
Abstract
Hedgehog signaling is essential for vertebrate development; however, less is known about the negative regulators that influence this pathway. Using the mouse P19 embryonal carcinoma cell model, suppressor of fused (SUFU), a negative regulator of the Hedgehog (Hh) pathway, was investigated during retinoic acid (RA)-induced neural differentiation. We found Hh signaling increased activity in the early phase of differentiation, but was reduced during terminal differentiation of neurons and astrocytes. This early increase in pathway activity was required for neural differentiation; however, it alone was not sufficient to induce neural lineages. SUFU, which regulates signaling at the level of Gli, remained relatively unchanged during differentiation, but its loss through CRISPR-Cas9 gene editing resulted in ectopic expression of Hh target genes. Interestingly, these SUFU-deficient cells were unable to differentiate toward neural lineages without RA, and when directed toward these lineages, they showed delayed and decreased astrocyte differentiation; neuron differentiation was unaffected. Ectopic activation of Hh target genes in SUFU-deficient cells remained throughout RA-induced differentiation and this was accompanied by the loss of Gli3, despite the presence of the Gli3 message. Thus, the study indicates the proper timing and proportion of astrocyte differentiation requires SUFU, likely acting through Gli3, to reduce Hh signaling during late-stage differentiation.
Introduction
Hedgehog (Hh) signaling plays pivotal roles in neural development, where Sonic Hedgehog (SHH) is essential in patterning the differentiation of motor neurons and interneurons in the neural tube [1] and inducing differentiation of cerebellar neurons and glial cells [2]. When Hh ligand is absent, the transmembrane receptor Patched (PTCH) inhibits the transmembrane protein Smoothened (SMO) from translocating to the plasma membrane of the primary cilium [3]. Inhibition allows the suppressor of fused (SUFU) to sequester full-length Gli transcription factors [4] or promotes Gli phosphorylation [5] and partial proteosomal degradation into a truncated repressor [6,7]. In the presence of Hh, however, PTCH is inhibited, which allows the SUFU-Gli complex to be recruited into the primary cilium [8]. SUFU dissociates from Gli allowing the translocation of Gli to the nucleus where it acts as a transcriptional activator [7].
In vertebrates, there are three Gli proteins, with Gli1 serving solely as an activator as Gli1 is an Hh target gene, present only after activation of the pathway [6]. Gli2 and Gli3 are either transcriptional activators or repressors, with Gli3 acting primarily as a repressor [6]. Thus, while Hh target gene transcription is tightly regulated through Gli, other proteins regulate Gli's. One of these is SUFU, a regulator of Gli processing that is key to Hh pathway activation. Through the reduced processing and stability of the Gli3 repressor [9], Sufu loss of function alleles cause constitutive expression of Hh target genes in the absence of an Hh ligand [10 –12].
Loss of Sufu in humans causes medulloblastoma tumor growth [13,14] and Nevoid Basal Cell Carcinoma Syndrome [14,15], while mouse embryos containing a targeted deletion of Sufu are embryonic lethal at E9.5 due to neural tube closure defects [11]. Furthermore, the loss of Sufu function in the mouse retina restricts retinal neuron differentiation [12] and in the cerebellum results in developmental delays in neuron differentiation [16] and cerebellum mispatterning [17].
In this study, an in vitro system utilizing the mouse P19 embryonal carcinoma cell line was employed to study when the Hh pathway is activated, its requirement during differentiation, and the effects of Sufu loss of function on neural cell fate. The cell line is readily differentiated into neural lineages in the presence of retinoic acid (RA) [18]. RA causes P19 cells to develop functional neurons and supporting astrocytes [19], and although this is well known, few studies have investigated the role of Hh signaling in this system [20,21]. Of these, one study showed the overexpression of a constitutively active Gli2 promotes neuronal differentiation [20], but another revealed the knockdown of Gli2 causes the same phenotype [21]. Thus, these and many unanswered questions remain, and although Hh signaling is precisely controlled in vivo, how SUFU is involved in determining neuron and astrocyte cell fate decisions is not known.
To address this shortfall, this study aims to better understand the essential role of SUFU in neuroectodermal differentiation. Using the P19 cell model and timeline investigations, we show when Hh signaling is required during RA-induced neural differentiation, and through genetic ablation of Sufu, demonstrate that there is an essential role for SUFU in astrocyte determination.
