH-KO/ARH patient-derived HLCs and control HLCs (Fig. 3D). However, albumin secretion and the metabolizing activity of CYP3A4 were significantly lower in wild-type iPSC-HLCs and ARH-KO/ARH patient-derived HLCs than in 12 h plated PHHs (PHH12) (Supplementary Fig. S7B), which is also consistent with previous studies by Boon et al. [28]. These results suggest that overall hepatocyte differentiation capacity is not impaired by the c.649 G > T LDLRAP1 mutation or the KO of the LDLRAP1 gene.
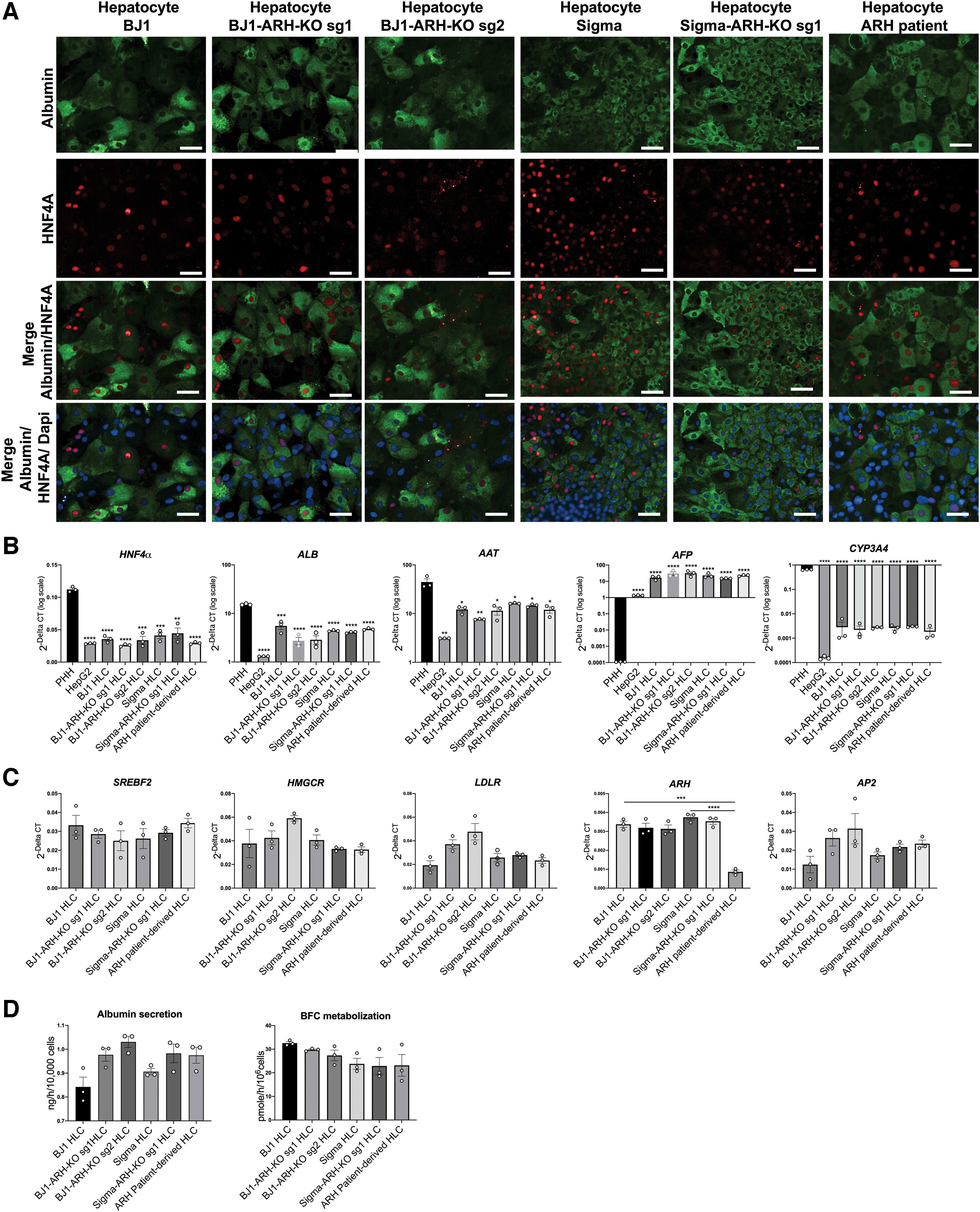
hiPSC differentiation into HLCs.
