
Abstract
Mesenchymal stem cells (MSCs) have been shown to be involved in bone injury repair. Programmed cell death 4 (PDCD4) is not only a tumor suppressor gene but also plays roles in the regulation of MSC function. The aim of the study was to uncover PDCD4 potential regulatory roles and mechanisms in the osteogenic differentiation and bone defect repair of MSCs. shRNA technique was used to knock down PDCD4 expression in umbilical cord-derived mesenchymal stem cells (shPDCD4-UCMSCs). Their phenotype was characterized by flow cytometry and the differentiation potential was verified. We found that PDCD4 knockdown did not affect the surface molecule expression of UCMSCs, but significantly enhanced their osteogenic differentiation and osteogenesis-related molecule expression. Mechanistically, glycogen synthase kinase-3β (GSK-3β) phosphorylation and β-catenin expression were significantly increased in shPDCD4-UCMSCs during the osteogenic differentiation process. The β-catenin inhibitor PNU-74654 reversed shPDCD4-increased osteogenesis and osteogenesis-related molecule expression. The results of animal experiments showed that shPDCD4-UCMSCs markedly improved the defect healing in rabbits. Our findings suggest that PDCD4 acts as a negative regulator of MSC osteogenic differentiation through GSK-3β/β-catenin pathway. Targeting PDCD4 may be a way to improve MSC-mediated therapeutic effects on bone injury.
Introduction
As multipotent progenitor cells, mesenchymal stem cells (MSCs) possess a high capacity of self-renewal and differentiation into different lineages, including neurons, lipocytes, osteoblasts, chondrocytes, and others [1]. These cells are also endowed with various biological functions, such as tissue repair and regeneration, in addition to immunomodulatory and anti-inflammatory activities. Due to these biological properties, MSCs are often incorporated into cellular therapy strategies for various diseases [2].
Tissue regeneration is a complicated process in which MSCs undergo a series of events, including adhesion, proliferation, migration, and differentiation, among which, MSC migration to injured tissues and inflamed areas plays a vital role in tissue regeneration [3]. MSC recruitment to such injured or inflamed areas is made possible through the release of multiple cytokines and chemokines by damaged tissues or apoptotic cells, which ensures the MSCs proliferate and differentiate where needed [4]. It has been found that intravenously infused MSCs could migrate to injured or inflamed tissues and exert therapeutic effects on bone injury [5,6].
Programmed cell death 4 (PDCD4) is a classical tumor-suppressing protein [7], which can also restrain the activation of transcription factors [8] and play an inhibitory role in a variety of solid tumors [9]. In recent years, it has been clear that PDCD4 is more than just a tumor suppressor, as it regulates the immune response in autoimmune or inflammatory diseases [10 –12].
MicroRNAs (miRNAs) are noncoding single-chain small RNAs that can specifically target mRNAs, thereby leading to their degradation or translation inhibition; Pdcd4 is the major target gene of miRNA-21. Usually, miRNA-21 is abundantly expressed in bone marrow stromal cells, and inhibiting miR-21 expression can induce a notable decrease in osteoclast activity and, thus, promote bone formation [13]. In contrast, miR-21 overexpression can promote the differentiation of MSCs into osteocytes or adipocytes [14]. In addition, PDCD4 inhibits the proliferation and differentiation potential of the adipose-derived MSCs (ADMSCs) [15]. These data indicate that PDCD4 may participate in the directional differentiation of MSCs into bone cells.
In this study, our results showed that PDCD4 is highly expressed in umbilical cord-derived mesenchymal stem cells (UCMSCs) and silencing PDCD4 expression significantly increased the osteogenic differentiation and bone defect repair of UCMSCs through glycogen synthase kinase-3β (GSK-3β)/β-catenin pathway. To our knowledge, this is the first report showing a role of PDCD4 in regulating osteogenic differentiation and bone defect repair of MSCs. Our study may lay the groundwork for prospective studies on bone destructive diseases and for developing more effective treatment approaches for bone injury or diseases using MSCs by targeting on Pdcd4 gene or on its related signaling pathways.
Materials and Methods
MSC culture and expansion
Umbilical cords were collected from healthy puerpera at Obstetrical Department of The Second Hospital, Cheeloo College of Medicine, Shandong University. Bone marrow was taken from patients with hypoferric anemia at Hematology Department, whereas subcutaneous skin and adipose tissue were taken from discarded tissues resected during surgery at the Department of Burns and Plastic Surgery. The specimens were collected as medical waste and the patients' oral consent was obtained. This research was authorized by the Ethics committee of The Second Hospital, Cheeloo College of Medicine, Shandong University (Application No. KYLL-2016(GJ)P-0011). MSCs were isolated and expanded using the method we previously described [16].
To extract UCMSCs and ADMSCs, the respective tissues were first rinsed with phosphate-buffered saline (PBS) and then shredded with scissors; this was followed by digestion by collagenase for 1 h and 0.25% ethylene diamine tetraacetic acid (EDTA)-trypsin (25200056; Gibco, Waltham, MA) for 30 min, after which the obtained cells were washed and filtered. In contrast, the bone marrow-derived MSCs (BMMSCs) and mononuclear cells were obtained from bone marrow using the human Lymphocyte Separation Medium (Ficoll, P8900; Solarbio, Beijing, China) and cultured in DMEM/F12 medium (SH30023.01B; HyClone, Logan, UT), containing 10% fetal bovine serum (10100147; Gibco), 20 ng/mL basic fibroblast growth factor (13256029; Invitrogen, Carlsbad, CA), and 100 IU penicillin and 100 μg/mL streptomycin (SV30010; HyClone) at 37°C and 5% CO2 for 3 days.
