
Abstract
Construction of many tissues and organs de novo requires the use of external epithelial cell sources. In the present study, we optimized the isolation, expansion, and characterization of porcine oral epithelial cells from buccal tissue (Buccal Epithelial Cells, BECs). Additionally, we tested whether key markers [cytokeratin 14 (ck14), p63 protein, and sonic hedgehog molecule (shh)] expression profiles are correlated with three buccal epithelial clone types. Two digestion methods of BECs isolation [Method 1, M1 (collagenase IV/dispase and accutase) and Method 2, M2 (collagenase IV/dispase and trypsin/EDTA)] were compared. Cells obtained by more effective method were further cultured to the third passage and analyzed. Holoclone-, meroclone-, and paraclone-like colonies were identified based on BEC morphology. Immunofluorescent staining was performed to compare selected markers for the indicated buccal clone types. Comparative analysis demonstrated the advantage of isolation using M1 over M2. Cells from the third passage exhibited average 92.73% ± 2.27% presence of ck14. Real-time polymerase chain reaction confirmed expression of tested genes [cytokeratin 8 (ck8), ck14, integrin β1, and p63]. The highest level of ck14, shh and p63, was observed for holoclones. The comparable ck14 expression was observed in the mero- and paraclones. Meroclones expressed significantly lower levels of shh compared with paraclones. The weakest p63 expression was observed in the paraclone-like cells. It was demonstrated that holoclones are the richest in shh (+) and p63 (+) stem cells and these cells should appear to be a promising alternative for obtaining epithelial cells for tissue engineering purposes.
Introduction
The oral mucosal epithelium is the barrier between the host and exterior environment. It is derived from two embryological sources. Buccal mucosa, vestibule, palate, gingiva, intraoral surfaces of the lips, and floor of mouth develop from ectoderm. The tongue mucosa is derived from endoderm [1 –4].
Moreover, the oral mucosa is organized in three tissue subtypes corresponding to their functions. The masticatory (tough) oral mucosa in the hard palate and gingiva shows adaptation to a mastication of the food. The lining (flexible) oral mucosa is less exposed to mechanical forces. This subtype of oral mucosa takes part in the speech and swallowing of the food. It is in the buccal mucosa, soft palate, intraoral surfaces of the lips, alveolar mucosa, and ventral surface of the tongue. The third subtype of the tissue is specialized oral mucosa. This is a mix of masticatory and lining oral mucosa on the dorsal surface of the tongue. Features associated with three subtypes of oral mucosa led to various cell differentiation [1 –4].
Masticatory and specialized subtypes of human oral mucosa are keratinized. In contrast to them, the human epithelium of flexible oral mucosa is nonkeratinized. It should be noted that the location of cell keratinization differs among mammalian species. For example, porcine flexible oral mucosa is keratinized [5]. Keratinized oral epithelium is built from cells of basal, spinous, granular, and corneal layers. Basal, spinous, intermediate, and superficial layers are parts of the nonkeratinized oral epithelium. The crucial proteins of all oral epithelial cells (OECs) are cytokeratins. These proteins make a cytoskeleton within the cells and provide for mechanical resilience. The extracellular spaces of oral epithelium are filled by ceramides, which regulate the permeability of the oral epithelial barrier. The stratified squamous epithelium of oral mucosa is in direct contact with the lamina propria, which is situated under. In this connective tissue blood vessels, structural fibers, nerves, minor salivary glands, and fibroblasts with other cells are placed [1 –4].
Engineering of many tissues and organs de novo requires using external epithelial cell sources. Isolated and expanded OECs have been previously used to reconstruct ocular surface [6 –12]. OECs can also be used for construction of tissues/organs for application in reconstructive urology. The concept of creating tissue-engineered urinary bladder wall by utilizing smooth muscle cells and urothelial cells (UCs), reported by Atala and coworkers cannot be used in cancer patients [13]. Therefore, there is a need to find a new UC source for construction of urinary bladder de novo [14 –16].
The current role of OECs includes seeding of the inner graft surface for urethral reconstruction. Bhargava and coworkers performed the first clinical trial on urethral reconstruction using de-epidermized dermal scaffold seeded with autologous fibroblasts and OECs from buccal mucosa [17]. More recently a few other trials using OECs seeding on a biodegradable scaffold for urethra construction were reported [18 –20]. Optimization of OEC isolation, expansion, and characterization is necessary to explore new applications of these cells in regenerative medicine. Unfortunately, data on in vitro culture conditions of OECs are sparse. Few protocols for OEC isolation from gingival mucosa and from buccal mucosa have been described in the literature [21 –26]. Buccal mucosa seems to be the promising source of OECs due to an easier way to obtain a small tissue biopsy. The type of OECs isolated from buccal mucosa is called Buccal Epithelial Cells (BECs).
Holoclone-, meroclone-, and paraclone-like colonies were identified in in vitro cultures of normal epithelial cells. Holoclones are richest in stem cells and these cells could be the option for tissue engineering approaches [16,27 –29]. Data on characterization of different buccal epithelial clone types by key buccal epithelial and stem cell markers have not been reported.
