
Abstract
The corneal endothelium (CE) is vital for the cornea to maintain its transparency. However, CE dysfunction occurs due to aging, intraocular surgery, trauma, dystrophy, etc. Corneal transplantation is the only method to clinically treat CE dysfunction; however, this treatment strategy faces the disadvantages of a global cornea shortage, graft failure, and severe side effects. There is a recognized need for a substitute for the CE. Stem cells are becoming increasingly common for the treatment of human diseases. In fact, several studies have documented the induction of corneal endothelial-like cells (CECs) from stem cells, but an ideal procedure has not yet been established. Thus, this study aimed at exploring a more efficient and robust differentiation method. We used a modified approach to differentiate induced pluripotent stem cells (iPSCs) into CECs. After the identification of differentiated CECs, the CECs were injected into the anterior chambers of the eyes of a rabbit model of bullous keratopathy. The rabbits were maintained in the eye-down position to ensure that the cells attached to the cornea. The results showed that corneal edema was alleviated in the rabbits injected with CECs compared with that in the rabbits belonging to the control group. This study extends the ability to differentiate iPSCs into CECs and provides a potential strategy for the treatment of reduced visual acuity caused by CE deficiency in the future.
Introduction
Corneal disease is a cause of blindness worldwide, particularly in some developing countries, and corneal endothelial disease accounts for a significant number of corneal disease cases. The corneal endothelium (CE), which is the innermost layer of the cornea and is derived from the neural crest, maintains the transparency of the cornea by functioning as a pump and a barrier. However, corneal endothelial cells decrease due to increasing age, intraocular surgery, trauma, and dystrophy. Dysfunction occurs when the endothelial cell density in the cornea decreases to less than 500/mm2, which leads to bullous keratopathy and can eventually result in persistent corneal edema and visual loss. Because corneal endothelial cells have little regenerative potential in vivo, the current strategy for CE dysfunction is corneal transplantation, which faces a global cornea shortage and some severe complications, including secondary glaucoma, graft failure, and rejection. Therefore, there is an urgent need to find a substitute for corneal endothelial cells.
During the past decade, several alternative strategies have been attempted, and these measures can be classified into three categories. The first involves the injection of corneal endothelial precursor cells, corneal endothelial cells, or blood endothelial progenitor cells into the anterior chamber of the eye [1 –5]. This strategy might be the most appropriate if a sufficient number of cells can be easily obtained. The second strategy is the construction of a tissue-engineered corneal endothelium (TECE) with corneal endothelial-like cells (CECs) derived from different kinds of stem cells and progenitors [6 –12]. After being seeded on a biosynthetic carrier or scaffold, the cell sheet is transplanted to replace the unhealthy CE. Although CECs can be induced, the differentiation method has not been fully developed, and robust and efficient differentiation methods still need to be explored. In addition, biocompatibility, stability, safety, high transparency, nontoxicity, and biodegradation are challenges related to the application of biosynthetic carriers and scaffolds in the TECE [13 –16]. The last strategy involves the promotion of corneal endothelial cells proliferation [17,18], but the cells have only expanded in vitro, which hinders the application of this approach. Taken together, the information mentioned earlier reveals that despite intense research efforts, a few strategies have entered clinical trials.
Although robust and efficient differentiation methods are lacking, stem cells with a self-renewal potential, unlimited proliferation and multipotent differentiation capabilities, and the ability to maintain their undifferentiated state are, indeed, an ideal source of CECs. Induced pluripotent stem cells (iPSCs) can be used for autologous therapy, because they can be derived from somatic cell reprogramming [19]. In addition, the application of iPSCs can avoid ethical conflicts faced by embryonic stem cells. Hence, the identification of effective iPSC differentiation methods is meaningful.
In the current study, we investigated a modified method for the differentiation of iPSCs to CECs based on previous studies. In addition, corneal edema was relieved by the injection of differentiated CECs into the anterior chambers of the eyes of a rabbit model of CE deficiency. This investigation extends the available methods for the differentiation of iPSCs to CECs and might constitute another step toward the clinical application of iPSCs for the treatment of CE deficiency.