On day 4, the unattached cells were removed along with the culture medium, while the adherent cells were replenished with fresh medium. When the cells reached a confluency of around 80%, they were digested with 0.25% EDTA-trypsin and subcultured. The fourth passage MSCs were used for our experiments.
Construction and transfection of lentivirus
A shRNA or overexpression sequence specific for Pdcd4 gene (NM_014456) was acquired from Shanghai GeneChem, Co., Ltd., China. The interference RNA sequences for human Pdcd4 (shRNA-1: GGTTTGTAGAAGAATGTTT or shRNA-2: ACCATTACTGTAGACCAAA) or the mock sequence (TTCTCCGAAC GTGTCACGT) were connected to the hU6-MCS-Ubiquitin-EGFP-IRES-puromycin frame to construct the GV248 lentiviral vectors and package lentivirus.
UCMSCs were seeded in a six-well plate with a density of 2 × 105 cells/well. The next day, the culture medium was replaced by opti-MEM (51985034; Gibco) containing lentiviral particles with a multiplicity of infection of 40. The cells were then divided into control (Mock-MSCs) and lentiviral infected (shPDCD4-MSCs) or overexpression PDCD4 (oePDCD4-MSCs) groups and incubated at 37°C and 5% CO2. After 12 h of infection, the medium was replaced by fresh DMEM/F12. Stable PDCD4-knockdown or PDCD4-overexpression MSCs were obtained by screening with 2 μg/mL puromycin (P8230; Solarbio) after 3 days of lentiviral infection.
Western blot analysis
The total proteins were extracted from UCMSCs lysed by RIPA buffer. The protein (50 μg per well) was separated by electrophoresis in SDS-polyacrylamide gel and transferred onto the polyvinylidene fluoride membrane (ISEQ00010; Millipore, Billerica, MA). Subsequently, the membrane was blocked in 5% skimmed milk [dissolved in Tris-buffered saline containing 0.1% Tween-20 (TBST)] for 2 h at 20°C–30°C, and then incubated overnight with rabbit or mouse anti-human monoclonal antibodies against PDCD4 (9535S; CST, Danvers, MA), photo-GSK-3β (5558S; CST), GSK-3β (12456; CST), β-catenin (8480S; CST), and β-actin (4970S; CST) (all 1:1,000) at 4°C, followed by goat anti-rabbit or anti-mouse IgG H&L (HRP) (ab97051, ab97023; Abcam, Cambridge, MA) for 1 h at 20°C–30°C.
After washing the membranes with TBST, we added Chemiluminescent HRP substrate (WBKLS0500; Millipore) to visualize and analyze the bands using the AlphaView-FluorChem Q system (ProteinSimple, San Francisco, CA). The relative protein expression was measured with the band density of Western blots using Image-Pro Plus software (Version 6.0; Media Cybernetics, Silver Springs, MD).
Real-time polymerase chain reaction
To verify the efficiency of PDCD4 knockdown, total RNA was extracted from the cells with Trizol™ Reagent (15596-026; Invitrogen) and then reverse transcribed into cDNA by the SuperScript™ IV First-Strand Synthesis System (18080-051; Invitrogen). Furthermore, real-time polymerase chain reaction (PCR) was performed with amplified cDNA under the ChamQ Universal SYBR qPCR Master Mix (Q711; Vazyme Biotech Co., Ltd., Nanjing, China) and specific primer pairs; the sequences of the forward and reverse primers are shown in Table 1. The amplified products were electrophoresed using 2% agarose gel. The expression of the target genes was calculated using the 2−ΔΔCt method and reported as the fold relative to the control group, after normalizing to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase expression.
The Primers Used for the Real-Time Polymerase Chain Reaction
GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GSK-3β, glycogen synthase kinase-3β; OCN, osteocalcin; OPN, osteopontin; PDCD4, programmed cell death 4; Runx2, Runt-related transcription factor 2.
Flow cytometry analysis
To determine the phenotype of UCMSCs, the cells were dissociated with 0.25% EDTA-trypsin, prepared into single-cell suspension, and counted. Then, the cell suspensions were divided into four tubes, each containing 2 × 105 cells.
Five microliters PE, PerCP, and adenomatous polyposis coli (APC) anti-human isotype control IgG1 antibodies (400112, 400150, 400120; Biolegend, San Diego, CA) were added to the first tube, respectively; 5 μL PE anti-human CD105 antibody (323206; Biolegend) and 5 μL APC anti-human CD19 antibody (302212; Biolegend) were added to the second tube; 5 μL PerCP anti-human CD73 antibody (344014; Biolegend), 5 μL PE anti-human HLA-DR antibody (307606; Biolegend), and 5 μL APC anti-human CD45 antibody (304012; Biolegend) were added to the third tube; while 5 μL PerCP anti-human CD90 antibody (328118; Biolegend), 5 μL PE anti-human CD11b antibody (301306; Biolegend), and 5 μL APC anti-human CD34 antibody (343510; Biolegend) were added to the last tube.
The cells were then incubated at 4°C in the dark for 30 min. After washing with PBS, the cells were resuspended, detected by flow cytometry (FACSAria III; BD Biosciences, Franklin Lakes, NJ), and analyzed by FlowJo VX10 software.
