
Abstract
Homozygous familial hypercholesterolemia (hoFH) is a rare disorder caused primarily by pathological mutations in the low-density lipoprotein receptor (LDLR), which disrupts LDL-cholesterol (LDL-C) metabolism homeostasis. hoFH patients are at extremely high risk for cardiovascular disease and are resistant to standard therapies. LDLR knockout animals and in vitro cell models overexpressing different mutations have proved useful, but may not fully recapitulate human LDLR mutation biology. We and others have generated induced pluripotent stem cells (iPSC) from hoFH patient's fibroblasts and T cells and demonstrated their ability to recapitulate hoFH biology. In this study, we present the generation and characterization of a cohort of seven hoFH-iPSC lines derived from peripheral blood mononuclear cells (PBMC) collected from four homozygous and three compound heterozygous patients. The hoFH-iPSC cohort demonstrated a wide range of LDLR expression and LDL-C internalization in response to rosuvastatin that correlated with the predicted pathogenicity of the mutation. We were able to confirm that hoFH-iPSC cohort were pluripotent by differentiation toward all three germ layers and specifically to hepatocyte-like cells (HLC), the cell with primary LDL-C metabolic regulatory control, by expression of hepatocyte markers. hoFH patient PBMC-derived iPSC recapitulate the LDLR dysfunction of their specific mutation. They were capable of differentiating to HLC and could be useful for early developmental studies, pharmacology/toxicology, and potentially autologous cell therapy.
Introduction
Familial hypercholesterolemia (FH) is a genetic deficiency caused primarily by mutations in the low-density lipoprotein receptor (LDLR) gene [1]. FH presents in three forms, heterozygous (one affected allele, htFH), homozygous (two affected alleles with the same mutation, hoFH), and compound heterozygous (two affected alleles with different mutations), the latter of which can be described as hoFH since the physiological outcome is essentially the same [2]. Heterozygous FH patients present with a range of LDL-cholesterol (LDL-C) of 300–500 mg/dL, while hoFH patient's LDL-C can approach as high as 1,000 mg/dL [3]. Because of this inability to regulate systemic LDL-C, hoFH patients develop accelerated cardiovascular disease and are particularly prone to coronary blockage and heart attack [4 –7].
FH prevalence for heterozygous and homozygous forms is estimated to be 1:200–300 and 1:300,000, respectively [8]. As htFH patients have one wild-type LDLR, standard interventions for hypercholesterolemia can be very effective [3]. However, hoFH patients are frequently much less responsive to these therapies and require more aggressive treatment to reduce LDL-C [9]. Since it is a monogenic disorder, gene therapy has long been of interest as treatment for hoFH [10], but still has not achieved a clinical success.
Induced pluripotent stem cells (iPSC) have been previously reprogrammed from hoFH patient cells, including fibroblasts and T cells [11 –16]. They have been shown to accurately recapitulate the LDLR and cholesterol dysfunction associated with hoFH [12 –14]. Fattahi et al. successfully transduced hoFH-iPSC derived from hoFH fibroblasts with a lentivirus to deliver a constitutively expressed LDLR transgene [17]. We have demonstrated restoration of LDLR feedback control in FH-iPSC using an episomal plasmid with 10 kb of upstream regulatory sequences [13,18] and were the first to successfully use clustered regularly interspaced short palindromic repeats/CRISPR-associated protein 9 (CRISPR)/Cas9 to directly repair the pathological homozygous mutation found in the LDLR gene and restore normal LDLR function [14].
In this study, we expand on our hoFH-iPSC work by reporting the reprogramming of peripheral blood mononuclear cells (PBMC) acquired from seven hoFH patients —four homozygous and three compound heterozygous. These cells recapitulated LDLR dysfunction and specifically differentiated to hepatocyte-like cells (HLC). hoFH-iPSC may be useful as models of (1) early development with dysfunctional LDLR [19], (2) the effect of statin or other cholesterol-lowering therapy in hoFH patients [20 –22], (3) differential cholesterol metabolism between normal and defective LDLR [14,15], as well as (4) a cell source for engineering liver-like tissues and patient-specific gene correction [14,23].