Absence of the LDLRAP1 protein in the ARH patient-derived HLCs and ARH-KO-derived HLCs
Western blotting analysis using an anti-LDLRAP1 N-terminal antibody demonstrated that the ARH patient-derived HLCs and the ARH-KO-derived HLCs had undetectable LDLRAP1 protein expression, whereas the iPSCs-derived HLCs from normal individuals (control) revealed a ∼34 kDa band, corresponding to the LDLRAP1 protein (Fig. 4). Thus, both ARH patient-derived HLCs and ARH-KO-derived HLCs did not express LDLRAP1, which indicates that the c.649 G > T LDLRAP1 mutation leads to the absence of LDLRAP1 protein expression in the ARH patient-derived HLCs.
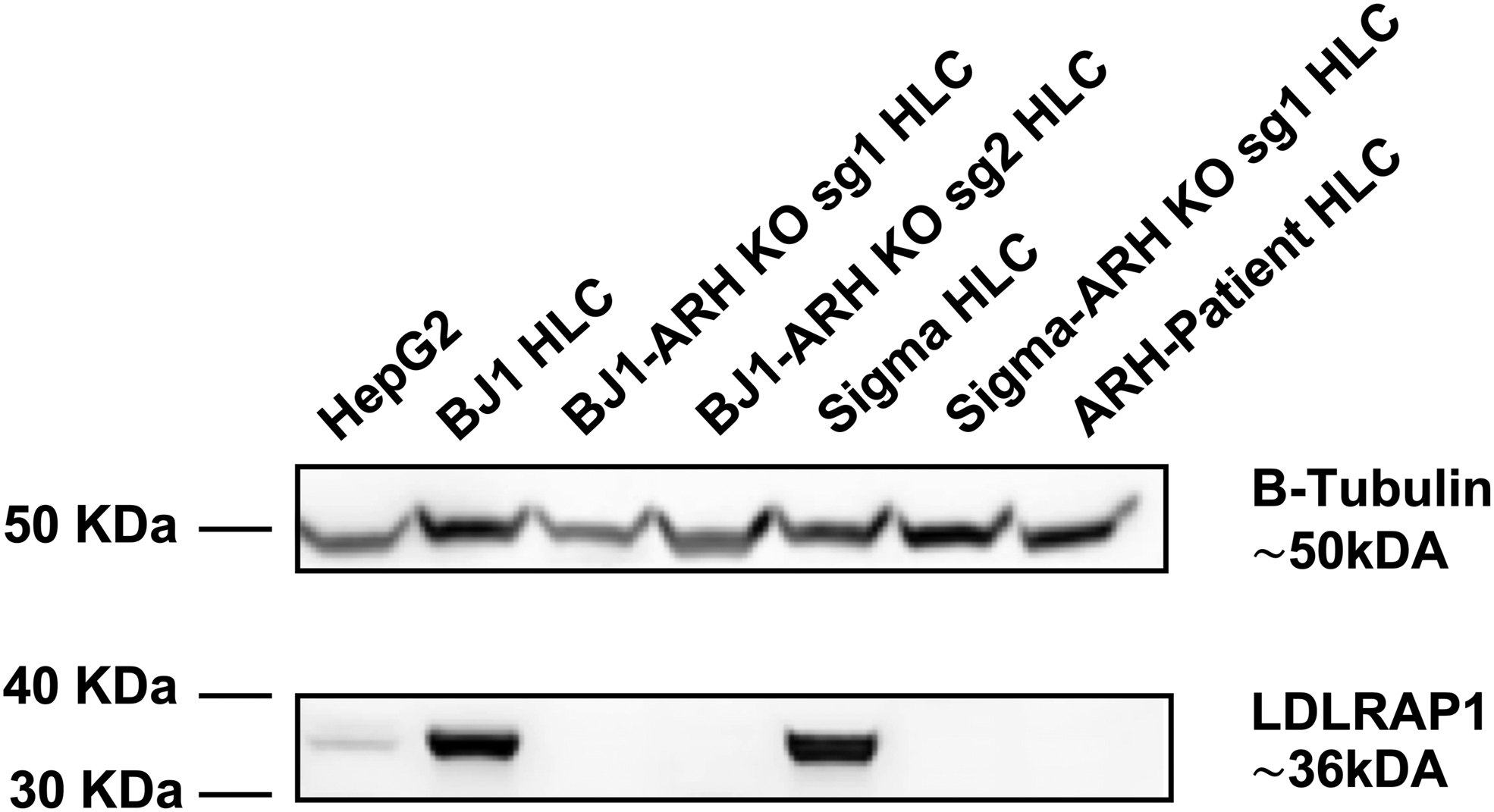
Expression of the LDLRAP1 protein in ARH patient-derived HLCs and ARH-KO-derived HLCs (BJ1-ARH KO sg1, BJ1-ARH-KO sg2, and Sigma-ARH-KO sg1 HLCs) and control subjects (BJ1 HLCs and Sigma HLCs). LDLRAP1 protein probed with an N-terminal LDLRAP1-antibody. B-Tubulin was used as a housekeeping protein (n = 3). LDLRAP1, LDLR adaptor protein 1.
