
Abstract
Mucin 1 (MUC1) is a transmembrane glycoprotein overexpressed in several cancer cells in which it regulates cell surface properties, tumor invasion, and cell death. Recently, we reported that MUC1-C, the C-terminal subunit of MUC1, is involved in the growth of mouse embryonic stem (ES) cells. However, the functional significance of MUC1-C in human ES cells remains unclear. In this study, we investigated the expression and function of MUC1-C in human ES cells. Based on reverse transcription-polymerase chain reaction, western blotting, and confocal microscopy following immunostaining, undifferentiated human ES cells expressed MUC1-C and the expression level decreased during differentiation. Inhibition of MUC1-C, by the peptide inhibitor GO201 that targets the cytoplasmic domain of MUC1-C (MUC1-CD), reduced cell proliferation and OCT4 protein expression, and promoted cell death. Moreover, the inhibition of MUC1-C increased the intracellular reactive oxygen species (ROS) levels and downregulated expression of glycolysis-related enzymes. These findings indicate that expression and function of MUC1-C are required for stem cell properties involved in cell proliferation, maintenance of pluripotency and optimal ROS levels, and a high glycolytic flux in human ES cells. In addition, forced overexpression of MUC1-CD increased the efficiency of reprogramming from fibroblast cells to induced pluripotent stem cells, suggesting that MUC1-C expression can contribute to the reprogramming process.
Introduction
Mucin 1 (MUC1) is a membrane-associated glycosylated protein that regulates adhesion, migration, and cell signaling in various cells [1]. The heavily glycosylated N-terminal subunit of MUC1 (MUC1-N) includes sea urchin sperm protein enterokinase and agrin (SEA) domain. As the SEA domain contains an autoproteolytic cleavage site, MUC1 is translated as a single protein, but is rapidly converted to MUC1-N and the C-terminal subunit of MUC1 (MUC1-C) [2]. MUC1-C is composed of a highly glycosylated extracellular domain, a transmembrane domain, and a multifunctional cytoplasmic domain [3]. Under normal conditions, two MUC1 subunits are associated as a heterodimeric complex through hydrogen bonds on the plasma membrane. However, the complex dissociates in response to stimulation with inflammatory cytokines through additional proteolytic cleavage events, and therefore, MUC1-N can be released from the cell surface [4].
Cytoplasmic domain of MUC1-C (MUC1-CD) contains a CQC motif, a highly conserved CQCRRK sequence, essential for MUC1-C homodimer formation. MUC1-C homodimerization is essential for nuclear localization and interaction with other factors [5]. Previous studies have shown that MUC1-C participates in intracellular signaling pathways and contributes to growth and survival of cancer cells [6 –9]. In addition, it binds to various transcription factors and regulates the expression of their target genes, including pluripotency factors in a number of cancer stem cells [10 –12]. MUC1-C also contributes to the signaling and stability of factors involved in intracellular reactive oxygen species (ROS) regulation, and regulates the expression of glycolysis-related genes in cancer cells [13].
Embryonic stem (ES) cells are established from the inner cell mass of blastocysts. ES cells can self-renew, maintaining an undifferentiated state when cultured in vitro, and generate three germ layers of the embryo [14]. Due to these characteristics, ES cells are considered suitable cell sources for cell therapy. In terms of metabolism, ES cells utilize the glycolysis process as a major source of adenosine triphosphate production, as in cancer cells [15]. Recently, studies on induced pluripotent stem (iPS) cells and reprogramming technology for disease modeling and drug development have been recognized as being among the most important approaches in regenerative medicine [16,17]. However, the detailed mechanisms of the reprogramming process are still being elucidated and problems still exist, such as low efficiency, genomic insertion, and tumorigenicity [18]. Therefore, it is necessary to develop useful methods that can improve the efficiency of iPS cell production and maintain the stability of iPS cells for safe iPS cell-based cell therapy. Accordingly, numerous studies are being conducted to overcome these problems.
We recently reported that MUC1-C contributes to the growth and redox balance in mouse ES cells [19]. However, the detailed function of MUC1-C in human embryonic stem (hES) cells remains unclear. This study was therefore carried out to confirm the function of MUC1-C in hES cells by investigating MUC1 expression and the effect of MUC1-C inhibition, and by analyzing the effect of MUC1-CD overexpression on reprogramming.