Experimental Procedures
Human subjects and human pluripotent cell approvals
We obtained written informed consent from seven hoFH patients enrolled in the Rogosin Institute's hoFH repository (NCT01109368). Collection of patient blood samples and generation of iPSC for hoFH-related research was approved by the Rogosin Institute and University of Louisville IRB (18.0296) and IBC (14–043) committees. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki.
PBMC isolation and cell reprogramming
Peripheral blood (10 mL) was collected from consented patients and shipped overnight to Louisville. Isolated hoFH-PBMC were reprogrammed to hoFH-iPSC using CytoTune 2.0 (ThermoFisher Scientific, Waltham, MA) according to the manufacturer's instructions.
Mutation confirmation by Sanger sequencing
Mutations were confirmed by the Center for Genetics and Molecular Medicine (CGeMM) DNA Core at the University of Louisville using Sanger sequencing of genomic DNA from hoFH-iPSC [14]. Primers were designed to cover ±250 bp surrounding the target mutation (Integrated DNA Technologies, Coralville, IA) (Supplementary Table S1), and the results were analyzed using SeqMan Pro (DNASTAR, Madison, WI).
Statistical analysis
LDLR protein levels, LDLR-3,3′-dioctadecylindocarbocyanine (DiI) signal regression analysis, and albumin (ALB) enzyme-linked immunosorbent assay results were quantified in Excel (Microsoft, Redmond, WA) and reported as average ± standard deviation. Box and whisker plots representing Median, 5th, 25th, 75th, and 95th percentiles, two-way analysis of variance (ANOVA), and Pearson correlation were performed in SigmaPlot (Systat Software, San Jose, CA) with statistical significance determined by P < 0.05.
Further Materials and Methods details can be found in the Supplementary Documents.
Results
Generation of patient-specific hoFH-iPSC cohort
The seven donor hoFH patient's LDLR genes were sequenced as part of the Rogosin Institute's repository to identify homozygous/compound heterozygous mutations (Table 1). For each patient, we state their initial LDL-C levels, sex, the mutation's classification, and accession number as reported in other publications [24], the Leiden Open Variation Database for LDLR and ClinVar database. Two patients, LR2 and LR5, shared the same homozygous c.2043C>A mutation, but were unrelated. All cohort mutations are either class 1 or 2, except LR7 allele 2, which is not determined, but likely class 3, 4, or 5 [24] since there is protein present, but none of it is immature. An outline of the effects of mutation class on LDLR processing is presented in Supplementary Table S2 [24].
FH Patient Donor Data
Unrelated, ∼100 kDa truncated protein.
LDL-C, low-density lipoprotein-cholesterol; LDLR, low-density lipoprotein receptor; LOVD3, Leiden Open Variation Database; NA, not available; ND, not determined.
Each patient's blood sample was processed for PBMC isolation, cryopreserved, and reprogrammed with Sendai virus (SeV)-based vectors as described in the Supplementary Methods. After approximately 10 manual passages, colonies were tested for pluripotency markers Oct4 and SSEA4 as well as retention of SeV (Fig. 1). All cell lines were positive for Oct4 and SSEA4, but negative for residual SeV. A low passage wild-type LDLR cell line (BL3) also reprogrammed with SeV was used as a positive SeV control (Fig. 1). Samples labeled with primary antibody isotype controls were prepared at the same time and show no detectable nonspecific labeling (Supplementary Fig. S1). All hoFH-iPSC lines were allowed to form embryoid bodies (EB), then differentiated toward each germ layer (ectoderm, mesoderm, and endoderm), and assessed for marker transcript expression (Supplementary Fig. S2).

Reprogrammed FH-iPSC are pluripotent and SeV free. PBMC from FH donor blood was reprogrammed to iPSC using SeV vectors in feeder free conditions. Single colonies were manually passaged 10 times then tested for expression of pluripotency markers and retention of SeV vectors. All seven FH-iPSC lines were positive for the pluripotency markers Oct4 and SSEA4 (AF488), but negative for SeV (AF594). Nuclear DAPI labeling confirms the presence of cells. An early passaged (p3) LDLR normal iPSC line, BL3, shows the presence of residual SeV along with negative isotype control, IgG (AF647). All images were acquired at the same settings and uniformly brightened to enhance visualization. Scale bar = 50 μm. DAPI, 4′,6-diamidino-2-phenylindole; FH, familial hypercholesterolemia; IgG, immunoglobulin G; iPSC, induced pluripotent stem cells; LDLR, low-density lipoprotein receptor gene; PBMC, peripheral blood mononuclear cells; SeV, Sendai virus.