Ck14 is a part of BEC signature phenotype. Buccal epithelial tissue is cytokeratin 14 (ck14)-positive throughout all cell layers [25]. p63 is expressed by basal epithelial cells, which are capable of self-renewal and differentiation into other cells [7,16,30]. Shh is a protein with an important role in mammalian embryogenesis. This molecule is expressed by epithelial stem cells [31].
The aim of this study was to optimize isolation and expansion of BECs from porcine buccal mucosa for tissue engineering applications and to provide the progress in characterization of BECs by an analysis of ck14 protein and selected genes [cytokeratin 8 (ck8), cytokeratin 14 (ck14), integrin β1, p63)]. We indicated for the first time three buccal epithelial clone populations and analyzed whether selected marker (ck14, p63, shh) expression profiles are correlated with these clones.
Materials and Methods
Animals and tissue collection
BECs were isolated from buccal tissues harvested from 20 male domestic pigs during a planned economic slaughter in a local slaughterhouse. Immediately after collection, buccal mucosa tissues were immersed in the DMEM/Ham's F12 medium (Dulbecco's modified Eagle's medium, HyClone, UT) supplemented with antibiotics (penicillin 100 U/mL, streptomycin 100 mg/mL, and amphotericin B 5 mg/mL; Corning, New York). After the transport to the laboratory, the tissues were placed under a laminar flow to maintain sterility and washed five times using fresh PBS (phosphate-buffered saline; Corning, New York) supplemented with antibiotics. Then, the tissue pieces were transferred into the Petri dish and a fresh medium supplemented with antibiotics was added. Under sterile conditions, the adjacent tissues (like fatty tissues) were removed. The obtained tissue containing epithelium and stroma was cut into pieces (2 cm2).
Histological and immunohistochemical staining
The buccal tissue was macroscopically inspected and fixed in 10% buffered formalin at pH = 7.2 for further evaluation. Formalin-fixed, paraffin-embedded tissue block was cut and stained with Hematoxylin–Eosin. Selected paraffin block was cut into 3–4-μm-thick sections, using a manual rotary microtome (Accu-Cut, Sakura Finetek, Torrance, CA) and then placed on extra-adhesive slides (SuperFrost Plus, Menzel-Glaser, Germany). Immunohistochemical staining was done according to standard protocols. The immunohistochemical procedure was standardized using a series of positive and negative control reactions. The primary rabbit monoclonal anti-ck14 antibody (Table 1) was used to test the expression of ck14 protein. Tissue sections were incubated with primary antibody for 1 h. The reaction was performed using of EnVisionFlex+ Anti-Mouse/Rabbit HRP-Labeled Polymer (Dako; Agilent Technologies, Inc., Santa Clara, CA) and 3,3′diaminobenzidine (DAB) for the localization of the antigen–antibody complex. The slides were counterstained in Mayer's Hematoxylin. Finally, tissue sections were dehydrated in alcohol gradient, cleared in xylene, and sealed with Dako mounting medium (Agilent Technologies, Inc.). The antibody-labeled slides were evaluated by two independent pathologists under a low-power ( × 20) ECLIPSE E800 light microscope (Nikon Instruments Europe, Amsterdam, Netherlands).
Antibodies Used for Buccal Epithelial Cells Characterization
Isolation protocols
Two methods described previously by other authors with a few modifications (Table 2) were compared with an elaborate, efficient, and reproducible method for the isolation and culture of porcine BECs. A total of 80 isolations (40 isolations/method) were performed, including 4 isolations from each tissue (2 isolations/tissue/method).
Protocols Used for Establishment of Oral Epithelial Cell Culture from Buccal Mucosa
EDGS, EpiLife® Defined Growth Supplements.
Tissue pieces (2 cm2) were incubated overnight (∼15 h) at 4°C in a conical tube containing 3 mL of collagenase IV/dispase II (3 mg/mL/4 mg/mL, Life Technologies, Carlsbad, CA) solution in the horizontal orientation to separate the epithelial layer from the stroma. Then, each tissue piece was transferred to the Petri dish containing an equal volume of DMEM/Ham's F12 medium supplemented with fetal bovine serum (FBS; Panbiotech, Aidenbach, Germany). The epithelial surface was mechanically scraped from the tissue piece using the blunt side of a scalpel. The epithelial sheet obtained was cut into smaller pieces. The suspension with undigested tissue pieces was transferred into the conical tube and centrifuged (300 g, 5 min). After the supernatant aspirating, the cell pellet with undigested epithelial pieces was resuspended in 3 mL of commercially available 1 × Accutase solution (Corning, Santa Barbara, CA) (Method 1, M1) or in 3 mL of 0.25% trypsin/0.02% EDTA (Biomed, Lublin, Poland) (Method 2, M2) and incubated for 15–20 min at 37°C. If digestion of epithelial pieces was not complete, it was extended by 5 min. After the enzyme inactivation, the BECs were carefully scraped with the blunt side of scalpel and the remaining tissue pieces were discarded. Then the suspension was filtered through 100 μm nylon cell strainer (Becton, Dickinson & Company, Franklin Lakes, NJ) and centrifuged at 300 g for 5 min. After cell resuspension in 1 mL of CnT-57 medium (Cellntec, CnT, Bern, Switzerland), they were counted using a Trypan Blue exclusion test.