Materials and Methods
Human ES cell culture
The hES cell line H9 (WiCell Research Institute, Madison, WI) was maintained on mouse embryonic fibroblast (MEF) cells in ES medium, including Dulbecco's modified Eagle's medium (DMEM)/F12 and 20% knockout serum replacement (Gibco/Life Technologies, Grand Island, NY), as described previously [20]. The hES cells were cultured in a feeder-free condition using plates coated with Matrigel (Corning, NY) and mTeSR™ medium (Stem Cell Technologies, Vancouver, BC, Canada) for experiments. The hES cells were dissociated into single cells using StemPro™ Accutase™ Cell Dissociation Reagent (Accutase; Gibco/Life Technologies), plated on a Matrigel-coated dish in mTeSR containing 10 μM Y-27632 (Calbiochem, San Diego, CA), and cultured for 2 days before specific experiments. To stimulate the differentiation of hES cells, cells in the MEF medium were treated with 10 μM retinoic acid (RA; Sigma-Aldrich, St. Louis, MO) for 14 days. The medium was changed daily. The study on hES cells was approved by the Internal Review Board of Chungbuk National University (no. 2020-0099).
Feeder cell preparation
When the MEF cells were grown to 80%–90% confluency, 10 μg/mL mitomycin C (Sigma-Aldrich) was added into culture plates for inactivation of the MEF cells, and they were incubated for 2 h. The culture medium was removed and MEF cells were washed with PBS, treated with TrypLE™ Express (Gibco/Life Technologies), and collected for immediate use or frozen storage. A day before use, MEF cells (7–8 × 105/plate) were plated into gelatin-coated 100 mm plates.
Treatment with MUC1-C inhibitor peptide
The cells were treated with control peptide CP1 (D-RRRRRRRRR-AQARRKNYGQLDIFP) or MUC1-C inhibitor GO201 (D-RRRRRRRRR-CQCRRKNYGQLDIFP) at the indicated concentrations for 24 h. CP1 and GO201 peptides were synthesized by AnyGen Co. (Gwangju, Republic of Korea) as described previously [21].
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated and reverse transcribed as described previously [22]. Quantitative PCR reaction was performed using a Rotor-Gene SYBR® Green PCR Kit (Qiagen, Hilden, Germany). The PCR reaction was performed for 10 min at 95°C for 1 cycle and 15 s at 95°C, 20 s at 58°C–62°C, and 15 s at 72°C for 40 cycles. The primer sequences used in the experiment are listed in Table 1. A standard PCR reaction was performed using 1 μL of the cDNA solution according to the following procedure: 20–30 cycles of denaturation for 60 s at 95°C, annealing for 60 s at 58°C, and elongation for 60 s at 72°C, as described previously [19,23]. The primer sequences used herein are also listed in Table 1.
List of Primer Sequences for Quantitative PCR and Standard PCR
MUC1, mucin 1; PCR, polymerase chain reaction.
Western blotting
Whole cell lysates were prepared and analyzed as previously described [20]. The antibodies to lactate dehydrogenase A (LDHA, catalog no. 3582), phosphoinositide-dependent kinase-1 (PDK1, catalog no. 3062), pyruvate kinase isozyme M2 (PKM2, catalog no. 4053), and β-ACTIN (catalog no. 3700) were purchased from Cell Signaling Technology (Danvers, MA). Antibody to octamer-binding transcription factor 4 (OCT4, catalog no. 611203) was obtained from BD Biosciences (San Jose, CA). The antibodies to NANOG (catalog no. 33759) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, catalog no. 32233) were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). The antibody to MUC1 (catalog no. ab109185) was from Abcam (Cambridge, MA). The density of each band was measured by software ImageJ and normalized to GAPDH or β-ACTIN.
Cell proliferation assay
The cells were placed onto a 96-well plate, incubated for 48 h, and then treated with CP1 or GO201 at the indicated concentrations for 24 h. Cell proliferation was measured using a Cell Proliferation ELISA, BrdU kit (Roche Diagnostics, GmbH, Mannheim, Germany), according to the manufacturer's instructions. BrdU incorporation was quantified based on the absorbance at 450 nm using a microplate spectrophotometer (BioTeK, Winooski, VT).
Measurement of ROS, apoptosis, and lactate production
The cells were treated with CP1 or GO201 at the indicated concentrations for 24 h. To measure intracellular ROS levels, cells were incubated with 10 μM 2′,7′-dichlorofluorescein diacetate (DCF-DA) (Molecular Probes, Eugene, OR) for 30 min at 37°C. The fluorescence images were obtained by fluorescence microscopy (Nikon, Tokyo, Japan), and the dissociated cells were analyzed by flow cytometry using FACSCalibur (BD Biosciences). For assessment of apoptosis, the cells were analyzed using an Annexin-V-FLUOS Staining Kit (Roche Diagnostics) and a FACSCalibur flow cytometer. For measurement of lactate, we used a lactate colorimetric assay kit (BioVision, Inc., Milpitas, CA) following the manufacturer's protocol. The absorbance at a wavelength of 450 nm was measured using a microplate spectrophotometer (BioTek).