UCMSC differentiation assay
The potential of UCMSCs for differentiation into osteoblasts, adipocytes, and chondrocytes were investigated using differentiation kits (05-440, 05-330/331/332, 05-220/221, BI, Kibbutz Beit Haemek, Israel) according to the manufacturer's instructions. Briefly, UCMSCs were diluted as single-cell suspensions, after which they were placed in 24-well plates (6 × 104 cells/well) with either osteogenic or adipogenic differentiation media for 14–21 days; the medium was replaced by fresh medium every 2–3 days with or without 100 μM PNU-74654 (HY-101130; MedChem Express, Monmouth Junction, NJ), an inhibitor of β-catenin. After that, osteoblasts differentiated from UCMSCs were stained by Alizarin Red S (C37C00150, BI), while adipocytes were stained by Oil Red-O (C37A00150, BI).
As for differentiation into chondrocytes, UCMSCs were seeded in 96-well U-bottom culture plates at 1 × 105 cells/well and then cultured in chondrogenic medium for 14–21 days; the medium was renewed every 3–4 days and chondrogenic differentiation was detected with Alcian Blue staining (C37B00150, BI). Finally, all plates were visually inspected and photographed under a microscopy, and the relative intensity was measured by Image-Pro Plus software.
Animal model
The male New Zealand white rabbits (3–6 months old, 2.9–3.4 kg weight) were used and housed in independent ventilation cages, with constant temperature (23°C) and humidity, and a 12-h light/12-h dark cycle. All animal experiments were approved by the Ethics committee of The Second Hospital, Cheeloo College of Medicine, Shandong University. The rabbits were randomly divided into three groups, with eight in each group. A circular bone defect with a diameter of 4 mm was prepared under the tibial nodule of rabbits by electric drill [17]. The hydrogel prepared by mixing Mock-UCMSCs or shPDCD4-UCMSCs and VitroGel™ 3D (TWG003; TheWell Bioscience, North Brunswick, NJ) was placed at the bone defect, while only hydrogel was placed in the negative group.
After 3 months, the bone defects were observed by IVIS Spectrum in vivo imaging system (Perkin-Elmer, Waltham, MA). The diameter of bone defect was measured by Image-Pro Plus software and the area was calculated by the following formula: area = π × (diameter/2)2. Bones with defects were separated, dissected along the sagittal plane, fixed by 4% polyformaldehyde, decalcified with EDTA decalcifying solution (E1171; Solarbio), and embedded in paraffin to prepare 8 μm paraffin sections.
Goldner trichrome staining
To stain the osteogenesis in the bone defects, the Goldner trichrome staining was performed on the bone sections using Goldner trichrome stain kit (G3550; Solarbio), according to the manufacturer's instructions. Following xylene deparaffination for 20 min, the paraffin sections were dehydrated with alcohol (100%, 95%, 90%, and 80%; each 5 min) and rinsed in water. After incubation in Weigert iron hematoxylin solution for 5 min, the sections were washed with running water for 1 min and differentiated in 1% acid alcohol solution for 3 s. Then the sections were stained in Ponceau acid fuchsin for 5 min, rinsed in 0.1% glacial acetic acid rapidly, and stained in 0.1% Orange G solution for 5 min until the collagen were decolorized, followed by washing in 0.1% glacial acetic acid for 10 s.
After that the sections were stained in light green solution for 5 min, differentiated thrice in 0.1% glacial acetic acid for 15 s, dehydrated in absolute ethyl alcohol, and mounted with neutral gum. Finally, the sections were observed under the microscope (Olympus, Tokyo, Japan).
Statistical analysis
The GraphPad Prism 6 software (La Jolla, CA) was used for statistical analyses. The comparison between groups was analyzed with unpaired Student's t-test and one-way analysis of variance. Data are represented by mean ± standard error of the mean (SEM); variations with a P value <0.05 were considered to have the statistically significant.
Results
PDCD4 is expressed in differently sourced MSCs
To verify the expression of PDCD4 in MSCs, we obtained MSCs from bone marrow, umbilical cord, and adipose tissue. The expression of PDCD4 in differently sourced MSCs was detected at mRNA and protein levels by real-time PCR and Western blotting, respectively. As shown in Fig. 1, PDCD4 expression was evident at both mRNA (Fig. 1A) and protein levels (Fig. 1B) in MSCs obtained from the bone marrow, umbilical cord, and adipose tissue, which indicated that PDCD4 expression in MSCs was not affected by the origin of these cells.