Cell culture
Cells were seeded in a density of 2 × 106 cells/cm2 and cultured in the commercially available CnT-57 growth medium (Cellntec) in standard conditions at 37°C with 5% CO2 atmosphere and 98% humidity. The medium was changed every second–third day. Cell morphology and growth were evaluated under inverted light microscope (Nikon Instruments Europe, Amsterdam, Netherlands). A potential presence of infections and any irregularities (such as changes in morphology or cell detachment) was analyzed.
Success rate of primary culture
Cell cultures that have reached at least 60% confluence during 10 days from the isolation day and showed morphology typical for epithelial cells were considered as successful. Any irregularities such as heterogeneous morphology, low attachment of the cells and infections were regarded as unsuccessful cultures. Success rate was calculated using a formula:
S—success rate [%]
n—number of successfully established cultures
N—number of all cultures.
Comparison of M1 and M2 effectiveness
Parameters such as: number of isolated cells, success rate of primary cultures and morphology were compared to select more effective method of establishment of a BECs primary culture. Cells obtained by this method were further characterized. BECs were cultured to the third passage. The cultures were detached from the growth surface with the use of commercially available 1 × Accutase solution (0.13 mL/1 cm2; Corning, Santa Barbara, CA).
Flow cytometry
BECs from the third passage were fixed in 1.6% paraformaldehyde (15 min, room temperature) and permeabilized with 60% methanol. The cells suspended in the methanol solution transferred to −20°C were incubated until analysis. Approximately 2 × 106 cells were used for analysis. Cells were washed twice in PBS, divided into four samples with 0.5 × 106 cells each, centrifuged for 5 min at 500 g, and suspended in the staining buffer (Becton, Dickinson & Company). The primary antibody (Table 1) was added to the labeling sample and rabbit monoclonal IgG to isotype the control sample. Then the cells were incubated at 4°C in the dark, washed twice in PBS, centrifuged for 5 min at 500 g, and suspended in the staining buffer again. After incubation with the secondary antibody (except unlabeled control) (Table 1) cells were washed and suspended in 150 μL of staining buffer each. Stained samples were immediately analyzed with the use of FACSCanto II flow cytometer (Becton, Dickinson & Company). A minimum of 10,000 events were collected. The obtained results were developed from three independent cultures. The percentage of cells containing ck14 was calculated using FlowJo software (Becton, Dickinson & Company).
Gene expression analysis
The expression of ck8, ck14, integrin β1, and p63 genes was analyzed by real-time polymerase chain reaction (PCR). The RNA was extracted using the RNeasy Mini Kit according to the manufacturer's protocol (Qiagen, Hilden, Germany). cDNA was synthesized from 500 ng of total RNA with the use of Transcriptor High-Fidelity cDNA Synthesis System (Roche Diagnostics, Basil, Switzerland). Quantitative real-time PCR was performed using PrimePCR™ primers (Bio-Rad, Hercules, CA) and LightCycler 480 SYBR Green I Master (Roche Diagnostics). The combination of the most stable reference genes (rpl4 and tbp) was selected with NormFinder methods [32]. The Roche LightCycler 480 software 1.5 (Roche Diagnostics) was used to perform advanced relative quantification analysis.
Identification of three clone types
BECs from the third passage from 15 cultures were seeded on 9 sterile glass coverslips in 12-well plates in a density of 2 × 104 cells/cm2 and cultured ∼5 days until formation of colonies. The cell morphology and growth were analyzed under an inverted light microscope (Nikon Instruments, Inc., New York). Three different clone types: holoclone-, meroclone-, and paraclone-like were identified based on their morphology. To characterize and compare the clones, the immunofluorescence staining was performed.
Immunofluorescence staining
Selected wells containing particular clone types (n = 10 per each clone type) were used for immunofluorescence staining to test whether ck14, p63, and shh are correlated with three buccal epithelial clone types. Wells were fixed by 15 min of incubation in 2% paraformaldehyde solution (Polysciences, Warrington, PA). After blocking with 4% bovine serum albumin (BSA; Sigma-Aldrich/Merck, Darmstadt, Germany) in PBS, the cells were stained for ck14, p63, and shh (Table 1). Next, secondary antibodies labeled with Alexa Fluor 488 were used (Table 1). The cells were counterstained with DAPI (Sigma-Aldrich/Merck). The slides were then mounted on Aqua-Poly Mount (Polysciences) and examined using a microscope (Leica Microsystems, Wetzlar, Germany). All acquisitions were obtained with the same exposure. Measurements of the fluorescence intensity of individual cells were performed using ImageJ-NIH (ImageJ Developers). Resulting data were used to calculate relative cell fluorescence (RCF) for ck14, p63, and shh:
RCF—RCF,
cs—cell surface,
fic—cell fluorescence intensity,
fib—background fluorescence intensity.
Statistical analysis
Statistical analysis was performed using Mann–Whitney test for comparison of cell number isolated from 2 cm2 sections using two different enzymatic methods, one-way ANOVA for comparison of cell number in passages, the first–the third, obtained from 1 cm2 growth surface. Comparison between three types of buccal epithelial clones were made using Kruskal–Wallis test and Dunn's post hoc. Data were analyzed by the GraphPad 8 Prism (GraphPad Software, San Diego, CA). Statistical significance was defined as P value <0.05.