Cell culture and establishment of stable cell lines expressing MUC1-C
Human neonatal fibroblast (BJ) cells were obtained from American Type Culture Collection (ATCC, Manassas, VA) and cultured according to the provided instructions. GP2-293 cells, a packaging cell line for the retrovirus preparation, were obtained from Clontech Laboratories, Inc. (Palo Alto, CA), and maintained in 10% fetal bovine serum containing DMEM. Human MUC1-CD was cloned into the retrovirus vector derived from murine stem cell virus (pMSCV). Retroviral vectors pMSCV and pMSCV-MUC1-CD along with pVSV-G were transfected into GP2-293 cells using Fugene HD (Promega, Madison, WI). Retroviruses were prepared as previously described [24]. The retroviral supernatant was applied to BJ cells with 8 μg/mL of polybrene (Sigma-Aldrich). To establish stable cell lines, the cells were selected in a medium containing 1 mg/mL G418 (Sigma-Aldrich) for 14 days.
Reprogramming of fibroblast cells
iPS cells were generated using a Cytotune 2.0 Sendai Reprogramming Kit (Invitrogen) following the manufacturer's protocol. Briefly, 2 days before transduction, control-BJ cells and MUC1-CD-BJ cells were plated in a six-well plate (1 × 105 cells/well). After 2 days, 70%–80% confluent BJ cells were transduced with Sendai viruses for 24 h. Seven days post-transduction, cells were harvested to prepare single cell suspensions and re-plated at a density of 1 × 105 cells/well onto MEF feeder cells.
Confocal microscopy
The cells cultured on glass cover slips in four-well plates were fixed with 4% paraformaldehyde and stained with anti-MUC1 (ab109185; Abcam), anti-OCT4 (611203; BD Biosciences), anti- TRA-1-60 (MAB4360), and anti-TRA-1-81 (MAB4381; Millipore, Billerica, MA) at 4°C overnight. The immunostained signal was detected with fluorescein isothiocyanate-conjugated anti-mouse IgG/IgM (BD Biosciences) or Alexa Flour 488-conjugated anti-rabbit or mouse secondary antibody (Invitrogen). The nuclei were stained with Hoechst 33258. The cell images were obtained with a confocal laser scanning microscope, LSM-880 (Carl Zeiss, Oberkochen, Germany).
Statistics
The results are presented as the mean ± standard deviation (SD) of data derived from at least three independent experiments. Statistical significance of differences between two groups was evaluated using a student's t-test. P < 0.05 was considered statistically significant.
Results
The level of MUC1-C decreased during early differentiation of hES cells
To investigate the expression of MUC1 in undifferentiated hES cells and differentiated cells, we determined the levels of MUC1 gene expression after RA treatment. Considering that the expression levels of OCT4 were drastically reduced (Fig. 1A), it was confirmed that RA induced differentiation of hES cells. The expression level of MUC1 mRNA significantly decreased at the initial stage of differentiation (Fig. 1A). Western blot analysis using the antibody detecting MUC1-C also showed that the protein level of MUC1-C declined significantly during the early differentiation (Fig. 1B). The expression levels of MUC1 mRNA and protein drastically decreased on the first day of differentiation and then slightly increased, but never recovered until 7 days of differentiation. The immunofluorescence staining analysis showed that MUC1-C was highly expressed in the undifferentiated cells compared to the RA-treated differentiated cells (Fig. 1C). These results suggest that MUC1-C expression is decreased at the early stage of differentiation and MUC1-C is involved in the regulation of hES cells.

Expression of MUC1 in hES cells. The hES cells were treated with 10 μM RA for the indicated period.