ARH patient-derived and ARH-KO-derived HLCs reiterate the functional defects of LDLRAP1 mutations
As the LDLRAP1 protein was absent in both ARH patient-derived and ARH-KO-derived HLCs, we next investigated whether LDL uptake was affected, using an LDL uptake assay. Consistent with the hypercholesterolemic phenotype of patients with LDLRAP1 loss-of-function mutations, the ability to take up LDL from the cell culture medium was significantly reduced in ARH patient-derived HLCs compared to control HLCs (Fig. 5). Likewise, in ARH-KO-derived HLCs, LDL uptake was significantly reduced. However, for both ARH patient-derived and ARH-KO-derived HLCs, low levels of LDL uptake persisted. These findings are in line with the findings of Rodriguez-Jimenez et al. [22]. They declared the presence of an additional mechanism for LDLR internalization in lymphocytes, which moderately compensates the absence of normal LDLRAP1 protein; and also, are consistent with the known generally milder phenotype of ARH patients than HoFH patients [3,31]. Therefore, ARH patient-specific iPSC-derived HLCs can be a suitable model to study LDLRAP1 mutations and the function of the encoded protein.
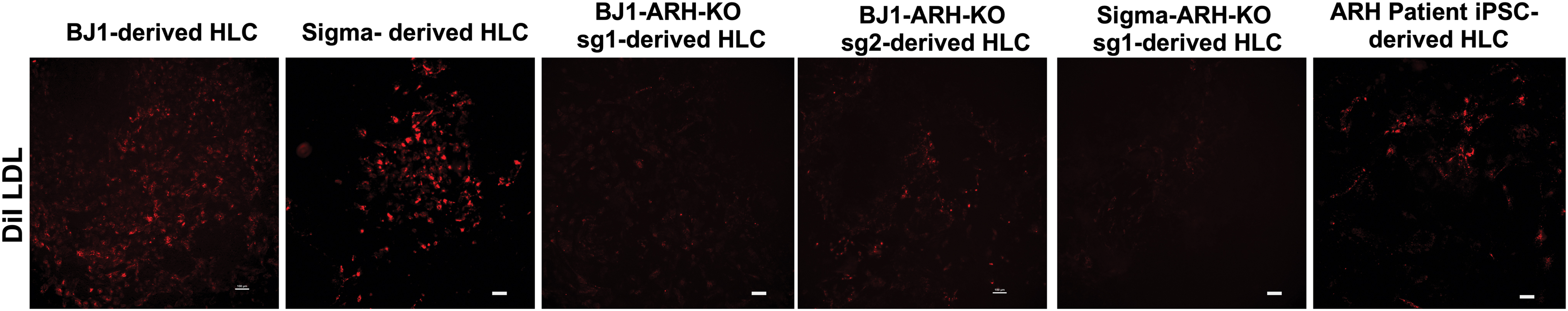
LDLR-dependent endocytosis of DiI LDL. Representative pictures of LDL uptake of control hepatocytes (BJ1 and Sigma-HLCs) and ARH−/− HLCs (ARH-KO HLCs and ARH-patient HLCs) detected by fluorescent microscopy (n = 3). Scale bars: 100 μm. Color images are available online.