Results
Histological and immunohistochemical analysis of buccal mucosa
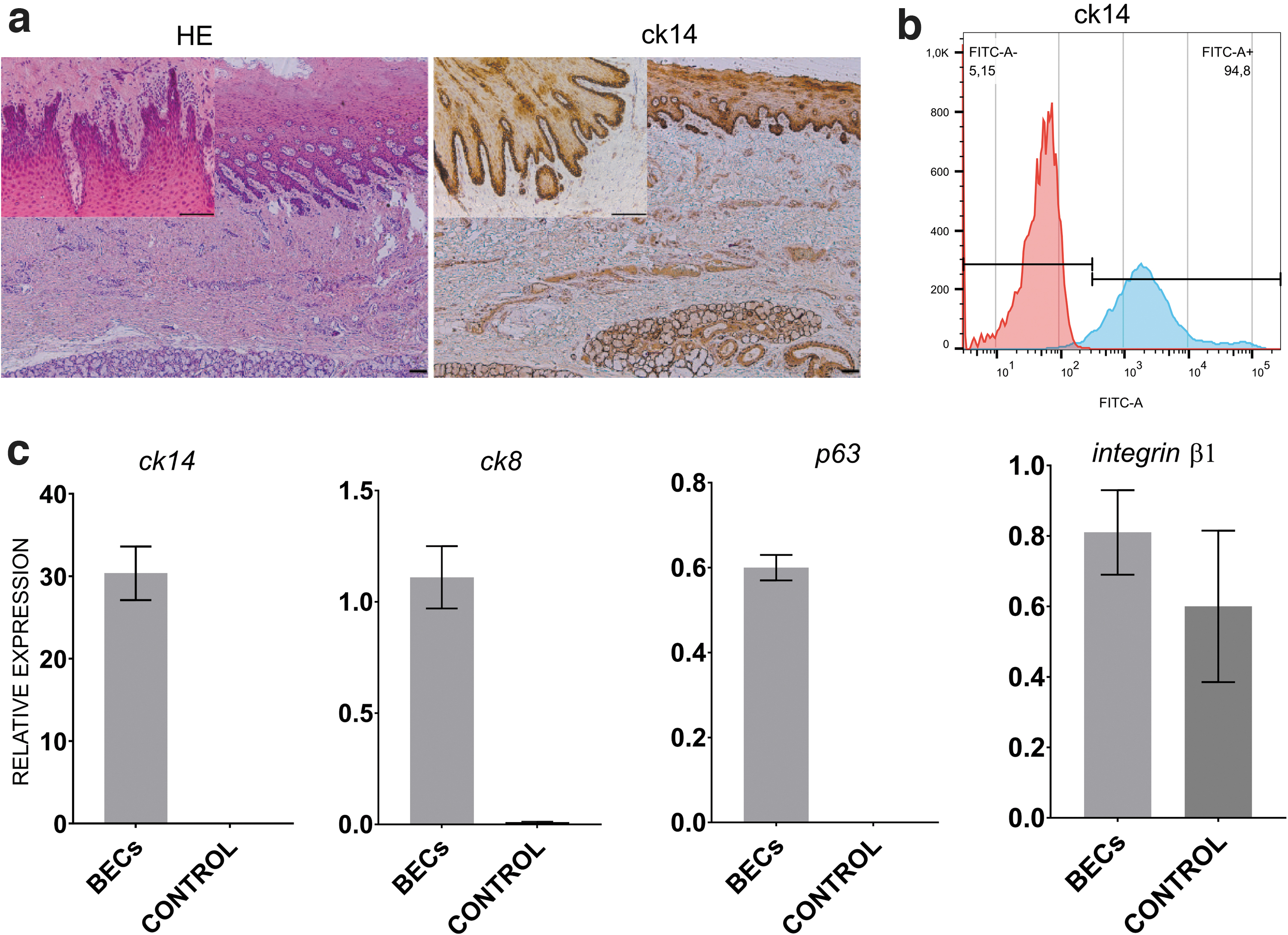
Histological and immunohistochemical analysis of buccal mucosa confirmed the presence of buccal epithelium layer (Fig. 1a). The expression of ck14 in the basal layers was higher than in the suprabasal layer (Fig. 1a).
Efficiency of BECs isolation
The average number of cells isolated using different methods is presented in Fig. 2a. The results showed that significantly higher number of cells was provided by M1 (524.375 ± 187.591), compared with M2 (264.167 ± 86.893, P < 0.01). M1 allows also to achieve higher success rate of culture establishment than M2 (45% vs. 35%, respectively, Fig. 2b). We discarded cultures in which infections were present (Fig. 2g), in which epithelial cells did not appear as a dominant population of attached cells (Fig. 2g’), and which did not reach 60% confluence during 10 days from the isolation day (Fig. 2g”).