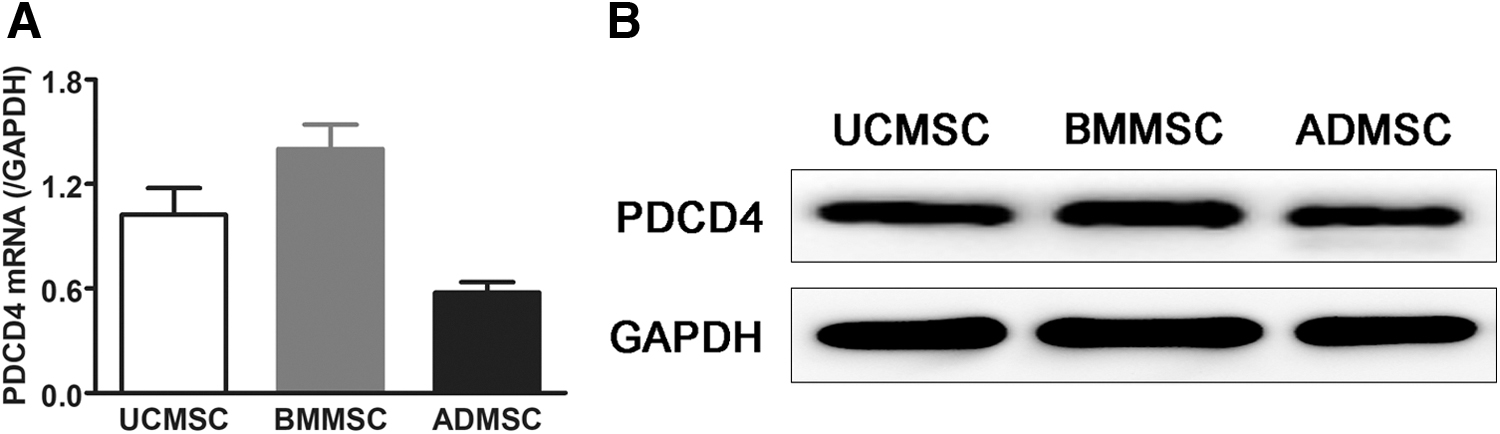
PDCD4 was expressed in MSCs from various sources. The total mRNA and proteins of UCMSCs, BMMSCs, and ADMSCs were extracted. The expression of PDCD4 was detected at the mRNA level
PDCD4 knockdown does not alter the phenotype of UCMSCs
To explore the role of PDCD4 in the biological function of MSCs, we infected UCMSCs with a shRNA lentiviral vector to knock down PDCD4 expression. After screening the positive cells with puromycin, we verified the knockdown efficiency by detecting PDCD4 expression using real-time PCR and Western blotting. As shown in Fig. 2, both shRNA-1 and shRNA-2 resulted in almost no PDCD4 expression, neither at the mRNA level (Fig. 2A) nor at the protein level (Fig. 2B), which indicated that both shRNAs had a high knockdown efficiency. shRNA-2 was applied for further experiments since it exhibited a slightly higher knockdown efficiency than shRNA-1.
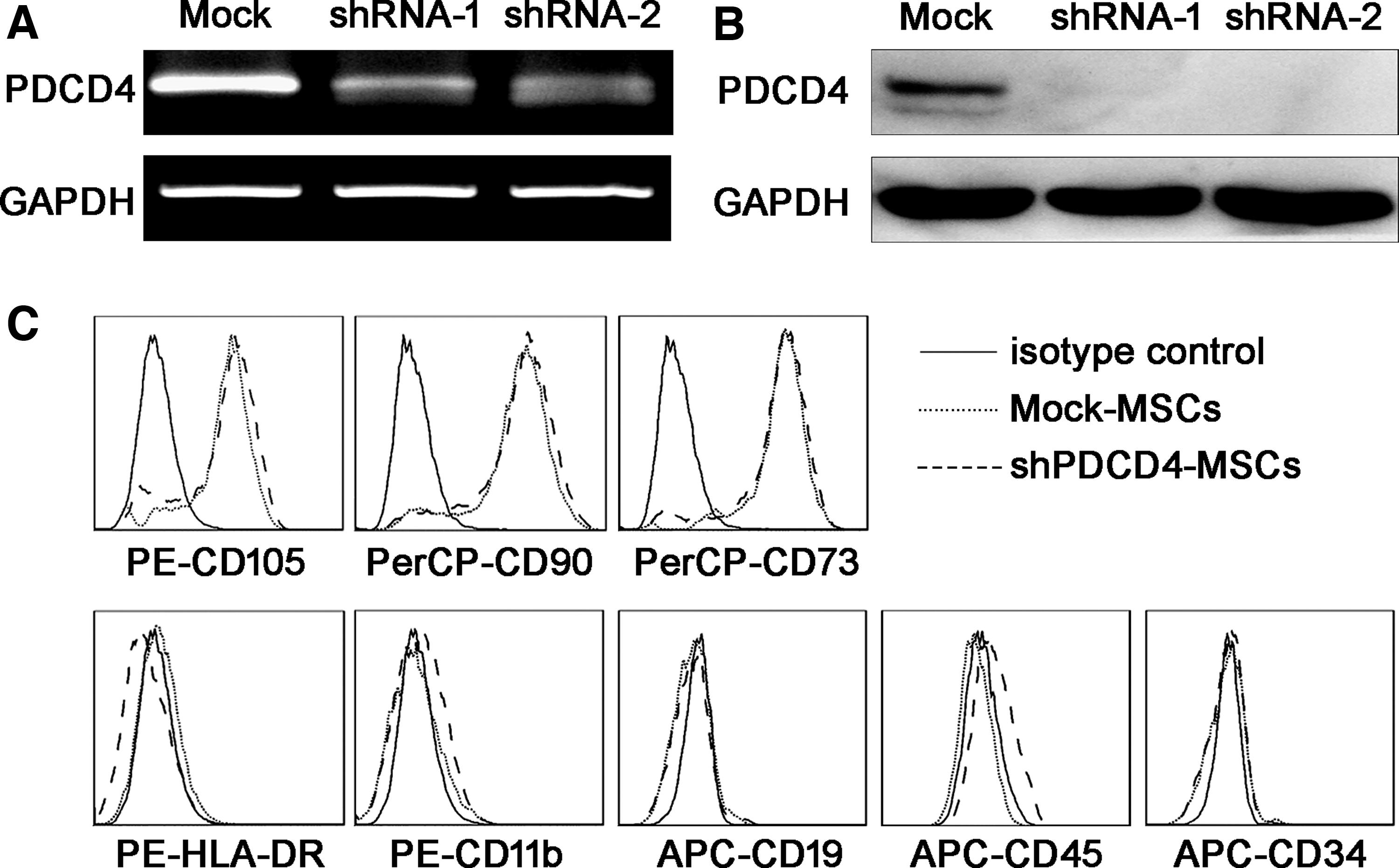
PDCD4 expression and surface marker profile were confirmed in UCMSCs after PDCD4 knockdown. Two different interference sequences (shRNA-1 and shRNA-2) along with a control sequence (Mock) were prepared into lentiviral vectors for PDCD4 knockdown. After screening by puromycin, PDCD4 expression at the mRNA and protein levels was detected by RT-PCR
After PDCD4 knockdown, we detected the expression of surface markers commonly used to identify UCMSCs by flow cytometry; there was no significant difference in the expression of neither positive (characteristic) MSCs markers (CD105, CD90, and CD73) nor negative markers (HLA-DR, CD11b, CD19, CD45, and CD34) (Fig. 2C) between control (Mock-MSCs) and shRNA-treated UCMSCs (shPDCD4-MSCs).