Materials and Methods
Cell culture and differentiation
P19 embryonal carcinoma cells (a gift from Dr. Lisa Hoffman, Lawson Health Research Institute, London, ON, Canada) and Sufu knockout (Sufu−/−, described below) P19 cells were cultured on adherent tissue culture plates in Dulbecco's modified Eagle medium (DMEM) containing 5% fetal bovine serum (FBS) and 1% pen/strep at 37°C and 5% carbon dioxide. Cells were passaged every 4 days or at 70% confluency, whichever occurred first. To induce neural differentiation, ∼1.05 × 106 cells were cultured on bacterial grade Petri dishes for 4 days in 0.5 μM RA to form embryoid bodies (EB) [19]. Next, EBs were resuspended and plated on adherent tissue culture dishes in the presence of 0.5 μM RA, for a total of 10, 14, or 17 days.
Untreated controls were grown as described, but without RA. For Smoothened Agonist (SAG; EMD Millipore) and Cyclopamine (Cyc; EMD Millipore) studies, cells were cultured as above, but in the presence of 0.5 μM RA plus 10 nM SAG or 10 μM Cyc, or 10 nM SAG alone during EB formation, and then replated as described above in the presence of 0.5 μM RA or were left untreated for the SAG-only treatment.
Cas9 plasmid preparation
The pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene plasmid: 62988) was used, where sgRNAs for Sufu (Supplementary Table S1) were cloned into the PX459 vector [22]. Briefly, sgRNAs were amplified and phosphorylated, the vector digested using the BbsI restriction enzyme, and vector and sgRNA were incubated at room temperature for 1.5 h of ligation. Ligated plasmids were transformed into competent bacteria, colonies were selected, and isolated plasmids were sequenced at the London Regional Genomics Centre (Robarts Research Institute, London, ON, Canada) using the U6 primer (Supplementary Table S1).
Knockout lines
PX459-sgRNA plasmid (2 μg) was transfected using 10 μL of Lipofectamine 2000 (Invitrogen). After 4 h, media were changed, and cells were grown to 70% confluency for clonal selection. Selection media containing 1 μg/mL puromycin were replaced every 24 h for 4 days, and then cells were grown in complete media without puromycin until ready to be passaged. Knockout genotypes were determined by collecting genomic DNA (Qiagen DNeasy® Blood & Tissue kit, 69504), and performing standard polymerase chain reaction [PCR; DreamTaq Master Mix (2 × ), Thermo Scientific] with Sufu-specific primers (Supplementary Table S1).
Amplicons were sequenced at the London Regional Genomics Centre and compared to wild-type sequences using pair-wise sequence alignment (Geneious 2021™). KO line no. 2 sequencing was examined using tracking of indels by decomposition (TIDE) analysis (
Overexpression lines
Sufu KO no. 2 was transfected with 2 μg of either 1436 pcDNA3 Flag HA (Addgene plasmid: 10792), a pcDNA Flag HA vector with the Sufu open reading frame cloned from pcDNA Su(fu) (CT no. 116; Addgene plasmid: 13857) [24] using BamHI (New England Biolabs) and XhoI (New England Biolabs) restriction enzymes, or hGli3 flag3x (Addgene plasmid: 84921) using Lipofectamine 2000 (Invitrogen). Cells were incubated for 4 h in the presence of plasmid, media were then changed, and cells were grown to 70% confluency. Transfected cells were treated with 500 μg/mL neomycin (G418) selection media every 48 h for 2 weeks.
Real-time reverse transcriptase PCR
To determine relative mRNA expression levels, total RNA was collected from RA-treated cells at various time points using QiaShredder (Qiagen) and RNeasy (Qiagen) kits. cDNA was made using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific) and amplification was performed using gene-specific primers (Supplementary Table S1). Quantitative reverse transcriptase PCR (RT-qPCR) was done using 500 nM of each reverse and forward primers, 10 μL of SensiFAST SYBR No-ROX Mix (Bioline), and 1 μL of cDNA template. Samples were analyzed using a CFX Connect Real-Time PCR Detection System (Bio-Rad) using the comparative cycle threshold method (2-ΔΔCt). Relative expression was normalized to L14 and untreated or wild-type untreated controls to determine fold change expression.
Immunoblot analysis
Cells were lysed in 2% sodium dodecyl sulfate (SDS) buffer containing 62.5 mM Tris-HCL pH 6.8, 10% glycerol, 5% mercapto-2-ethanol, and 1 × Halt Protease Inhibitor Cocktail (Thermo Scientific; 1:200). Proteins were separated using SDS-polyacrylamide gel electrophoresis, and then transferred to polyvinylidene difluoride membranes (Bio-Rad, 1620177). Membranes were probed with primary antibodies to β-III-tubulin (1:1000; Cell Signaling Technology), GLI3 (1:1000; R&D Systems), SUFU (1:1000; abcam), Glial Fibrillary Acid Protein (GFAP; 1:1000; Invitrogen), and β-actin (1:10,000; Santa Cruz Biotechnology) and then with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies (1:10,000, Sigma). Signals were detected using the Immobilon® Classico Western HRP Substrate (Millipore) and imaged using a Chemi Doc Touch System (Bio-Rad).