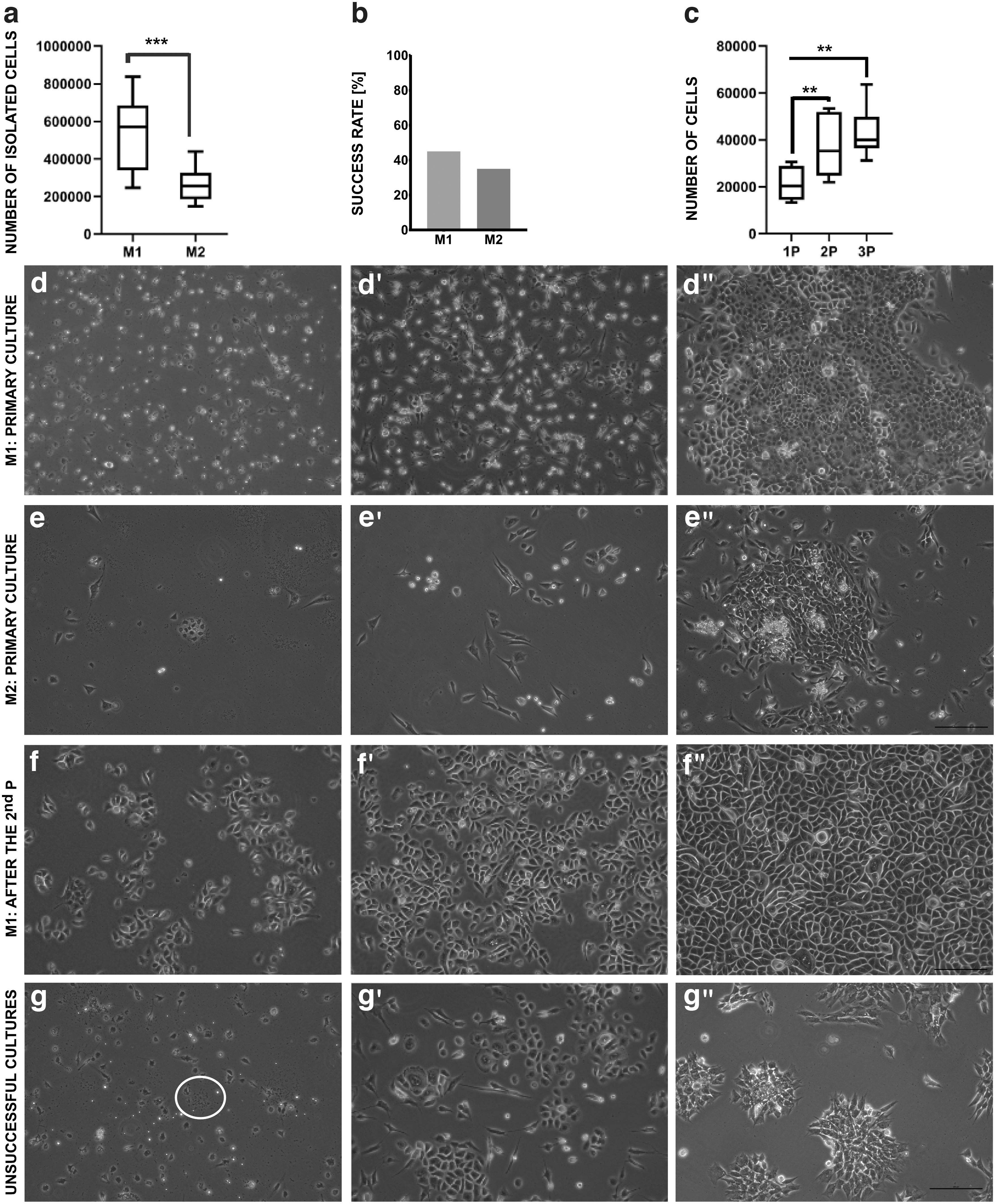
Morphological analysis
The morphological analysis of BEC cultures established by both methods is presented in Fig. 2. In the first 2–3 days of the culture only single cells attached to the surface of the culture flasks. During subsequent days of culture, attached cells changed the shape from small round to flat (Fig. 2d–d”, e–e”). Cells that did not adhere to culture flask surface were successfully removed with every medium change. Heterogeneous cultures were regarded as a failure (Fig. 2g’). Before the first passage, cells have often grown in islands of different sizes (Fig. 2d”, e”). After the first–third passage, cells form a characteristic epithelial “cobblestone” type pattern (Figs. 2f–f”, and 3a). The morphology of cultured cells indicated their epithelial character (Figs. 2f–f” and 3a). Most of the cultures established by M2 did not reach the confluence suitable for the passage (70%) (Fig. 2e”, g”). Contamination of BEC culture by fibroblasts was rarely observed after the first passage of effectively isolated BECs (Fig. 2f–f”).
Comparison of M1 and M2 effectiveness
It was demonstrated that M1 is a more effective method of establishment of the primary culture of BECs than M2 based on the comparison of isolation results and the success rate of established primary culture (Fig. 2a, b).
Analysis of BEC numbers after the first, the second, and the third passage
Using an optimizing isolation protocol for BECs, we obtained and cultured cells until the third passage. The average number of BECs detached from 1 cm2 growth surface after the first, the second, and the third passage are shown in Fig. 2c. The second passage resulted in higher number of cells detached from 1 cm2 compared with the first passage. Similarly, the third passage resulted in approximately twofold increase in comparison to the first passage. The statistical significances between results for the first and the second, the first and the third were observed (P < 0.01). There was no statistical significance between the second and the third passages (P = 0.37).