PDCD4 knockdown enhances osteogenic differentiation of UCMSCs
Furthermore, we investigated the effect of PDCD4 on the osteogenic, adipogenic, and chondrogenic differentiation potential of UCMSCs. As shown in Fig. 3A, shPDCD4-MSCs exhibited significantly enhanced osteogenesis than that in Mock-MSCs. However, there was no significant difference between the two groups in adipogenic and chondrogenic differentiation. On day 14 of osteogenic differentiation, we harvested the UCMSCs from the two groups and extracted their mRNAs; we then proceeded to evaluate the mRNA expression of molecules related to osteogenic differentiation by real-time PCR. Notably, Runt-related transcription factor 2 (Runx2), osteopontin (OPN) and osteocalcin (OCN) were significantly upregulated in shPDCD4-MSCs compared with those in Mock-MSCs during their osteogenic differentiation (Fig. 3B).

The osteogenic differentiation of UCMSCs was increased by PDCD4 knockdown.
PDCD4 knockdown promotes Wnt pathway activation during osteogenic differentiation of UCMSCs
The classical Wnt pathway is one of the most important pathways during MSC osteogenic differentiation. The phosphorylation of GSK-3β promotes the activation of β-catenin, which initiates the expression of osteogenic differentiation-related factors, such as Runx2, among others. Thus, we examined the expression of GSK-3β and β-catenin, at both mRNA and protein levels, in Mock-MSCs, shPDCD4-MSCs, and these two groups during osteogenic differentiation.
The results showed that GSK-3β expression was no significant difference in the four groups (Fig. 4A), whereas β-catenin mRNA expression was significantly increased in the knockdown group compared to the mock group (Fig. 4B) during osteogenic differentiation. As for the protein expression, Western blotting analysis revealed that both GSK-3β phosphorylation and β-catenin expression were higher in shPDCD4-MSCs than in Mock-MSCs during osteogenic differentiation (Fig. 4C).

PDCD4 knockdown promoted the activation of Wnt pathway during osteogenic differentiation of UCMSCs. Mock-MSCs and shPDCD4-MSCs were induced toward the osteogenic lineage using a differentiation kit according to the manufacturer's instructions. On day 14 of osteogenic differentiation, the two groups of UCMSCs were extracted total mRNA and protein, respectively. The expression of GSK-3β
The β-catenin inhibitor PNU-74654 reverses PDCD4 knockdown-induced osteogenesis of UCMSCs
To confirm that PDCD4 regulated UCMSC osteogenesis through the GSK-3β/β-catenin pathway, a β-catenin inhibitor, PNU-74654, was used during osteogenic differentiation. Our results showed that the osteogenesis of siPDCD4-MSCs was significantly higher than in Mock-MSCs; however, the addition of PNU-74654 effectively reversed the increase in osteogenesis induced by PDCD4 knockdown (Fig. 5A). Moreover, 100 μM PNU-74654 could inhibit the expression of β-catenin, and the increased expressions of osteogenesis-related molecules Runx2, OPN, and OCN by PDCD4 knockdown were also reversed by PNU-74654 (Fig. 5B).