Inhibition of MUC1-C reduces cell proliferation, promotes cell death, and suppresses expression of pluripotency markers in hES cells
MUC1-C is involved in the formation of the MUC1 homodimer through the CQC motif, and MUC1-C homodimerization directs nuclear localization and interaction with various effectors [21]. GO201 is a cell-penetrating peptide inhibitor that contains the CQC motif directly binding to the MUC1-C at the CQC motif [21]. To investigate the function of MUC1-C in hES cells, we examined the change in properties of hES cells after treatment with GO201 and CP1, where CQC was replaced with AQA. In hES cells, the size and shape of the colony provide information about the pluripotent states. Undifferentiated hES cells show typically round, flat, and compact colonies with well-defined edges [25]. We observed that the GO201-treated cells had a smaller colony size, and the increased concentration of GO201 resulted in irregular colony edges and increased cell death compared to the CP1-treated cells (Fig. 2A). Next, we performed a BrdU proliferation assay. As shown in Fig. 2B, the BrdU incorporation was significantly decreased in GO201-treated cells compared to CP1-treated cells. Moreover, the number of alkaline phosphatase-positive colonies significantly decreased after treatment with GO201 compared to CP1 treatment (Fig. 2C). The annexin V/PI staining showed that GO201 treatment increased apoptosis of hES cells in a concentration-dependent manner (Fig. 2D). Considering that CP1 also increased apoptosis of hES cells at a concentration of 10 or 20 μM, CP1 appears to have nonspecific toxic effects at a high concentration. We then investigated the expression levels of OCT4 and NANOG, which are representative pluripotent markers in ES cells, and observed that GO 201 significantly decreased OCT4 expression at the protein level (Fig. 2E). These results reveal that MUC1-C inhibition results in overall changes of hES cells in terms of growth, survival, and OCT4 expression, leading to a loss of ES cell properties.
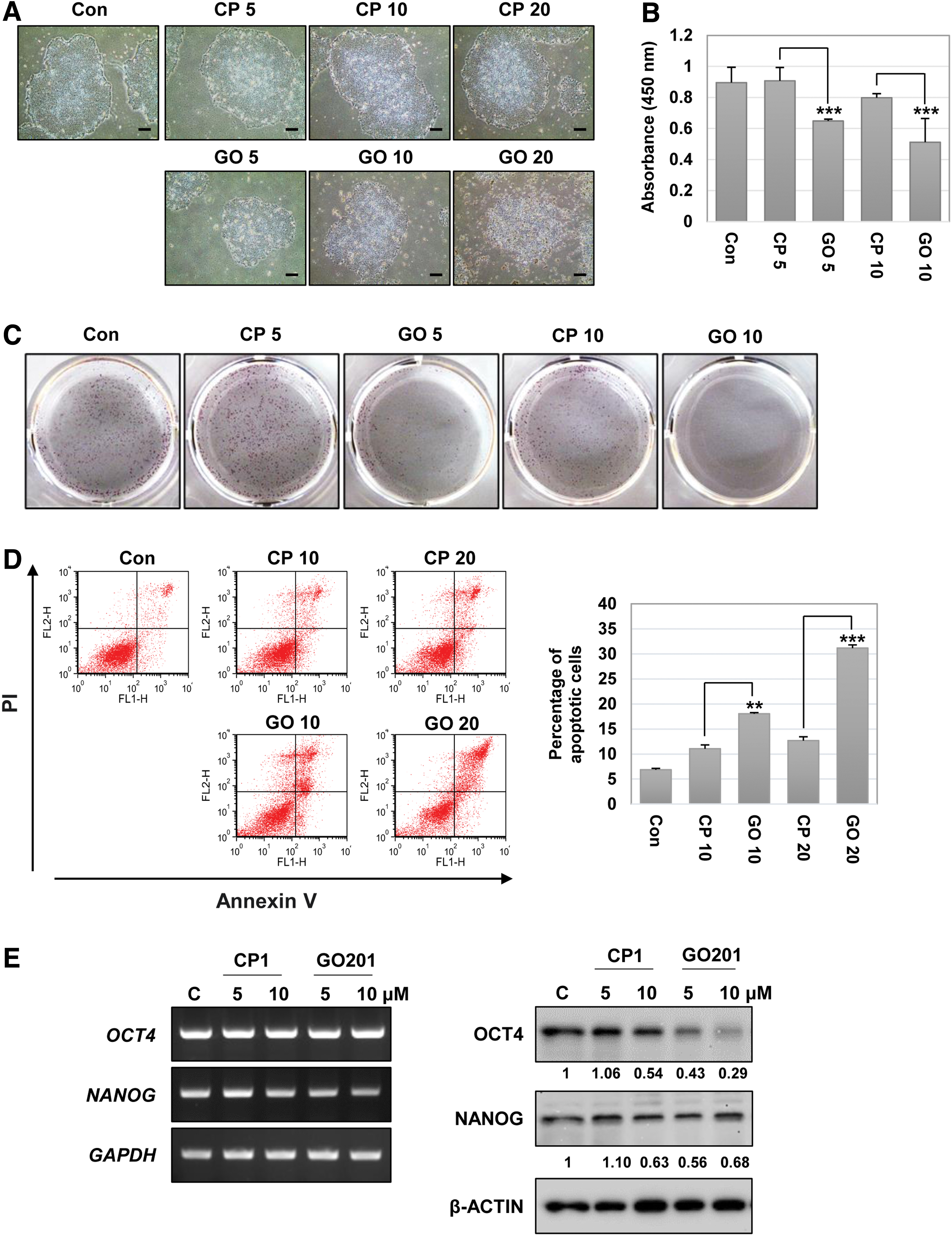
Effect of MUC1-C inhibition in hES cells. The hES cells were incubated with 5–20 μM CP1 or GO201 for 24 h.