Cell phenotype and expression of selected genes
BEC phenotype from the third passage was confirmed in all analyzed cultures. The cells from the third passage exhibited average 92.73% ± 2.27% expression of ck14 (Fig. 1b). Real-time PCR analysis confirmed expression of tested genes. Results obtained for BECs were compared with control (adipose-derived mesenchymal stromal cells, AD-MSCs, from the third passage) (Fig. 1c). The obtained results indicated successful isolation and expansion of BECs (Fig. 1).
Morphological characterization of buccal epithelial clones
The morphology of BECs after the third passage indicated the presence of holoclone-, meroclone-, and paraclone-like cells. The morphological analysis of buccal epithelial clones from the third passage is presented in Fig. 3a. Holoclone-like colonies were formed by the smallest cells (Fig. 3a). Meroclone-like clones had intermediate size (Fig. 3a). Paraclone-like colonies were formed by the biggest cells (Fig. 3a). Finally, we obtained 10 wells for each type of clones to stain and analyze.

Expression of selected markers on buccal epithelial holoclonal, meroclonal, and paraclonal cells
The strongest expression of ck14 and shh was observed in the holoclones (201.4 ± 78.52 and 9.23 ± 3.26, respectively; P < 0.05) (Figs. 3b–5). The comparable ck14 expression was observed in the mero- and paraclones (P > 0.99; Figs. 3b, and 4). Meroclones expressed significantly lower levels of shh (2.52 ± 0.94) compared with paraclones (4.31 ± 1.3; P < 0.001; Figs. 3b, and 5). The highest expression of p63 (1.50 ± 0.51) was observed in holoclone-like cell colonies (Figs. 3b, and 6). Meroclone-like cells expressed significantly lower levels of p63 (1.42 ± 0.48). The weakest p63 expression was observed in the paraclone-like cells (0.99 ± 0.30; P < 0.0001) (Figs. 3b, and 6).

Representative fluorescence signals of ck14, DAPI, and merge in BECs after the third passage. Fluorescent microscopy; objective magnification × 40; scale bar = 100 μm.
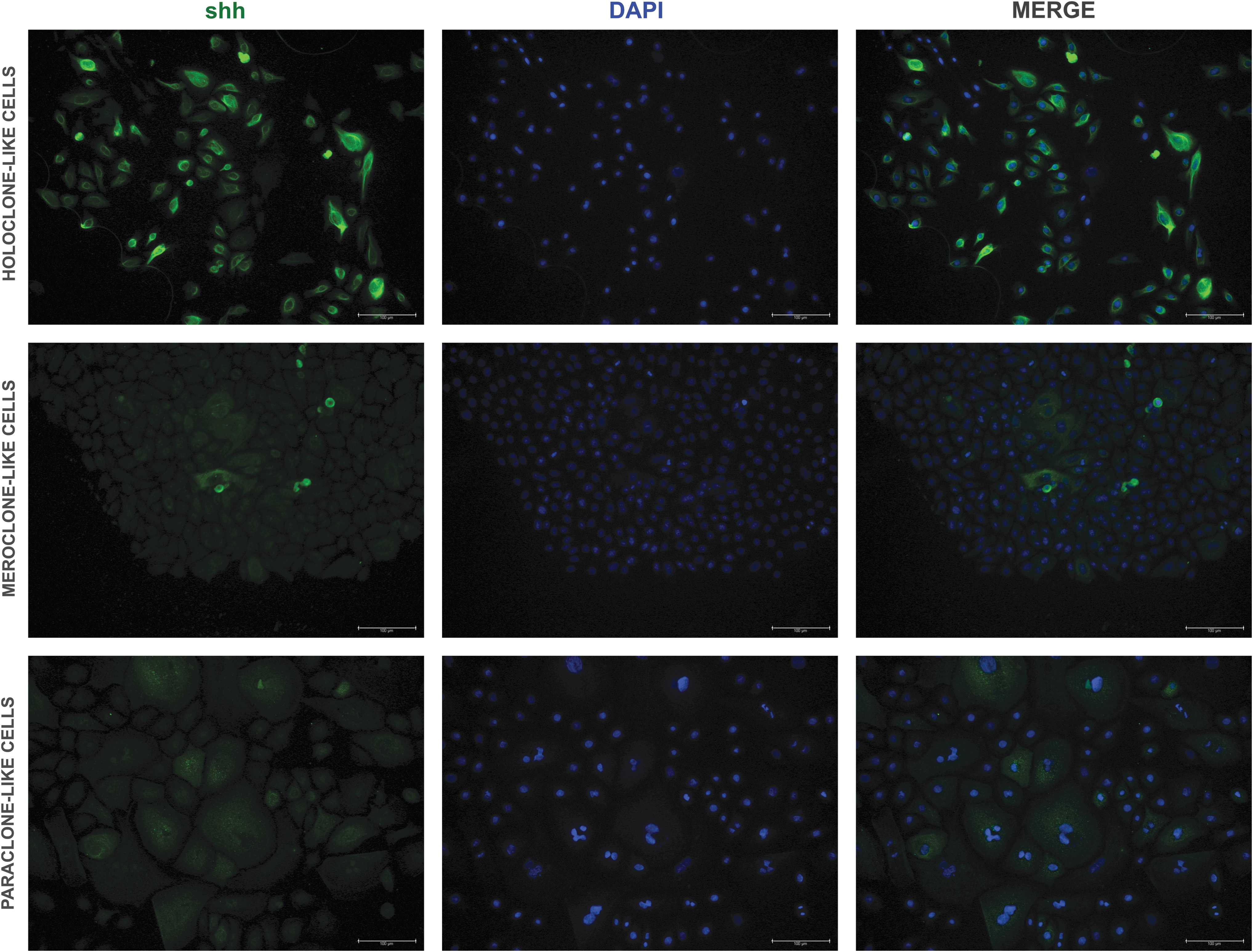
Representative fluorescence signals of shh, DAPI, and merge in BECs after the third passage. Fluorescent microscopy; objective magnification × 20; scale bar = 100 μm.