Cohort hoFH-iPSC mutation confirmation and LDL-C internalization
After establishing the hoFH-iPSC cohort cell lines, we next confirmed the reported genetic mutations. Primer pairs (Supplementary Table S1) were designed for ∼250 bp on either side of the target mutation locus (Fig. 2) and amplicons subjected to Sanger sequencing. All mutations were confirmed as well as surrounding sequences as we have demonstrated previously [14].
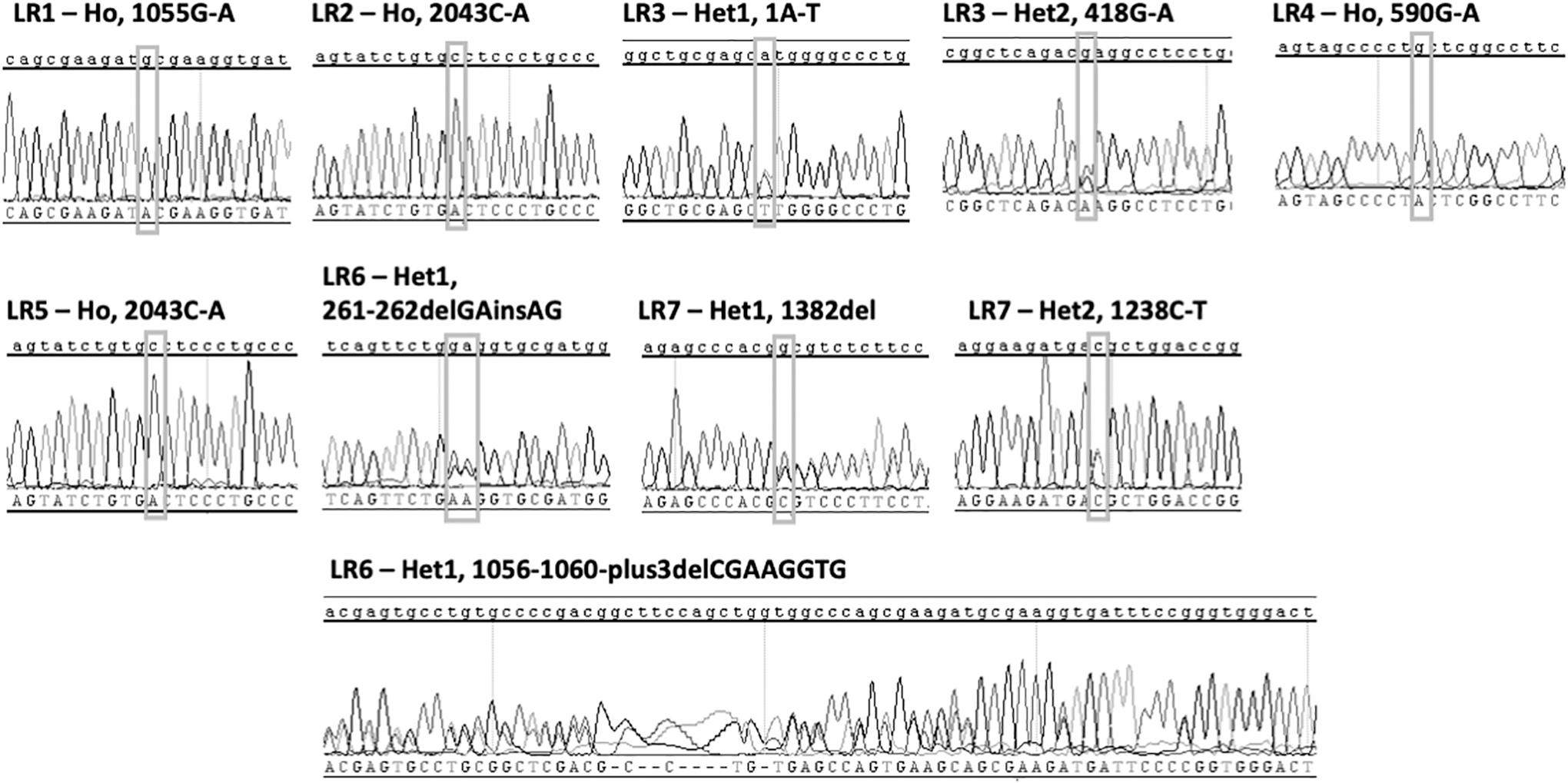
FH-iPSC mutation confirmation. After reprogramming, FH-iPSC mutations and the surrounding gene 250 bp up and downstream were confirmed by Sanger sequencing.