Materials and Methods
Human iPSC culture
The human iPS cell line that was purchased from Cellapy company (Beijing, China) and generated from renal epithelial cells in urine using Sendai virus reprogramming was cultured on plates coated with Matrigel (BD354277; Corning) and in Pluripotency Growth Master 1 (PGM1) (CA1007500; Cellapy) medium. Ethylenediaminetetraacetic acid (EDTA) (CA3001500; Cellapy) was used for the passaging of iPSCs every 4–5 days. The cells were incubated at 37°C in a humidified atmosphere containing 5% CO2. The medium was changed every day.
Differentiation of iPSCs to CECs
To achieve differentiation to CECs, the neural crest cells (NCCs) should be induced first. The NCCs were prepared by adapting the procedure created by Wang A [20]. Briefly, iPSCs were cultured in PGM1 medium for at least one passage to adapt to the culture system. Once 80% confluence was reached, the cells were detached by EDTA, plated into ultra-low-detachment plates (Costar), and grown as embryonic body-like floating cell aggregates in basic culture medium for 5 days. The basic culture medium contained 80% Dulbecco's modification of Eagle's medium-F12+GlutaMAX™-1 (Gibco), 20% knock-out serum (Gibco), 1% nonessential amino acids (Gibco), 0.1 mM β-mercaptoethanol (Gibco), and 8 ng/mL bFGF (PeproTech). The cell aggregates were then allowed to adhere to Matrigel-coated plates in NCCs induction medium consisting of StemPro™ neural supplement (Thermo Fisher Scientific), 20 ng/mL basic fibroblast growth factor (bFGF; PeproTech), 20 ng/mL epidermal growth factor (EGF; R&D), and basic culture medium. After 7 days, colonies with rosette structures could be observed on the plate surface, were mechanically harvested, and were then cultured in suspension for 7 days. The cell aggregates were then replated onto Matrigel-coated plates with NCCs induction medium until confluence or passage. Thereafter, the induced neural crest stem cells (NCSCs) could be harvested for the subsequent experiments.
For the induction of CECs from the NCSCs, we modified previously published protocols [21 –24]. Briefly, the NCSCs were changed to CEC induction medium and cultured for 7 days. The CEC induction medium contained 8 ng/mL bFGF (PeproTech), 0.1 × B-27 supplement (Gibco), 10 ng/mL recombinant human platelet derived growth factor-BB (R&D), 10 ng/mL recombinant human Dickkopf-related protein 2 (R&D), 1 μM SB431542 (PeproTech), 2.5 μM Y27632 (Sigma), and basic culture medium. The medium was changed every day. After 7 days, the medium was transformed to CEC maintenance medium consisting of DMEM/F12, 8% fatal bovine serum (Gibco), 5 ng/mL EGF (R&D), and 20 ng/mL nerve growth factor (PeproTech) until confluence or passage was reached.
RNA extraction and real-time PCR
The cells were collected, and total RNA was extracted by using the TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer's recommended protocol. The RNA concentration was determined by using a Nanodrop Spectrophotometer (Nanodrop Technologies, Wilmington, DE). The primer sequences are provided in Supplementary Table S1. A ReverTra Ace qPCR RT Kit (TOYOBO, Japan) and SYBR-Green PCR Master Mix (TOYOBO) were used for the detection of gene expression with an ABI 7500 Sequence Detection System (Life Technologies, NY). The gene expression levels were normalized to the expression of GAPDH. The relative expression level was calculated by using the 2−▵▵Ct method. Three independently repeated experiments were performed.
Immunofluorescence staining
The cells were fixed in 4% paraformaldehyde for 20 min, incubated with 0.5% Triton X-100 for 10 min, washed with PBS, and blocked with goat serum for 30 min at room temperature. The cells were then incubated overnight at 4°C with appropriate antibodies diluted in PBS with 1% BSA. The next day, the cells were rinsed three times with PBS and incubated with the corresponding secondary antibody, including Alexa Fluor™ 594 donkey anti-mouse IgG (H+L) (Thermo Fisher Scientific) and Alexa Fluor 488 goat anti-rabbit IgG (H+L) (Thermo Fisher Scientific), for 50 min at room temperature. The nuclei were stained with DAPI. Fluorescent images were obtained by using a fluorescence microscope.