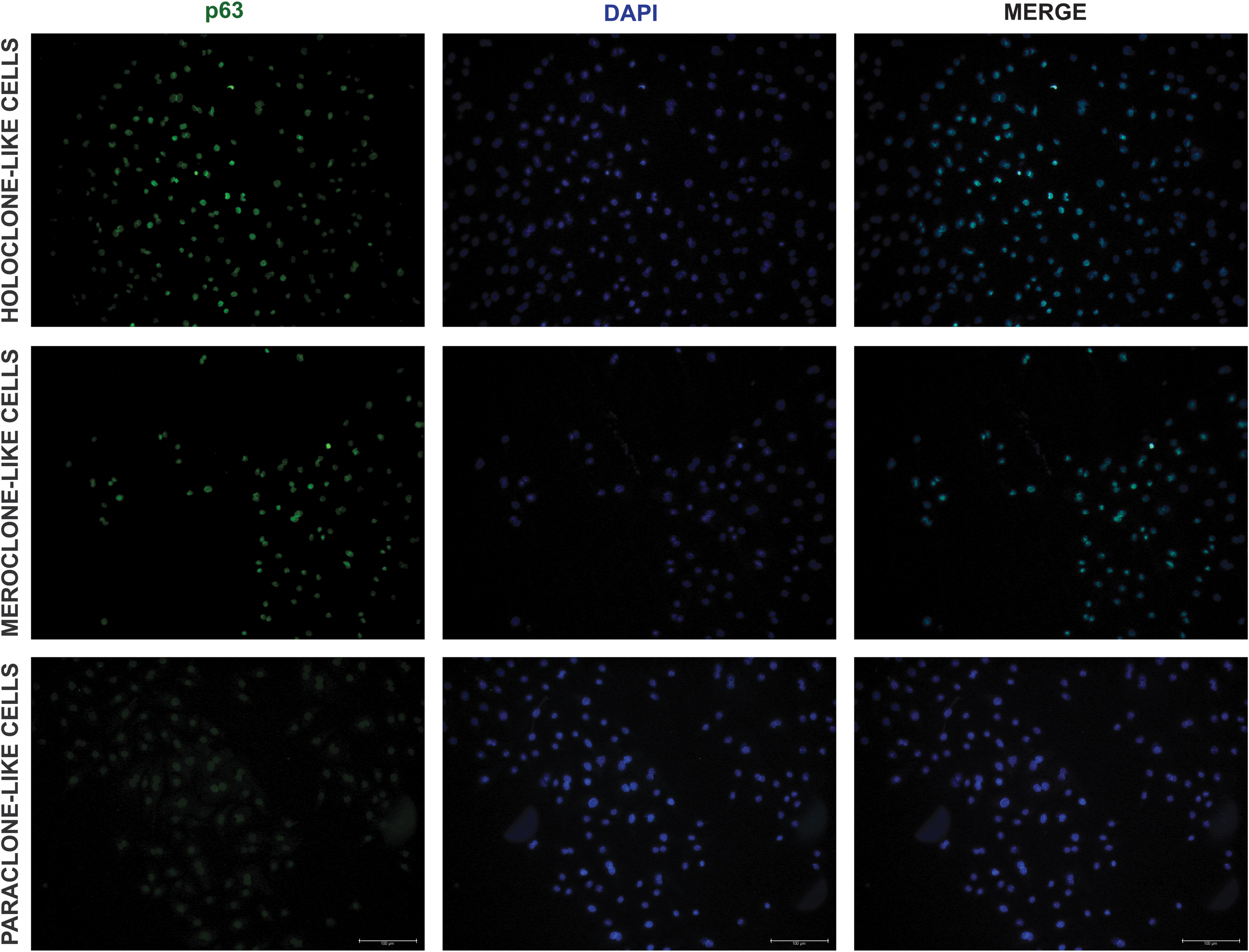
Representative fluorescence signals of p63, DAPI, and merge in BECs after the third passage. Fluorescent microscopy; objective magnification × 20; scale bar = 100 μm.
Discussion
Primary cultures of different epithelial cell types were successfully established using a variety of methods [33,34]. Until now, no study has completely defined the conditions of the porcine BEC primary culture establishment. We have optimized the BEC isolation and expansion protocols for tissue engineering application [21,35]. Recently Tait et al. presented an alternative method to obtain porcine BECs with a growth-arrested primary feeder layer and using pooled Human Platelet lysate [36].