We next investigated the ability of the derived hoFH-iPSC to internalize LDL-C (Fig. 3 and Supplementary Fig. S3) as described in Supplementary Methods. We used BL3 to establish fluorescence acquisition settings used for all cell lines (Fig. 3A). As expected, the brightest DiI signal was acquired from the BL3 cells. Qualitatively, LR1 and LR7 showed about half the ability to internalize LDL compared to BL3, while all other lines had minimal detectable DiI-LDL internalization. Representative images of the fluorescence for samples that included 50 × unlabeled LDL are presented in Supplementary Fig. S3.

FH-iPSC line LDL-C internalization.
Quantification of the fluorescence signal with and without unlabeled LDL, that is normalized to the cell count (ie, number of nuclei) is presented as box and whisker plots in Fig. 3B, while Fig. 3C is the normalized specific fluorescence (ie, fluorescence without unlabeled LDL minus fluorescence with unlabeled excess LDL). One-way ANOVA analysis indicates that there is a statistically significant difference in the sample means with statin treatment (P ≤ 0.05). When compared with BL3 control cells, DiI fluorescence intensity is significantly different for LR2, LR3, LR4, and LR5 (P ≤ 0.05), but no significant difference was detected comparing BL3 to LR1, LR6, and LR7, with P = 0.41, 0.06, and 0.40, respectively, although LR6 P-value suggests it trends toward a difference.
When comparing this data with the clinical data in Table 1, it demonstrates that hoFH LDLR mutation-specific effect on expression and function can be accurately modeled in these patient-derived cells.
Characterization of hoFH-iPSC cohort LDLR expression
We then investigated the LDLR protein expression [120 kDa (immature) vs. 160 kDa (mature)] under the same culture conditions used previously (Fig. 4). BL3 LDLR was quantified by densitometry units as 11.8 and was completely represented by the mature isoform.
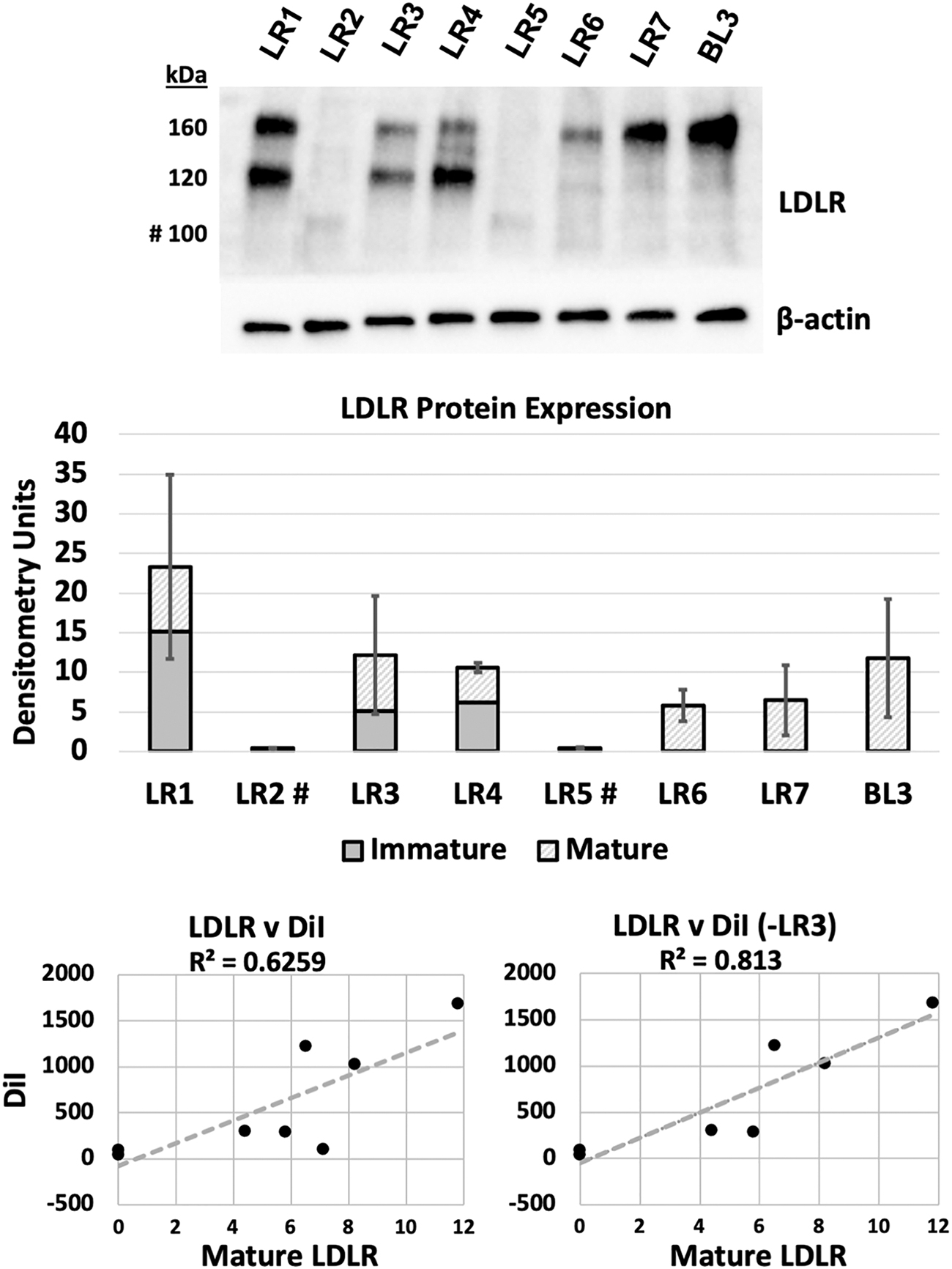