Discussion
Several studies have used hiPSCs to model FH linked to mutations in LDLR [32 –34] and PCSK9 [30]. We here demonstrate the potential of patient-specific iPSC-derived HLCs to also model the autosomal recessive form of FH. We generated iPSCs from fibroblasts of an ARH patient, with the newly described LDLRAP1: c.649G>T mutation [18]. These iPSCs were differentiated toward HLCs to determine whether they would be a suitable model to study LDLRAP1 biology. We demonstrated that LDLRAP1: c.649G>T HLCs do not express the LDLRAP1 protein and that LDL uptake activity in these ARH patient-derived HLCs was significantly reduced, and this to the same extent as in ARH-KO-derived HLCs generated from isogenic normal iPSCs.
More than 70% of LDL is cleared from the blood circulation through the hepatic LDLR [1]. Therefore, studying the role and function of LDLRAP1 in ARH disease using ARH patient-derived HLC model is likely to be much more clinically relevant, compared with studying the role of this protein in the existing cellular models such as hepatoma cells [19,20], fibroblasts [10,21], and lymphocytes [16,22]. For this purpose, we generated iPSCs from fibroblasts of a 17-year-old female Iranian patient who was genetically verified recently by our team as an ARH patient, with a novel variant c.649G>T in LDLRAP1 gene [18]. The clinical manifestations of the patient were mimicking the most severe form of genetic hypercholesterolemia LDLR-HoFH: appearance of cutaneous xanthomas since the age of 10 years; levels of LDL-C before treatment between levels observed in LDL receptor-negative and receptor-defective HoFH patients (720 mg/dL at the age of 11); presence of bilateral corneal arcus; and premature atherosclerotic cardiovascular disease from 13 years of age. Even though she received treatment with a high dose of rosuvastatin plus ezetimibe (60 mg/day and 10 mg/day, respectively), the patient presented with severe cardiovascular involvement, aortic valve stenosis, and supravalvular aortic stenosis, which is a rare finding in ARH patients.
As the control (Sigma and BJ1-derived HLCs) and the ARH patient-derived HLCs are from genetically unrelated individuals, which might influence the outcome of the functional comparison, we also created three ARH knockout lines from the control BJ1 and Sigma iPSCs, using the CRISPR-Cas9 technology (termed BJ1-ARH-KO sg1, BJ1-ARH-KO sg2, and Sigma-ARH-KO sg1). Results from the isogenic knockout lines compared with parent wild-type lines confirmed the differences seen between the control (Sigma and BJ1-derived HLCs) and ARH patient-derived HLCs, therefore validating the results seen in the ARH patient-derived HLCs.
Specifically, we differentiated the ARH patient-derived iPSCs and the ARH-KO iPSCs into HLCs and demonstrated that expression of hepatic markers and hepatocyte-specific functions, such as albumin secretion and BFC metabolization, were comparable to normal control iPSC-derived HLCs. Similar percentages of HNF4α-positive cells were observed in all differentiated HLCs (±70%–75%), except in Sigma-ARH-KO sg1 HLCs (±55%). However, as no difference in HNF4α-positive cells was observed in BJ1-HLCs and BJ1-ARH-KO sg1 HLCs, we believe that this difference is not caused by the KO of LDLRAP1 itself. Furthermore, as we observed via RT-qPCR and functional assays, Sigma-ARH-KO sg1 HLC did express other hepatocyte markers to a similar level as the other differentiated progeny and had similar levels of ALB secretion and BFC metabolization. Therefore, we believe that overall hepatocyte differentiation is not impaired and thus, the LDLRAP1 variant c.649G>T or the complete knockout of LDLRAP1 does not affect the ability of the iPSCs to generate HLCs. However, in line with the current state of the art [35], gene expression and functional analysis demonstrated that the HLCs still remain not fully mature [35 –37]. Furthermore, we found that only trace amounts of LDLRAP1 mRNA could be detected in the ARH patient-derived HLCs, in which both alleles contained a nonsense mutation in exon 7, suggesting that the mutation leads to the formation of an instable mRNA. In contrast to the LDLRAP1 mRNA in ARH patient-derived HLCs, the levels of LDLRAP1 mRNA were not significantly reduced in the ARH-KO-derived HLCs, in which both alleles contained CRISPR/Cas-mediated frameshift mutations.