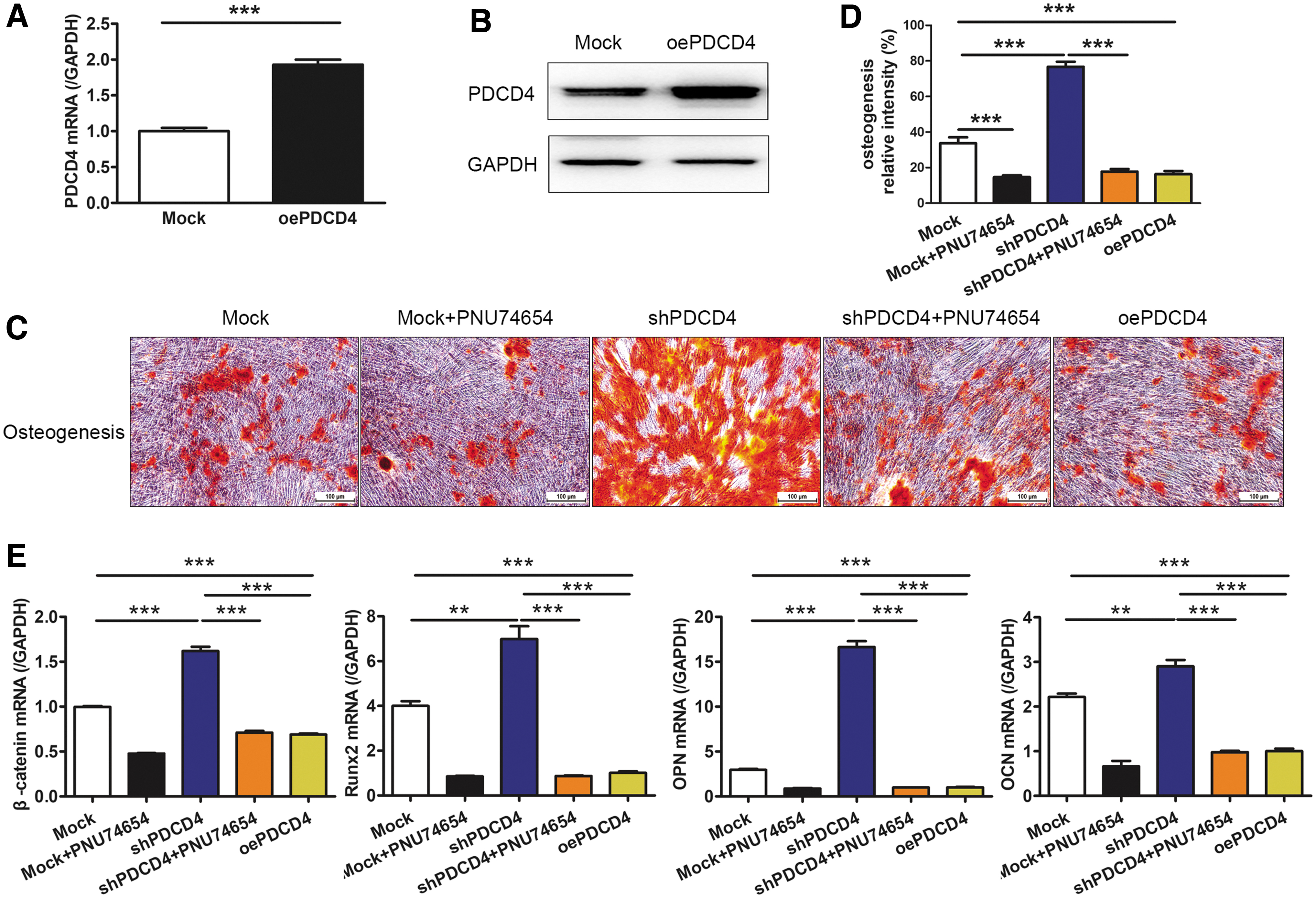
The β-catenin inhibitor PNU-74654 and oePDCD4 reduced the PDCD4 knockdown-induced osteogenesis in UCMSCs. A PDCD4 sequence and a control sequence (Mock) were prepared into lentiviral vectors for PDCD4 overexpression. After screened by puromycin, PDCD4 expression at the mRNA and protein levels was detected by RT-PCR
UCMSCs with PDCD4 knockdown promote bone defect reconstruction in vivo
To demonstrate that PDCD4-knockdown UCMSCs have a stronger ability for bone defect repair in vivo, a rabbit bone defect model was established and treated with Mock-UCMSCs and shPDCD4-MSCs. After 3 months, microcomputed tomography (CT) imaging of small animals showed that, compared with the negative group, both Mock and shPDCD4 groups had promoted repairing the bone defect, in which shPDCD4-UCMSCs had more significant effect on reconstruction and the area of bone defect decreased clearly (Fig. 6A, white arrowhead showing defect site).