MUC1-C is involved in the regulation of ROS levels and glycolysis in hES cells
To evaluate the effect of MUC1-C inhibition on redox balance, we performed a DCF-DA assay in GO201-treated or CP1-treated hES cells. As shown in Fig. 3A, the fluorescent intensity was substantially increased in GO201-treated cells in a concentration-dependent manner compared to the CP1-treated cells. Similar results were obtained by further measurement using fluorescence-activated cell sorting analysis (Fig. 3B). We then investigated the expression of various glycolysis-related genes at the mRNA levels and observed that the expression of LDHA and PDK1 was significantly downregulated by MUC1-C inhibition (Fig. 3C). The results at the protein level showed that the expression of PDK1 and PKM2 was clearly reduced by GO201 treatment compared to the control group (Fig. 3D). Expression of LDHA was slightly reduced by GO201 at the concentration of 5 μM, although no difference was found between GO201 and CP1 at the concentration of 10 μM. In addition, the amounts of lactate produced in GO201-treated cells were significantly reduced compared to CP1-treated cells (Fig. 3E). These results suggest that normal MUC1-C function is associated with the expression of glycolytic enzymes and contributes to the maintenance of stable glycolysis in ES cells.
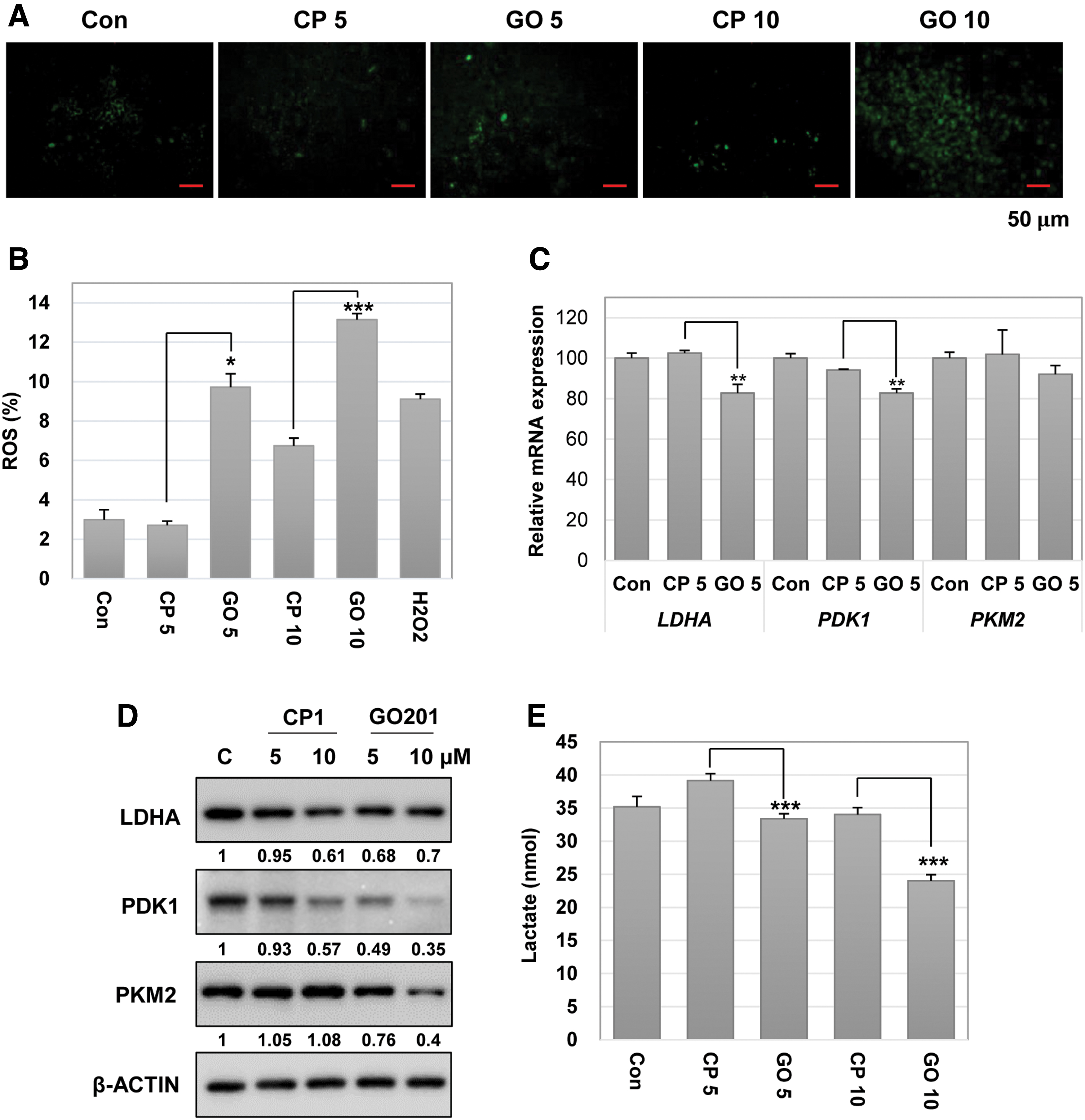
Inhibition of MUC1-C leads to an increase in ROS levels and a decrease in the glycolytic enzyme expression in hES cells. The hES cells were incubated with CP1 or GO201 at 5 or 10 μM for 24 h.
MUC1-CD promotes reprogramming of human fibroblast cells to iPS cells