Characterization of FH-iPSC LDLR response to statin treatment. FH-iPSC were treated overnight in LPDS media supplemented with 5 μM Rosuvastatin then processed for LDLR protein and transcript expression. A representative western blot of LDLR shows the pattern of truncated immature (100 kDa), immature (120 kDa), and mature (160 kDa) LDLR expression for each FH-iPSC line. BL3 is a functionally normal LDLR iPSC line. β-actin expression for the same blot is used as a loading control. Unitless densitometry quantification of immature (solid gray), mature (hatch), and total (gray+hatch) with SD of total LDLR protein. Scatter plots of all FH-iPSC and exclusion of LR3 as a DiI outlier were generated by plotting mature LDLR versus specific DiI fluorescence and analyzed by linear regression goodness-of-fit (R 2), Pearson correlation coefficient (r) and P-value (see text). n = 3. SD, standard deviation.
Patient LR1 is classified as a 2B mutation with 15.1 and 8.2 U of immature and mature LDLR, respectively.
Patients LR2 and LR5 have the same class 2A mutation indicating that all protein is retained in the endoplasmic reticulum (ER) in an immature form [24]. Unlike the other clearly class 2 mutants (LR1/3/4), c.2043C>A (FH-Lebanese) results in an immature truncated protein of ∼100 kDa [25]. We also note here the significantly reduced protein level as previously reported in studies using fibroblasts (FH 264) [25].
Patient LR3 is a compound heterozygote with class 1 and class 2B mutations. Patient LR3's LDLR protein was quantified as 5.1 and 7.1 U, immature and mature, respectively. Since the class 1 mutation should not produce protein, what is detected here should be from the 2B allele.
Patient LR4 is a homozygous class 2B mutation where we quantified 6.2 and 4.4 U, immature and mature LDLR, respectively. Similar to LR1, there was more immature protein, indicating accumulation of misfolded protein, but being class 2B, some ability for translated LDLR transport and processing to the mature state.
Patient LR6 is a compound heterozygote, with both alleles designated as class 1, indicating no protein translation. We quantified 0 and 5.8 U, immature and mature, respectively, which indicates that some protein processing is occurring, but its functionality is unclear.
For patient LR7, allele 1 is class 1, and while allele 2 mutation class is not specifically identified in the literature, the western blot results showing 0 and 6.5 U of immature and mature, respectively, suggest that it is not class 1 (since protein is present) or 2 (since no immature protein is present).