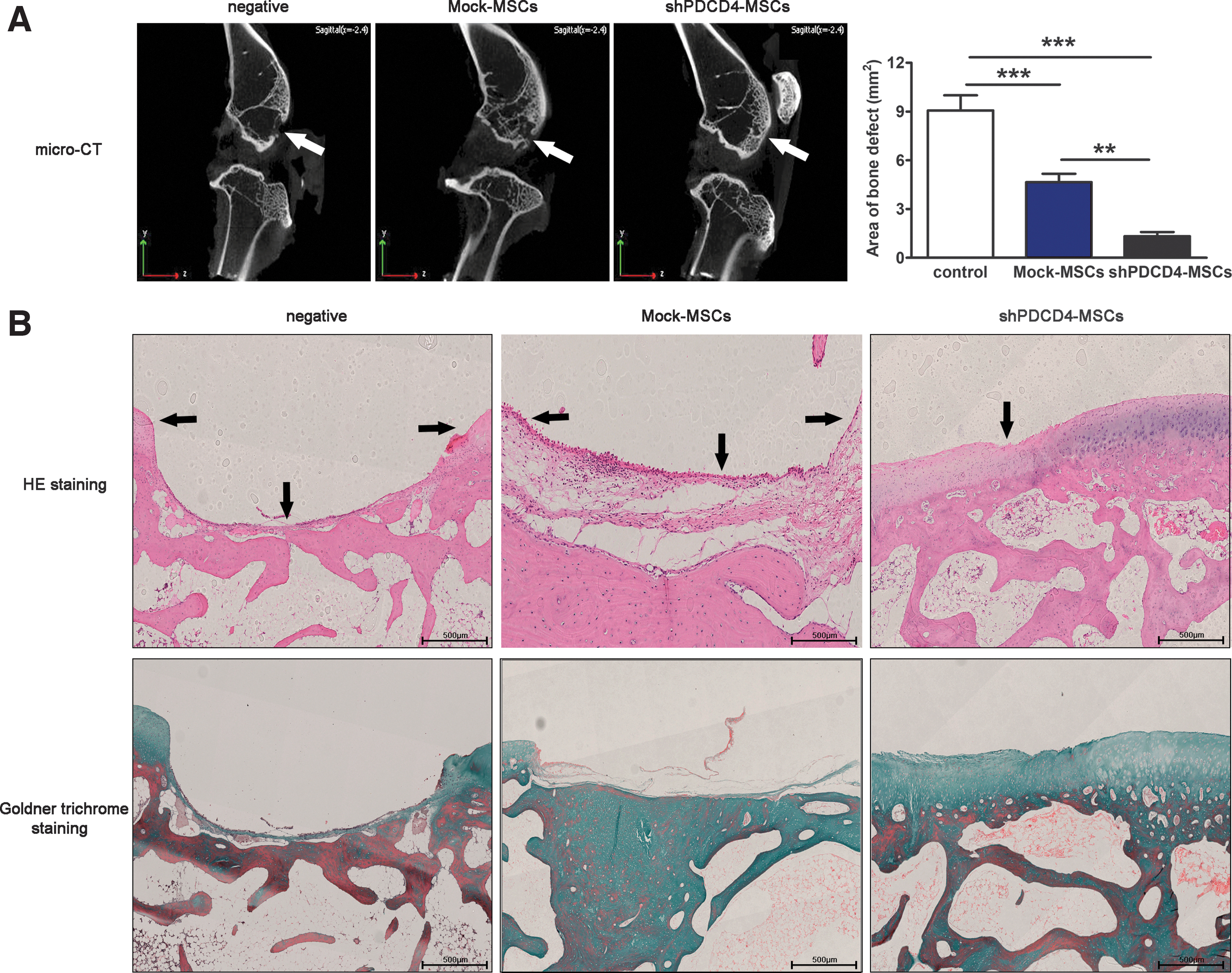
The deficiency of PDCD4 in UCMSCs can promote the repair of bone injury in vivo. A circular bone defect with a diameter of 4 mm was prepared under the tibial nodule of rabbits. The complex prepared by mixing Mock-UCMSCs or shPDCD4-UCMSCs and hydrogel was placed at the bone defect, while only hydrogel was placed in the negative group.
Paraffin sections of bone with defect were prepared and the bone composition was shown by hematoxylin and eosin and Goldner trichrome staining. The results showed that the defect area showed a trend consistent with Micro-CT results, and blue mineralized bone and red nonmineralized bone emerged from the defect site, and the defect area showed a trend consistent with Micro-CT results (Fig. 6B, black arrows indicate the bottom and highest point of the defect).
Discussion
PDCD4 is a well-known apoptosis-related gene that binds with eukaryotic cell translation initiation factor 4α (eIF4α) to inhibit translation. It was later found to be downregulated, or even absent, in various human malignant tumor tissues, indicating its tumor-suppressive role [18]. Hilliard et al. found that Pdcd4 −/− mice resisted experimental autoimmune encephalomyelitis and type I diabetes, and indicated for the first time that PDCD4 might be involved in the regulation of inflammation and immune response [10]; our group also reported that PDCD4 was involved in the regulation of multiple inflammatory diseases [11,12,19]. Recent studies have noted downregulated PDCD4 expression in adipose tissue-derived MSCs during adipogenesis [20].
PDCD4 exhibits a cardioprotective role and might also regulate the secretion of MSC-derived exosomes [21]. Furthermore, as a direct target of miR-21, PDCD4 was previously demonstrated to regulate the differentiation and function of osteoclasts [22]. And miR-21 could also modulate the adipogenesis and osteogenesis of MSCs through ERK-MAPK pathway, by targeting Sprouty2 (SPRY2), but not PDCD4 [23]. The above data suggest that PDCD4 may play roles in regulating the biological function of MSCs, but the role of PDCD4 in osteogenic differentiation and bone defect repair of MSCs is still unclear.
In this study, we found that MSCs from various human tissue sources expressed PDCD4. Because UCMSCs are simply acquired and innocuous to donors compared with bone marrow and ADMSCs (BMMSCs and ADMSCs), we mainly use UCMSCs in our experiments. We found that PDCD4 knockdown did not affect the expression of surface molecules in UCMSCs and, thus, did not induce any direct phenotypic change in these cells. However, osteogenic differentiation of shPDCD4-MSCs was significantly increased compared to the control group, which highlighted a novel role of PDCD4 in the regulation of osteogenic differentiation of MSCs. Thus, we further investigated the mechanism underpinning this phenomenon in vitro.
MSCs have the potential of self-renewal, proliferation, and multidirectional differentiation. They can differentiate into a variety of adult cells, such as osteoblasts, chondrocytes, hepatocytes, and cardiomyocytes, under different induction conditions; thus, it is no surprise that they are considered ideal seed cells for repairing damaged tissues. During bone injury, the stress response leads to the release of different inflammatory factors in vivo, which tend to accumulate in the injured location. In turn, this high concentration of cytokines and chemokines [eg, macrophage inflammatory protein-1α (MIP-1α), monocyte chemotactic protein 1 (MCP-1), C-X-C Motif Chemokine 7 (CXCL7), etc.] in the injured region effectively recruits MSCs, where MSCs undergo osteogenesis and secrete cytokines to repair the bone injury [24].