Na+/K+ ATPase activity detection
Na+/K+ ATPase activity assay kit (Solarbio, China) was used for detection of Na+/K+ ATPase activity. The whole process was referred to the manufacturer's recommended protocol. After sample preparation, enzymatic reaction, and determining the production of siderophores, the tubes were measured at 660 nm with a spectrophotometer (BioRad).
Fluorescence-activated cell sorting
The cells were collected with EDTA, blocked with 1% bovine serum albumin for 30 min, incubated with the primary antibody for 45 min, followed by staining with a fluorescence-labeled secondary antibody for 30 min. After washing, fluorescence-activated cell sorting (FACS; BD Biosciences, San Jose, CA) was performed.
Animal experiments
All the animal experiments were conducted at the Animal Experimental Center of the First Affiliated Hospital of Harbin Medical University and were approved by the Harbin Medical University Animal Ethics Committee in accordance with the guidelines of the Association for Research and the Vision and Ophthalmology statement for the Use of Animals in Ophthalmic and Vision Research as well as the principles of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. New Zealand white rabbits weighing 2.0–2.4 kg were used for the animal experiments. The rabbits were divided randomly into two groups: a control group and a CEC injection group (n = 6). Only the right eyes of the rabbits were used for the experiment. A CE defect model was established according to a previous study with minor modifications [3]. Briefly, under systemic and topical anesthesia, the area of the CE was marked with a 7.0-mm trephine to lightly score the epithelial surface. The anterior chamber was filled with a viscoelastic substance to avoid chamber collapse during the subsequent procedures. Approximately 7 mm of the rabbits' CE was removed with a 30-gauge silicone soft-tipped cannula (ASICO, IL). The anterior chamber was then washed three times with Hanks balanced salt solution. A total of 2 × 107 differentiated CECs were injected into the anterior chamber in the CEC injection group, whereas an equal volume of Hanks balanced salt solution was injected in the control group. The rabbits were placed in an eye-down position for the next 12 h. All the operations were performed by the same experienced ophthalmologist. After the operation, the corneas were photographed under slit-lamp, anterior segment optical coherence tomography (AS-OCT) (Optovue) and a corneal confocal microscope (Zeiss, Germany). The central corneal thickness was measured with an ultrasound pachymeter (Quantel, France).
Histological analysis
Twenty-one days after the operation, the eyes were removed, and the corneas were excised and immersed in 4% paraformaldehyde. After gradient dehydration in sucrose, the tissues were embedded in OCT compound (SAKURA). Then, frozen sections were prepared and subjected to immunofluorescence staining. Briefly, the sections were permeabilized with fixation in cold acetone for 15 min, rinsed three times with PBS, and incubated with goat serum, the appropriate antibody, and DAPI as previously described.
Statistical analysis
The data from each group are presented as the mean ± SD. Significant differences were determined by Student's t test (two-tailed) and two-way ANOVA by using GraphPad Prism software (version 6.0; La Jolla, CA). P values <0.05 were considered statistically significant.
Results
Differentiation and identification of NCSCs
Before corneal maturation, the NCCs were required during normal corneal growth development. We obtained NCSCs by using a procedure reported by Wang A [20]. A monolayer of cultured NSCSs with a spindle appearance is shown in Fig. 1A. The expression levels of NCSCs marker genes were verified by real-time PCR. The p75 and vimentin mRNA expression levels were higher in induced NCSCs than in iPSCs (Fig. 1B p75, P < 0.05; vimentin, P < 0.01 vs. iPSCs). Immunofluorescence was used to determine the localization of proteins, including NSE, p75, and vimentin. As shown in Fig. 1C, the induced NCSCs were positive for NSE, p75, and vimentin, confirming their identification. In addition, NCSCs homogeneity was confirmed by a flow cytometry analysis of p75. Overall, 78.8% p75-positive NCSCs can be obtained in our experiment (Fig. 1D).