As inhibition of MUC1-C with GO201 reduced stem cell properties in hES cells, we then employed MUC1-CD to increase MUC1-C signaling and investigated a potential role of MUC1-C signaling in reprogramming. First, we generated two stable cell lines, control-BJ cells harboring a control empty vector and MUC1-CD-BJ cells expressing exogenous MUC1-CD (Supplementary Fig. S1A, B). MUC1-CD-BJ cells showed a higher growth rate compared to the control-BJ cells harboring the empty vector (Supplementary Fig. S1C). The control-BJ cells and MUC1-CD-BJ cells were then reprogrammed by the Sendai virus reprogramming system (Fig. 4A). Recombinant Sendai virus vectors contain the four Yamanaka factors OCT4, SOX2, KLF4, and c-MYC, which do not integrate into the host genome and transiently replicate in the cytoplasm to make iPS cells without transgenes [26]. Twenty days after transduction, we found that larger, rounder, and denser colonies were generated in the MUC1-CD group compared to the control group (Fig. 4B). We further performed an alkaline phosphatase assay and quantified reprogramming efficiency using the iPS colonies obtained at 28 days post-transduction (Fig. 4C, D). The MUC1-CD-BJ cells had about 1.7-fold higher reprogramming efficiency compared with the control-BJ cells. We screened alkaline phosphatase-positive colonies, and individual colonies were mechanically isolated into 12-well plates for colony expansion. To confirm that the obtained iPS cells have normal properties of iPS cells, we then measured the expression of the pluripotency markers OCT4, TRA-1-60, and TRA-1-81 by use of immunofluorescence staining. iPS cells generated from MUC1-CD-BJ cells abundantly expressed pluripotent factors such as OCT4, TRA-1-60, and TRA-1-81, similar to hES cells and general iPS cells even after five passages (Fig. 4E). These results suggest that the enforced expression of MUC1-CD can promote reprogramming of human fibroblast cells to iPS cells.