Since cells require mature LDLR to internalize LDL-C by endocytosis, we calculated the correlation between mature LDLR protein detected by western blot with specific DiI fluorescence signal (Fig. 4).
The mean mature LDLR and DiI signal were graphed in a scatter plot and the R 2 linear curve fit determined to be 0.6259. Since we found that patient LR3's cells internalized low levels of DiI-LDL for the amount of mature LDLR, we treated it as a potential outlier and recalculated the goodness-of-fit R 2 = 0.813. For each of these conditions, the Pearson correlation coefficient (r) and P-value were calculated to be r = 0.791, P < 0.02 and r = 0.902, P < 0.01, for all data points and less LR3, respectively. These data demonstrate the dysfunctional nature of the LDLR in these patients and provide further evidence that the derived cells can replicate the mutation-specific receptor-mediated endocytosis pathology.
hoFH-iPSC HLC differentiation
The persistent excessively elevated LDL-C found in hoFH patients leads to accelerated development of atherosclerosis and particular susceptibility to coronary blockage and heart attack [6]. Since the liver is the primary organ responsible for LDL-C homeostasis [26], we sought to differentiate our newly derived hoFH-iPSC cohort specifically to HLC [13,14].
We utilized the three-stage differentiation protocol reported by Hay [27] and collected samples between days 3 and 6 of Stage 3. We utilized quantitative polymerase chain reaction to examine the expression of three genes specifically associated with hepatocytes, α-fetoprotein (AFP), ALB, and hepatocyte nuclear factor 4a (HNF4A) [28 –30], and expression was normalized to glyceraldehyde 3-phosphate dehydrogenase [31] (Fig. 5). All cohort hoFH-iPSC lines and the LDLR wild-type BL3 expressed these hepatocyte-specific genes, providing evidence of capability to differentiate to endoderm and the HLC lineage [13,14,27].
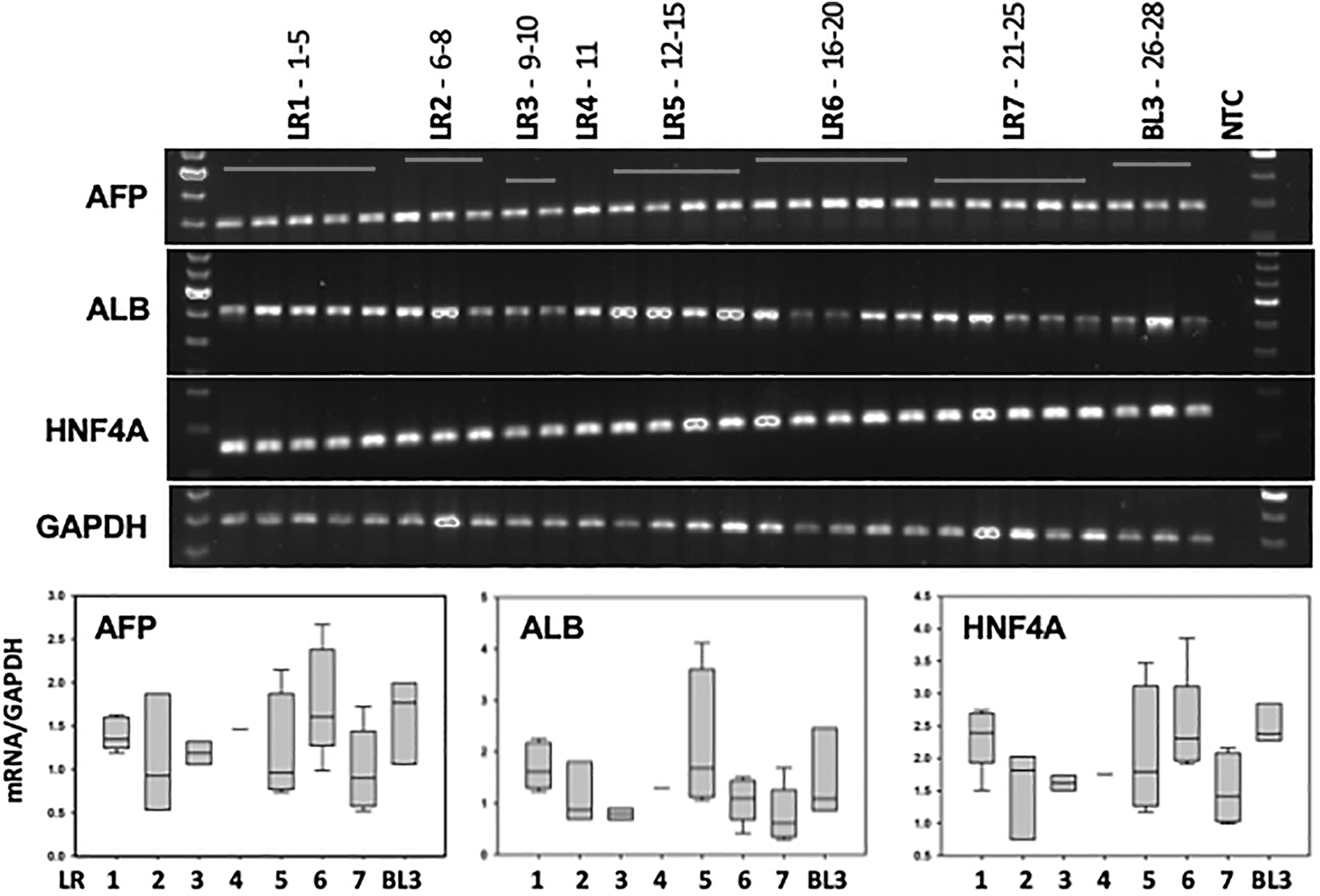
FH-HLC hepatocyte marker transcript expression. FH-iPSC and BL3 control were differentiated to HLC and processed for qPCR transcript expression of hepatocyte-specific markers AFP, ALB, and HNF4A with GAPDH loading control. Quantification of normalized transcripts presented in the box plots represents median, 5th, 25th, 75th, and 95th percentiles. AFP, α-fetoprotein; ALB, albumin; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; HLC, hepatocyte-like cells; HNF4A, hepatocyte nuclear factor 4a; qPCR, quantitative polymerase chain reaction.
Discussion