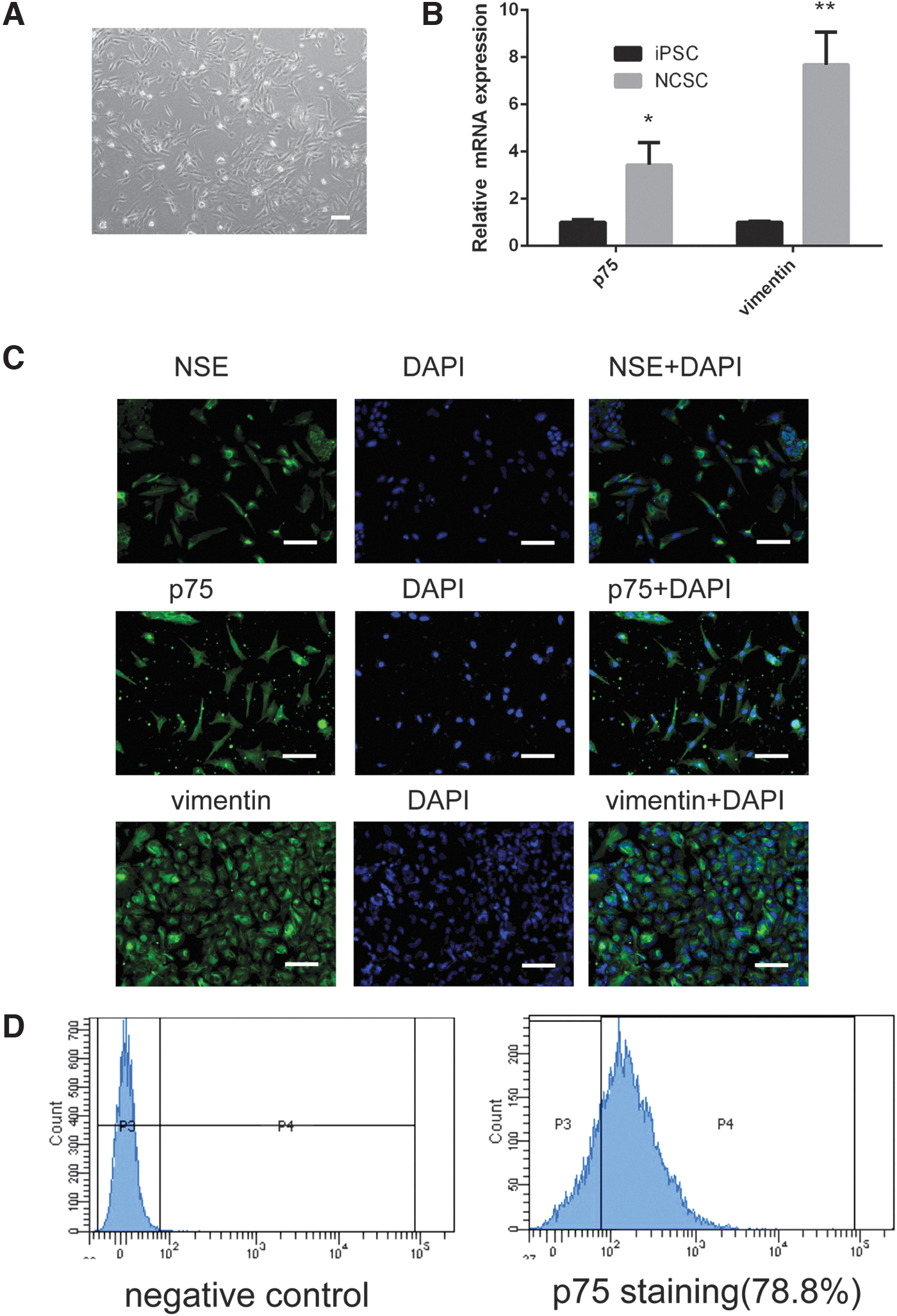
Characterization of NCSCs derived from iPSCs.