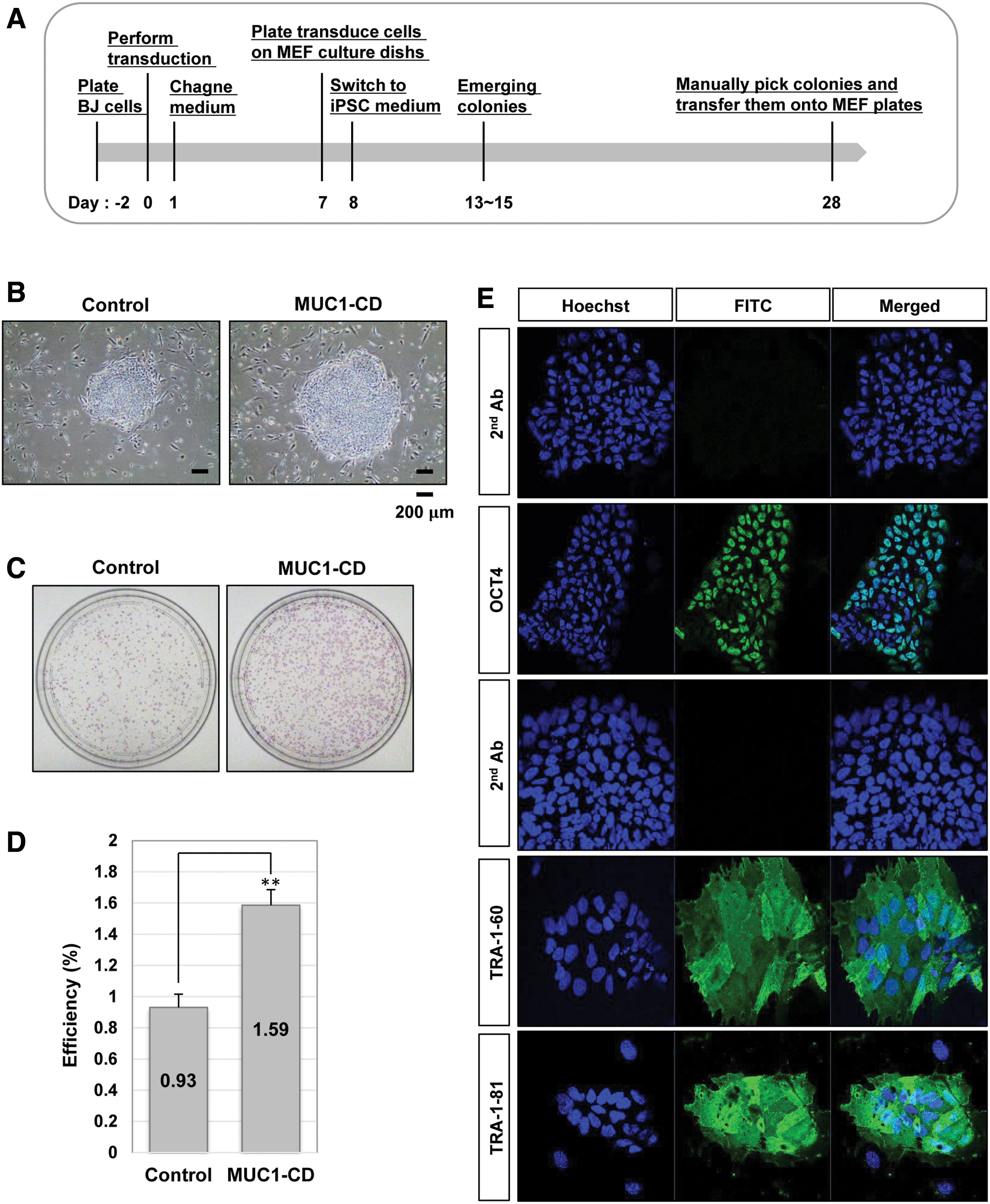
Overexpression of MUC1-C increases the efficiency of iPS cell generation.
Discussion
MUC1-C is overexpressed in diverse carcinomas and regulates the expression of pluripotency factors [12,27]. Given that pluripotent cells have properties common with cancer cells, it is worthwhile to investigate the role of MUC1-C in ES cells. In this study, we found that expression and normal function of MUC1-C are essential for the maintenance of hES cell properties and MUC1-C might be applicable to the reprogramming process of fibroblasts to iPS cells.
Regarding the role of MUC1-C in hES cells, it was previously shown that expression of MUC1*, a cleavage form of MUC1-C containing 45 amino acids of the extracellular domain, in contrast with 58 amino acids of the normal form, is associated with hES cell properties [28]. MUC-1* functions as a growth factor receptor associating with NM23-H1. Dimeric NM23-H1 and divalent anti-MUC1* antibody supported growth and pluripotency of hES cells independent of basic fibroblast growth factor (bFGF) signaling, and therefore, these molecules might be applicable for efficacious hES and iPS cell culture [28,29]. However, detailed intracellular effects of MUC1-C function on hES cells other than pluripotency were not investigated in aforementioned studies.
In this study, we provided more information regarding the role of MUC1-C in metabolism and redox control. To check the expression level of MUC1-C, we used an antibody recognizing the C-terminal region of MUC1-C and found that the expression level of MUC1-C is higher in the hES cells, possibly including MUC1* as a major form, compared to that of differentiated cells. Expression of MUC1-C induces the pluripotency factor expression (OCT4, SOX2, NANOG, and MYC), and drives stemness in mouse colorectal cancer and human neuroendocrine prostate cancer [11,12]. Silencing of MUC1-C in human triple-negative breast cancer was associated with suppression of OCT4, SOX2, KLF4, and MYC [30]. Our results showed that functional inhibition of MUC1-C with GO201 in hES cells significantly decreased the proliferation and induced cell death even in the presence of FGF signaling. Furthermore, GO201 treatment increased the ROS level and decreased OCT4 protein expression. These results suggest that MUC1-C participates in the regulation of survival and cell death of hES cells by regulating pluripotency marker expression and controlling optimal ROS levels.