Igarashi et al. isolated rabbit BECs using dispase II at 4°C for 16 h and trypsin/EDTA at 37°C for 10 min [24]. Calenic et al. isolated human gingival epithelial cells using collagenase IV/dispase at 4°C for 24 h and trypsin/EDTA at 37°C for 30 min [21,37]. Oda and coworkers isolated human gingival epithelial cells using dispase II at 37°C for 3–4 h and trypsin/EDTA at 37°C for 5 min [23]. Hustler and coworkers used the protocol by Oda to human BECs isolation [23,25]. Methods available in the literature used for isolation of OECs differ in cell/tissue source, digestive enzyme type, and isolation conditions, which makes result comparison difficult.
In this report, we used fragments of porcine buccal mucosa for BEC isolation. Large animal models significantly promote development of medical research, including tissue engineering approaches. It was shown that nonkeratinized canine and porcine buccal mucosa are more like human compared with other species [5].
In this study, we compare two other enzymatic digestion protocols [21,35]. The obtained results showed an advantage of the combination of collagenase IV/dispase and Accutase over the combination of collagenase IV/dispase and trypsin/EDTA in the number of isolated cells (Fig. 2a, b). This effect could be caused by the aggressive action of trypsin.
Commercially available serum-free CnT-57 medium was used for BEC culture. The addition of this medium provides the appropriate environment for homogeneous growth of epithelial cells and guide their proliferation [35]. Using of the serum-free medium is particularly important because it provides the possibility to use in clinical practice.
It is worth highlighting the fact that success rate for establishment of human cell cultures should be higher in comparison to results obtained in this report, in which a lot of attempts to establish porcine OEC cultures were unsuccessful. One of the main obstacles were microbiological contaminations and heterogenous cultures (Fig. 2g–g’).
Examined cells expressed ck8, ck14, integrin β1, and p63 genes and had a high level of ck14 protein (Fig. 1b, c). The cytokeratins have been earlier proved as the characteristic OEC markers, which could be preserved in in vitro cultures [3,25]. Presumably, there are stem cell niches in all types of oral epithelium [1,6,38,39]. p63 protein and integrins were used as epithelial stem cell markers by several authors. Integrins are essential adhesion receptors, which interact with ligands outside the cell, and signal transduction events occur this way. p63 and integrins are important for cellular proliferation [6,16,34,40,41]. Our results indicated that p63 and integrin β1 genes expression profiles allow for efficient stem cell enrichment from buccal tissue, as in other epithelial types (Fig. 1c).
Three types of clones (holoclones, meroclones, and paraclones) were identified in cultures of various epithelial cells [16,27 –29]. It is believed that holoclones with the highest proliferative potential are formed by stem cells [27]. In our previous study, we provided additional evidence that three UC populations with different properties can be isolated from the bladder.16 In the present report, we analyzed BEC morphology to identify three different clone types. The phenotypic differences between cells in the established cultures indicated the presence of holoclone-, meroclone-, and paraclone-like cells (Figs. 3–6).
The presence of holoclones rich in p63 (+) epithelial stem cells is especially important for tissue engineering approaches. It was reported that from the whole cell population, holoclone-like cells will have the greatest possibility to transdifferentiate [28,29]. That is why p63 was chosen to test whether there are differences in its distribution between indicated clone populations. Data on in vitro culture systems to expand p63(+) BECs population and to maintain their unique characteristics are sparse [21,42]. Methodological advances, such as performed in this study, will help to improve p63 (+) BEC culture establishment (Figs. 3, and 6).
Shh molecule belongs to the hedgehog signaling pathway. It was revealed that a dental epithelium expresses shh unlike the nearby oral epithelium [43,44]. In the present study, cells in all three types of clones were shh (+) with the highest level in holoclone-like cells. The keratin 14 promoter can enhance shh expression [45,46]. This finding can explain the results of our study, which indicated coexpression of both shh and ck14 in all three types of BECs (Figs. 3–5).
Shh is one of the key signaling pathways, which is involved in urinary bladder organogenesis. It was found that interactions of differentiating urothelium and bladder mesenchyme are necessary for bladder organogenesis. The induction of bladder mesenchyme to differentiate into smooth muscle requires the presence of bladder urothelium. Shh is involved in epithelial–mesenchymal interactions [47 –52]. In our previous research, we indicated for the first time that hedgehog (hh) signaling pathway was upregulated by AD-MSCs during healing of tissue-engineered urinary bladder [53]. In that study, better development of smooth muscle layer within cell seeded grafts was found. In our opinion, it was based on enhanced smooth muscle proliferation mediated by hh cascade. Results obtained in the present study indicated for the presence of shh (+) cell population in all three types of BECs. It could contribute to application of BECs in tissue engineering in urology. These cells appear to be a promising alternative for obtaining UCs from external urinary bladder sources.
Conclusions
The purpose of this study was to develop conditions for the isolation and growth of BECs and to characterize and compare the buccal epithelial clone populations. We obtained simple and repeatable method of BEC isolation and provided the progress in the characterization of these cells. The development of this method could contribute to application of BECs in tissue engineering. Additionally, we confirmed that three BECs types with differing features can be isolated from the buccal tissue. Holoclone-like cells are richest in stem cells and should be used in further studies, including transdifferentiation attempts toward UCs.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by a research task within the framework of the statutory activities no 296.