Differentiation and identification of CECs
After obtaining NCSCs, the next challenge was to differentiate NCSCs to CECs. We were unable to obtain CECs according to previously reported chemically defined methods in our study, so we modulated them slightly, and the results are shown in Fig. 2. Differentiated CECs displayed a cobblestone-like appearance (Fig. 2A). The levels of CE-related gene expression were verified by real-time PCR. As shown in Fig. 2B, the mRNA expression levels of CE-related genes, including ATP1A1 (P < 0.05), N-cadherin (P < 0.05), SLC4A4 (P < 0.05), and ZO-1 (P < 0.05), but COL8A2 (P > 0.05), were increased in CECs compared with iPSCs and NCCs, even though the expression levels of some of these markers in the CECs remained lower than those in the corneal endothelial cell line B4G12. The localization of the proteins was determined by immunofluorescence. The border of the CECs was stained with antibodies against Na+/K+ATPase, ZO-1, and N-cadherin, and the results were consistent with previous literature [9,10]. In addition, we performed an Na+/K+ATPase activity assay to determine the functional role of CECs. The Na+/K+ ATPase activity in CEC was 0.092 ± 0.029 U/mg prot, in B4G12 was 0.164 ± 0.014 U/mg prot, and in iPSC and NCC was close to zero. These results indicate the identification of CECs.
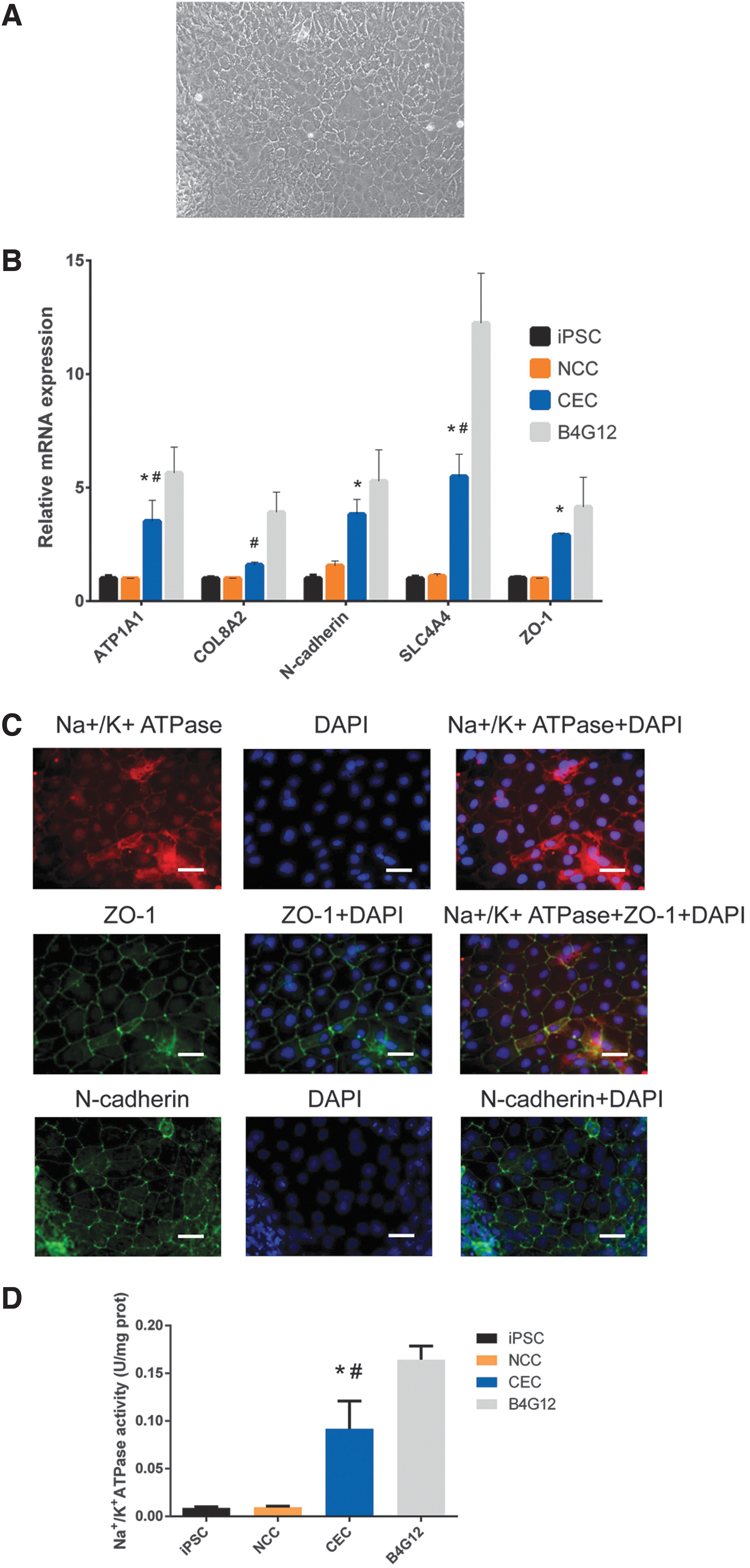
Characterization of CECs derived from NCSCs.