LDLRAP1 has an important role in LDL-C uptake in hepatocytes, as it mediates LDL-LDLR internalization by simultaneously binding of its N-terminal PTB domain (amino acid 48–175) to the cytoplasmic tail of LDLR, Clathrin Box module (amino acid 212–216) to clathrin proteins, and AP-2-binding module (amino acid 248–279) within its C-terminal to the β2-adaptin subunit of AP-2 [9,10]. The LDLRAP1 variant c.649G>T has a new stop codon in codon 217 in exon 7 just downstream of the Clathrin Box domain. As such a premature stop codon in the LDLRAP1 gene might create an N-terminal truncated protein [22], C-terminal truncated proteins, or no protein at all [11,16,17], we assessed presence of LDLRAP1 by western blotting. These studies demonstrated that HLCs derived from the ARH-iPSC and from all ARH-KO iPSCs did not express the LDLRAP1 protein.
To validate the c.649G>T in the ARH patient-derived HLC model, we tested whether the cells would phenocopy the clinical feature of decreased LDL uptake. We demonstrated that in line with the clinical characteristics, LDL uptake activity was significantly reduced in the ARH patient-derived and ARH-KO-derived HLCs compared to healthy control HLCs; however, the activity was not completely abolished. To the best of our knowledge, there is no previous report of LDL uptake in ARH patient-derived HLCs. However, consistent with our results, a recent report by Rodriguez-Jimenez et al. described that LDLRAP1:c.1A>G lymphocytes had a reduced, but not completely absent, uptake of fluorescent LDL compared to control lymphocytes [22]. Furthermore, they compared the LDL uptake in the LDLRAP1:c.1A>G lymphocytes with lymphocytes from a LDLR-defective patient who has a homozygous missense mutation in the cytoplasmic domain of LDLR, which directly interacts with LDLRAP1 and consequently prevents the interaction of the LDLR with the LDLRAP1 protein. Interestingly, similar to the LDLRAP1:c.1A>G lymphocytes, LDL uptake in these LDLR-defective lymphocytes was not completely inhibited. Reduced LDL uptake has also been demonstrated in lymphocytes of 28 ARH patients with different types of mutations in the LDLRAP1 gene [16]. Sirinian et al. [19], who transfected with an siRNA against LDLRAP1 gene, demonstrated that LDL internalization was reduced by ∼80%, due to the depletion of LDLRAP1. In line with previous reports [38,39], our findings suggest that although LDLRAP1 is the main LDLR endocytic adaptor for efficient LDL uptake in hepatocytes, other mechanism(s) may also be involved in LDL internalization. The ARH-iPSC lines generated in this study and the possibility of creating HLCs from these cell lines therefore constitute an interesting novel tool to identify these additional mechanism(s) for LDLR internalization, which partially compensates for the absence of a normal LDLRAP1 protein.
Conclusion
We developed a novel cell model to study the expression of the LDLRAP1 protein and LDLR activity by creating ARH patient-derived HLCs carrying a nonsense mutation c.649G>T in exon 7 of the coding sequence of LDLRAP1. This variant results in complete absence of the LDLRAP1 protein in the hepatocyte progeny and significantly reduced but not completely abolished LDL uptake. Additionally, we created three independent isogenic ARH-KO iPSC lines and subsequently ARH-KO-derived HLCs, as a phenocopy of the LDLRAP1: c.649G>T ARH patient-derived HLCs, which showed a similar reduction in LDL uptake. The molecular mechanism(s) of the only partial loss of LDL uptake remains unknown, but the creation of the hepatocyte model described here should enable future studies to identify possible other mechanism(s) involved in LDL uptake, aside from LDLRAP1.
Confirmation Statement
Each listed author confirms that their research is supported by an institution that is primarily involved in education or research.
Footnotes
Acknowledgments
We would like to thank Ruben Boon, Niels Vidal, and Rodrigo Madeiro da Costa for their discussions and technical assistance. Finally, we gratefully thank Prof. Etienne Sokal (Pediatric Gastroenterology and Hepatology, Université Catholique de Louvain) for providing the primary human hepatocytes.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was financially supported by KU Leuven Stem Cell Institute, Tabriz University of Medical Sciences Department of Molecular Medicine, and Royan Institute for Stem Cell Biology and Technology. Tine Tricot was funded by the research Foundation-Flanders (FWO) (1185918N).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.