There are numerous transcription factors involved in osteogenesis, among which, Runx2, a member of the RUNX family of transcription factors, is the major regulator of osteoblast differentiation and bone formation, and whose expression is induced early in the osteogenic differentiation process, particularly within the first week [25]. Runx2 promotes the expression of critical genes for osteogenic differentiation, such as OCN and OPN; OCN, secreted only by osteoblasts, is the most abundant noncollagenous protein, accounting for 1%–2% of matrix proteins and regulates the rate and extent of bone formation [26], whereas OPN, another important noncollagenous protein in the bone matrix, participates in bone remodeling; it contains a special cell binding motif through which it can connect cell adhesion molecules to the extracellular matrix in mineralized bone [27]. OCN and OPN are markers of late-stage osteogenic differentiation and are exclusively expressed in mature osteoblasts [27]. Expression of these genes hallmarks MSC differentiation into osteoblasts.
Receptor Activator of Nuclear Factor-κB (RANK) is expressed in osteoclast precursors or progenitor cells and osteoclast membranes. Osteoprotegerin (OPG) is a soluble decay receptor of RNAK ligand (RANKL), and mainly expressed on the membrane of osteoblasts. OPG can promote osteogenesis by blocking the interaction between RANK and RANKL. The relative level of RANKL/OPG is a key factor to determine the balance between osteogenesis and osteoclasts. Research has shown that inhibition of miR-21 expression in MSCs could downregulate the ratio of RANKL/OPG and reduce the activity and proliferation of osteoclasts, which were beneficial to the repair of bone destruction [28]. The proportion of RANKL/OPG was increased by targeting PDCD4 in miR-21-deleted mice, which increased the bone destruction [22].
Therefore, we speculate that PDCD4 has the potential function of regulating MSC osteogenic differentiation. In this study, the expression of osteogenic marker molecules, such as Runx2, OPN, and OCN, was significantly increased as a result of PDCD4 knockdown during osteogenic differentiation. However, the expression of OPG and RANKL had no significant differences (data not shown). Overall, our results confirmed the regulatory role of PDCD4 in the osteogenic differentiation of MSCs. Next, the mechanism of PDCD4 regulating the expression of osteogenic molecules will be further investigated.
The Wnt pathway is an important signaling pathway that plays a crucial role in bone mass regulation by promoting the osteogenic differentiation and proliferation of MSCs. When this pathway is not initiated, a degradation complex is formed by scaffold proteins, such as APC protein, GSK-3β, and β-catenin, resulting in the degradation of β-catenin. However, the binding of Wnt ligands to their receptors activates the signaling cascade; GSK-3β is phosphorylated and is, thus, liberated from the degradation complex, resulting in the inactivation of the complex. In turn, β-catenin is activated and translocated to the nucleus and initiates the transcription of downstream target genes, such as Runx2, thereby promoting the osteogenic differentiation of MSCs [28]. Hao et al. indicated that the activation of Wnt/β-catenin pathway mediated by miR-21/Pdcd4 axis may aggravate hypoxia injury in cardiomyocyte cell line H9c2 cells [29]. The data suggested that PDCD4 could induce the activation of Wnt pathway, but its effect on the activation of Wnt pathway during osteogenic differentiation of MSCs is unclear.
In this study, we found that phosphorylated GSK-3β and β-catenin were upregulated in shPDCD4-UCMSCs during osteogenic differentiation; notably, the levels of these components were normalized after β-catenin inhibition by PNU-74654 or oePDCD4. Therefore we thought that PDCD4 knockdown might activate the phosphorylation of GSK-3β by certain pathways, following the dissociation from β-catenin complex, and increase the expression of β-catenin, to promote the osteogenic differentiation of UCMSCs. On the other hand, endogenous PDCD4 may inhibit the transcription of β-catenin mRNA by inhibiting transcription factors. Our results were contrary to the activation of Wnt pathways by PDCD4 in H9c2 cells, which might be due to the different role of PDCD4 in different models and cells through different mechanisms.
Recently, a number of studies have confirmed the safety and efficacy of human UCMSCs in nonautologous transplantation of rabbit models, in which there was no evidence of a tissue or cell rejection reaction in any specimen [30]. Our animal experiments had also proven that UCMSCs could promote the repair of bone injury in vivo. The mechanism may be to promote autologous MSC osteogenic differentiation or angiogenesis by paracrine-related cytokines and vesicles [31]. However, the detailed mechanisms required further studies, which represented a limitation of this research.
To sum up, in this study, we revealed a novel role of PDCD4 in the regulation of osteoblastic differentiation of MSCs through GSK-3β/β-catenin pathway. These data suggest that targeting of PDCD4 or its related signaling pathways may significantly improve MSC treatment efficacy against bone injury or bone destruction diseases. Undoubtedly, further studies are warranted to address such notions.
Footnotes
Acknowledgments
We would like to thank Editage for English language editing.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Natural Science Foundation of China (No. 81600176), the Natural Science Foundation of Shandong Province (No. ZR2016HB71), the Science and Technology innovation project of Shandong Province (No. 2017GSF18136 and No. 2018GSF118034), and Rongxiang Regenerative Medicine Foundation of Shandong University (2019SDRX-05).