Differentiated CECs promote corneal transparency in vivo
Subsequently, CECs derived from NCSCs were used for the treatment of bullous keratopathy. After the operation, corneal edema was obviously decreased in the CEC injection group compared with the control group, as shown by slit-lamp photographs and AS-OCT images (Fig. 3A, B). In addition, we measured the central corneal thickness of these two groups on postoperative days 1, 3, 7, 14, and 21. No difference in the central corneal thickness was found between these two groups on the first postoperative day (control group 879.3 ± 153.4 μm vs. CEC injection group 855.5 ± 145.3 μm). However, a significant difference was observed between the groups starting from postoperative day 3. The central corneal thickness in the CEC injection group reached a peak on postoperative day 3 (1,086.7 ± 116.4 μm), and this thickness was thinner than that in the control group (1,456.1 ± 195.7 μm, P < 0.05). On postoperative days 7 (P < 0.001), 14 (P < 0.001), and 28 (P < 0.05), the central corneal thickness in the CEC injection group showed a gradual decrease and was significantly thinner than that in the control group (Fig. 3C). In addition, the cellular morphology of the corneal innermost layer was investigated in vivo by corneal confocal microscopy. The morphology of the corneal innermost cells in the CEC injection group was detected, whereas the control group did not exhibit this because of corneal edema. The cells in the CEC injection group were arranged in a mosaic pattern and were smaller and less regular than those in the native rabbit corneas (Fig. 3D). Finally, frozen sections of the corneas were prepared, and the sections were subjected to immunofluorescence staining for Na+/K+ ATPase and ZO-1. The immunostaining results demonstrated