Previous studies have reported that MUC1-C participates in a process to protect cells from cell death induced by various stresses [31,32]. MUC1-C contributes to maintaining cellular redox balance and increases the expression of antioxidant enzymes in cancer cells [21,33 –35]. ES cells rely primarily on glycolysis for energy supply and maintain low ROS levels compared to somatic cells because they have immature mitochondria [36]. Therefore, an increase in the ROS level by GO201 treatment may indicate the possibility that major metabolic activities are switched from glycolysis to oxidative phosphorylation by the functional interruption of MUC1-C.
MUC1-C, as a transcription co-activator, regulates the expression of genes involved in the metabolic pathways in cancer cells. MUC1 enhances the expression of several glycolytic genes, such as HK2, GLUT1, and PDK1, in human pancreatic cancer cells, and contributes to the regulation of glycolysis by directly interacting with PKM2 in mouse fibroblast cells and human breast cancer cells [37,38]. PDK1, an essential glycolytic enzyme, regulates self-renewal, differentiation, and reprogramming in hES cells [39,40]. PKM2 is a glycolytic enzyme and is abundantly expressed in rapidly proliferating cells such as ES cells and cancer cells. PKM2 is known to enhance pluripotency in human and mouse ES cells and to promote somatic cell reprogramming [41,42]. Consistent with these results, we observed that inhibition of MUC1-C decreases the expression of glycolytic enzymes (LDHA, PDK1, and PKM2) in hES cells. These results suggest that MUC1-C may contribute to the maintenance of pluripotency by regulating the expression and activity of glycolytic enzymes in hES cells.
Since iPS cells were established in 2006, various methods have been used to reprogram somatic cells to pluripotent cells, but the efficiency is low and takes about 3–4 weeks for iPS colonies to appear. To overcome these limitations, it is necessary to discover a new strategy. Exogenous MUC1-CD is known to function as an active form of MUC1-C when overexpressed in cancer cells [10,43]. BJ cells expressing MUC1-CD showed a higher growth rate compared to control BJ cells harboring an empty vector. As high proliferation rates are known to enhance iPS cell reprogramming [44] and MUC1-C function is required for pluripotency factor expression, we evaluated the possibility to apply MUC1-C to somatic cell reprogramming and found that expression of MUC1-CD increased the efficiency of iPS cell generation. Although we did not perform additional functional assays such as teratoma assays and differentiation assays for the characterization of the obtained iPS cells, it is clear that MUC1-C plays a positive role in the generation of iPS cells.
Recently, we reported the importance of MUC1-C in mouse ES (mES) cells [19]. The inhibition of MUC1-C reduced cell growth, increased cell death, and disrupted ROS balance in both human and mouse ES cells. On the other hand, there were differential effects in human and mouse ES cells. MUC1-C inhibition decreased the OCT4 expression level in hES cells, but not in mES cells [19]. Inhibition of MUC1-C changed the cell cycling phenotype in mES cells [19], but not in hES cells (data not shown). It is known that distinct signaling pathways are involved in the pluripotency of mouse and human ES cells. The FGF pathway activating MEK/ERK and PI3K/AKT is implicated in the maintenance of hES cells, whereas the LIF/STAT3 signaling pathway is essential for mES cells [45 –47]. Furthermore, hES and mES cells show a difference in the expression levels of cell cycle regulators [48,49]. These differences between hES and mES cells may be related with the different responses of the cells to MUC1-C inhibition. Taken together, our results provide additional information on the regulation of cell growth and pluripotency maintenance in hES and mES cells through MUC1-C.
Conclusion
Expression and function of MUC1-C were required for cell proliferation, maintenance of pluripotency and optimal ROS levels, and a high glycolytic flux in hES cells. MUC1-CD overexpression enhanced the efficiency of reprogramming, suggesting a positive role of MUC1-C in the generation of iPS cells.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was supported by grants from the National Research Foundation (NRF) of Korea funded by the Ministry of Science and ICT (2017M3A9B4065302, 2021R1A2C1006767) and by the Basic Science Research Program through the NRF funded by the Ministry of Education (2019R1A6A3A01092179) in the Republic of Korea.
Supplementary Material
Supplementary Figure S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.