We and others have generated hoFH-iPSC from fibroblasts [11 –14,16,17] and T cells [15]. Here, we report using hoFH PBMC. The relevant findings of this report are that (1) iPSC can be derived from patient's PBMC in blood samples collected from hoFH patients with a variety of LDLR mutations and shipped from a distant site, (2) the derived cohort of hoFH-iPSC retain characteristic LDLR-mediated LDL-C metabolism, and (3) hoFH-iPSC cohort differentiate to HLC, the primary parenchymal cell responsible for systemic LDL-C homeostasis regulation.
All seven patients had LDL-C levels >500 mg/dL, with LR1 and LR6 presenting with 815 mg/dL and 907 mg/dL, respectively. A non-FH patient without vascular disease or other risk factors would be considered for prescription therapy when the LDL-C exceeds 160 mg/dL [3]. These hoFH patients had three to six-fold greater LDL-C than individuals with high LDL-C and fully functional LDLR. htFH patients, possessing one functional allele, can generally be successfully treated to the recommended LDL-C goal of 70–100 mg/dL with lifestyle changes and medications that increase LDLR function: statins, ezetimibe, and PCSK9 inhibitors [3,32,33]. The limited effectiveness and adverse side effects of current therapies for hoFH patients without functional LDLR motivates the search for alternative treatment approaches.
FH has been studied for decades and was the principal disease model for dissecting receptor-mediated endocytosis, leading to a Nobel award [34]. Knowledge of FH and LDL-C metabolism has advanced as a result of research in patients, cells (FH fibroblasts, mutant LDLR overexpression) and animal models (LDLR knockout in mice, rat, and pig). The derivations of different patient-specific hoFH-iPSC lines provide unique models to further our understanding of hoFH pathology and cholesterol metabolism. We demonstrate here that the hoFH cell cohort retained the pathological mutations and reduced receptor-mediated LDL-C internalization that correlated with the expression of mutant-mature LDLR protein. These cells have the great advantage to provide an unlimited source for further experimental manipulation.
It is estimated that roughly 50% of the over 1,700 pathological mutations are class 2 [24,35]. Five out of the seven in this cohort have a predicted class 2 mutation in one or both alleles. These mutations are unique from the other four classes in that the mutation additionally causes synthesis of nonfunctional immature LDLR due to protein misfolding in the ER [24].