that the innermost layer of the cornea can be stained with antibodies against Na+/K+ ATPase and ZO-1 (Fig. 3E). These results reveal that the innermost layer of the cornea is composed of CECs derived from iPSCs. Overall, these results indicate that differentiated CECs could promote corneal transparency in a rabbit model of bullous keratopathy.
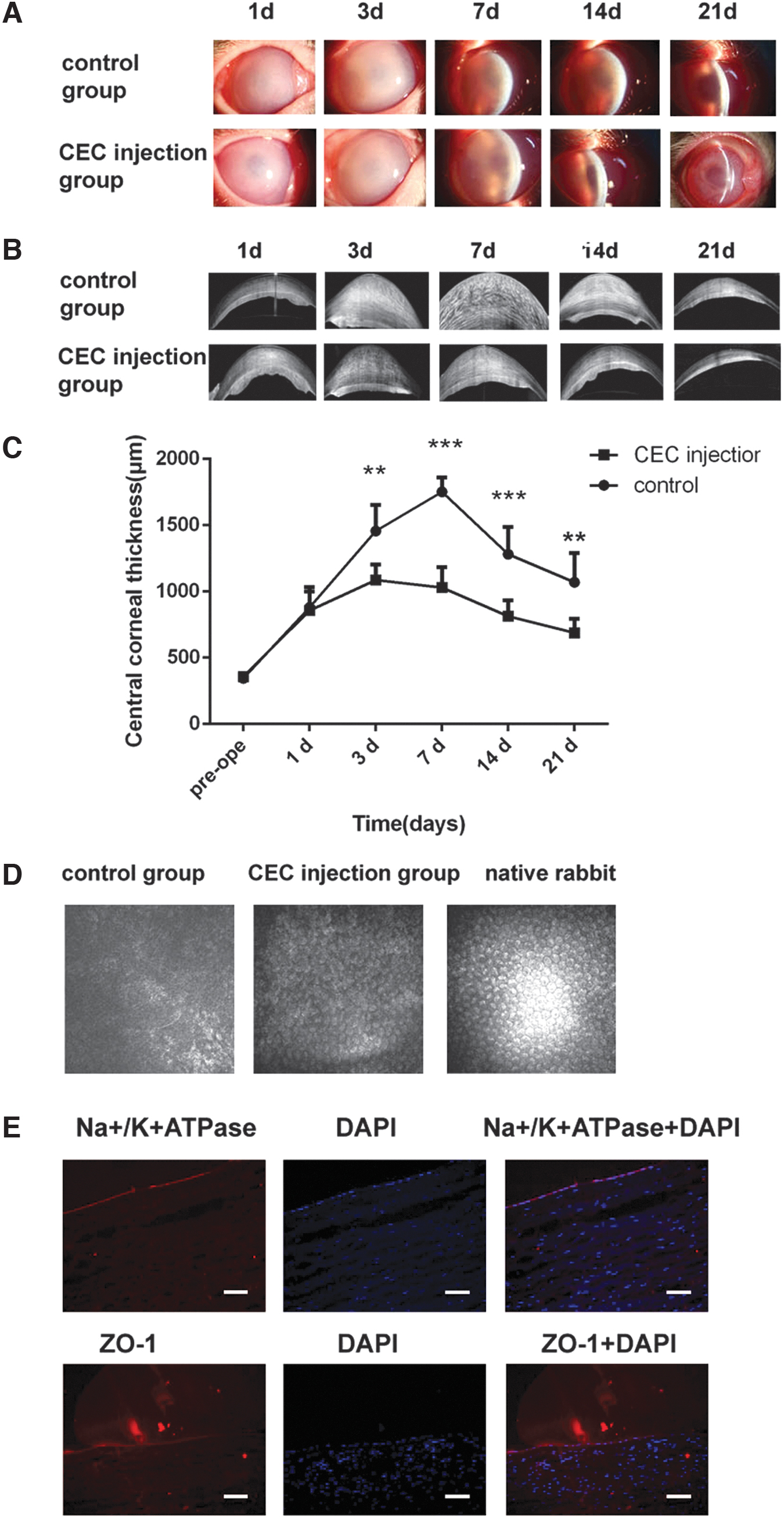
The intracameral injection of iPSC-derived CECs relieved corneal edema in a rabbit model of bullous keratopathy.
Discussion
Although several stem cell studies have attempted to treat CE deficiency, a few of the strategies can be applied to clinical settings. Thus, further studies should be performed. In the current study, NCSCs derived from iPSCs were differentiated into CECs by using a modified method. Further, we injected differentiated CECs into the anterior chambers of the eyes of a rabbit model of bullous keratopathy and found that corneal edema could be relieved. This study extends the findings of stem cell therapy for CE dysfunction.
Most of the previous studies on NCCs induction focused on dual Smad inhibitors, including BMP and TGFβ inhibitors [21,23 –26]. A few studies have used Wang's method, which is another differentiation method that depended on StemPro supplementation combined with bFGF and EGF. It was revealed that 98.3% p75-positive NCSCs can be obtained by using Wang's method [20]. In addition, induced NCSCs homogeneously expressed NCSC markers [20], which confirmed the homogeneity of these NCSCs. Therefore, we selected this method for NCSCs induction. In our experiments, the feasibility and availability of this method were further validated. We obtained NCSCs with positive p75, NSE, and vimentin expression from iPSCs, which provide favorable conditions for subsequent experiments.