In our previous publication [14] and in this study, statin treatment of the iPSC caused an accumulation of immature (120 kDa) LDLR protein. In cell models using class 2 overexpression in Chinese hampster ovary cells [36,37], misfolded immature LDLR caused ER stress and induced the unfolded protein response (UPR). In contrast, we found that FH-iPSC and HLC from class 2 FH-Piscataway did not show induction of ER stress or UPR when treated with low-dose statin [38]. The Watanabe Heritable Hyperlipidemic rabbit line was the only class 2 animal model [39], but is no longer available and to our knowledge, no examination of misfolded immature LDLR accumulation, and ER stress was ever undertaken. Therefore, class 2 hoFH-iPSC and HLC derivatives could be useful to determine the effects of LDLR misfolding and if they pose additional clinical challenges.
Although reduced LDLR function is not embryonic lethal, very little is known about the effects of LDLR mutations on early development [20]. As another example of how iPSC could be used to understand the effects of drugs on LDL and cholesterol metabolism, the effects of statins could be tested on embryonic stem cells and EB to determine their effects on development and epigenetic modifications of LDLR function.
Liver hepatocytes are the only known cells to regenerate on a massive scale. Paradoxically, when hepatocytes are isolated for culture, they rapidly lose their phenotype and senesce [40]. Derived hepatocytes from a source like iPSC have the potential for generating unlimited quantities for experimentation or therapy, but producing fully mature hepatocytes in culture is a challenge [41]. The protocol used here is reported to produce progressively mature hepatocytes over a total of 18 days [27]. Here, we demonstrate that the cohort hoFH-iPSC differentiated to express markers specific to hepatocytes, HNF4A, AFP, and ALB [27] supporting their potential use to model FH biology in human-derived HLC.
We were the first to report the use of CRISPR/Cas9 genome editing to correct LDLR dysfunction in hoFH-iPSC and HLC [14]. We directly repaired the homozygous 3 bp deletion in exon 4 of the LDLR gene demonstrating that CRISPR/Cas9 can completely correct the pathological mutation to restore feedback controlled LDLR-mediated LDL-C metabolism [14]. This is a clear example of using CRISPR/Cas9 genome editing to precisely insert a specific DNA sequence to normalize dysfunctional LDLR activity.
It is possible that future technological advances will allow use of such cells for therapy. They can also be used for testing CRISPR/Cas9 components for their efficiency and specificity in an individual patient. This could allow for direct delivery of customized CRISPR/Cas9 editing tools to the hoFH patient's liver. Although this approach is more complex than delivery of a constitutively expressed LDLR transgene (NCT03018678), successful delivery would permanently repair the dysfunctional LDLR gene with restoration of feedback control of gene expression. Although statin therapy would likely still be necessary, which would artificially induce LDLR expression, this might be preferable to long-term constitutive overexpression that cannot be downregulated.
A recent report generated transgenic hoFH-iPSC utilizing CRISPR/Cas9 to insert an LDLR transgene controlled by the hAPOA2 promoter into the AAVS1 site of class 1 hoFH-iPSC and successfully produced HLC that demonstrated restored feedback control of LDLR synthesis in response to statin [16]. However, although the AAVS1 site is frequently used as a “safe harbor” for transgene insertion, the long-term effects of disrupting the PPP1412C gene [42] and of driving LDLR expression with the hAPOA2 promoter and Woodchuck hepatitis virus Posttranslational Regulatory Element are not known. Clearly, more research is needed to develop gene-modified iPSC as effective and safe therapy for FH.
Conclusion
The treatment of hoFH patients is very challenging due to limited effectiveness of therapeutic options, high costs associated with treatment and decrease in quality of life [43 –45]. Derived hoFH-iPSC, such as presented here, can be useful for addressing many of the issues related to hoFH biology as well as development of cell and gene therapies. Our current toolset has the potential to advance novel approaches to this complex lipid disorder.
Footnotes
Acknowledgments
The authors thank the FH donors for permitting the collection and experimental use of their cells.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
Supported by National Institutes of Health grant NIH 1R21EB0022185 and Jewish Heritage Fund for Excellence (to N.L.B.).
Supplementary Material
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.