Several reports have shown that B27, PDGF-BB, and DKK2 can be used to successfully differentiate the NCCs to CECs. However, we were unable to replicate that in our experiments. It is possible that this phenomenon was due to different methods used for inducing the NCCs and different NCCs states. Hence, we revisited the approach for CECs differentiation. Through a literature review, it was found that TGFβ signaling was involved in CE development with complicated mechanisms. As mentioned by Ittner, lens-derived TGFβ signaling controls the development of the CE and trabecular meshwork by regulating FOX C1 expression [27]. TGFβ signal inactivation might cause corneal endothelial dysplasia. However, Cassandra and Flügel-Koch reported that TGFβ1 overexpression blocked normal development of the anterior chamber, including the CE [28]. Based on the findings mentioned earlier, the detailed role of TGFβ signaling in CE development remains unclear, and intervention with TGFβ expression during CEC differentiation might play a role. Promotion or inhibition of TGFβ expression in CEC differentiation medium was thus applied, and our results showed that TGFβ inhibition could help to obtain CECs and that TGFβ overexpression caused fibrosis, which is consistent with the previous finding that TGFβ contributes to endothelial-to-mesenchymal transition of corneal endothelial cells [29,30]. Our finding is also in agreement with the reports of Zhao, who induced CECs from NCSCs by suppressing TGFβ and ROCK signaling [25]. These results suggest that TGFβ inhibition might be one of the switches for CEC differentiation. However, there were differences between the in vivo and in vitro experiments. How to regulate TGFβ signaling in detail in the differentiation process needs further research.
Kinoshita et al. reported that the injection of human CECs into the anterior chamber aided the recovery of corneal transparency in patients with bullous keratopathy [31]. Peh and colleagues also demonstrated that the injection of corneal endothelial cells is an efficient strategy for the alleviation of corneal edema in a rabbit model of bullous keratopathy [3]. Our findings revealed that the intracameral injection of iPSC-derived CECs relieved corneal edema in rabbits with CE deficiency. These results show great promise for the feasibility of clinically treating patients suffering from bullous keratopathy with an intracameral injection of CECs. Compared with conventional corneal transplantation or TECE transplantation, intracameral injection is, indeed, an easy and minimally invasive method for curing corneal dysfunction and might replace conventional corneal transplantation or TECE transplantation in the future. In addition, iPSC-derived CECs represent an unlimited source, which can solve the lack of primary cultured corneal endothelial cells. In the next step, the optimal time for injection and minimum effective cell dose will be further explored.
Corneal transplantation is the most successful form of tissue transplantation in humans, with a less than 10% rejection rate. The rejection is characterized with severe corneal edema, corneal neovascularization, and conjunctival hyperemia. Actually, a very low amount of corneal neovascularization was found over the course of the experiment. Maybe it is because of the use of steroid eye drops that can help to reduce corneal neovascularization and conjunctival hyperemia after the surgery. In addition, corneal edema appeared when the CE was removed. Hence, it is difficult to determine whether corneal edema was due to rejection. In future studies, this aspect will be further investigated.
Other limitations regarding the present study should be noted. The presence of an intact Descemet's membrane (DM) was necessary for the success of the animal experiments. Corneal endothelial cells cannot attach to denuded corneal stroma, as has been proven by Peh [3]. This finding indicates that an intracameral injection of CECs is only suitable for CE deficiency-related diseases with a complete and intact DM. In addition, other animals, such as monkeys, might be more suitable for animal experiments because rabbit corneal endothelial cells can proliferate evidently in vivo. In addition, the homogeneity of injected cells was evaluated based on their morphology under a phase-contrast microscope. The FACS should be used for cell selection in the following experiment.
Conclusion
This study has shown a novel approach for differentiating iPSCs to CECs, and we found that the injection of CECs into the anterior chamber was beneficial for relieving corneal edema in a rabbit model of bullous keratopathy. Thus, this method might constitute a possible strategy for the treatment of reduced visual acuity caused by CE deficiency in the future.
Footnotes
Acknowledgment
The authors would like to thank all the participants involved in this work, and all members of the authors' laboratory who offered assistance in the present study.
Author Disclosure Statement
All authors declare that there are no conflicts of interests associated with the article.
Funding Information
This work was supported by the National Natural Science Foundation of China [81671844 and 81970776], Heilongjiang Academy of Medical Sciences Research and Transformation Special Fund [CR 201809].
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.