Immunofluorescence
Cells were differentiated, as stated previously, on coverslips coated with poly-L-lysine hydrobromide (Sigma), fixed with 4% paraformaldehyde for 10 min, and permeabilized in 0.2% Triton-X-100 for 10 min at room temperature. Coverslips were incubated for 30 min in 1% bovine serum albumin, 22.52 mg/mL glycine, and 0.1% Tween-20, and then overnight in a humidity chamber with β-III-tubulin (TUJ1, 1:400; Cell Signaling Technology) and GFAP (1:500; Invitrogen). Incubation in secondary antibodies (1:400), Alexa Fluor™ 660 goat anti-mouse IgG (Invitrogen) and Goat anti-Rabbit IgG Alexa Fluor Plus 488 (Invitrogen), was done in the dark for 1 h at room temperature. Coverslips were mounted onto microscope slides using Slowfade™ Gold antifade reagent with 4′,6-diamidino-2-phenylindole (Invitrogen) and cells were examined using a Zeiss AxioImager Z1 Compound Fluorescence Microscope at the Integrated Microscopy Facility, Biotron (Western University, London, ON, Canada). Images were analyzed using ImageJ.
Flow cytometry
Cells, differentiated as stated previously, were washed with ice-cold PBS without magnesium or chloride [PBS(−/−); Thermo Fisher] and dissociated with 0.25% Trypsin. Trypsin was deactivated with DMEM/F12 (Thermo Fisher) containing 10% ES-grade FBS (Thermo Fisher), and cells were centrifuged at 300 g for 5 min and reconstituted in PBS(−/−). Cells were counted using a trypan blue assay, separated to ∼1.0 × 106 cells per tube, and washed in Flow Cytometry Staining Buffer[(10% FBS in PBS(−/−)].
Cells were fixed using 4% formaldehyde in PBS(−/−) for 10 min at room temperature, washed with ice-cold PBS(−/−), and permeabilized with 0.2% Triton-X-100 in PBS(−/−) for 10 min. Cells were incubated with or without 1 μL of GFAP Monoclonal Antibody (GA5), Alexa Fluor 488, eBioscience™ (Invitrogen), per million cells for 1 h at room temperature, and then washed with PBS (−/−), strained using a 40 μm strainer (Falcon™), and analyzed using a BD FACSCanto II flow cytometer at the London Regional Flow Cytometry Facility (Robarts Research Institute). Data were analyzed using FlowJo (BD). Flow cytometry gating strategy is reported in Supplementary Fig. S1.
Results
RA and cell aggregation induce P19 neural differentiation
To induce neural differentiation cells were treated for 4 days in 0.5 μM RA to form EB, which were then plated for a total of 10 days to form neurons or 17 days to form astrocytes (Fig. 1A). Differentiation was confirmed with RT-qPCR of NeuroD1, NeuroG1, and Ascl1 increasing and subsequently decreasing expression (Fig. 1B). As expected [25], the neuronal marker, β-III-tubulin, was detected following RA treatment at 10 days (Fig. 1C) and coinciding with prominent signals for GFAP, an astrocyte marker, which increased by day 17 (Fig. 1C). Neither marker was prominent in cells undergoing spontaneous differentiation in the absence of RA (Fig. 1C), and together confirmed that neurons and astrocytes differentiated by RA.

RA is required for P19 cell neural differentiation.
Hh signaling is activated early in neural differentiation
Since reports note that Shh signaling is vital in vertebrate neural differentiation [1 –3], RT-qPCR was used to explore Hh component expression throughout RA treatment of P19 cells. Hh ligands Dhh, Ihh, and Shh were significantly upregulated after 1 day of RA treatment (P < 0.001, 0.01, and 0.05, respectively), as was Gli1 (P < 0.001), a Hh target gene, before returning to basal levels (Fig. 2A). Expression of Ptch1, Ptch2, Smo, Gli2, Gli3, and Sufu (Fig. 2A) was also confirmed. Since Gli3 can serve as an activator or repressor [6], Gli3 levels were explored throughout RA-induced differentiation.
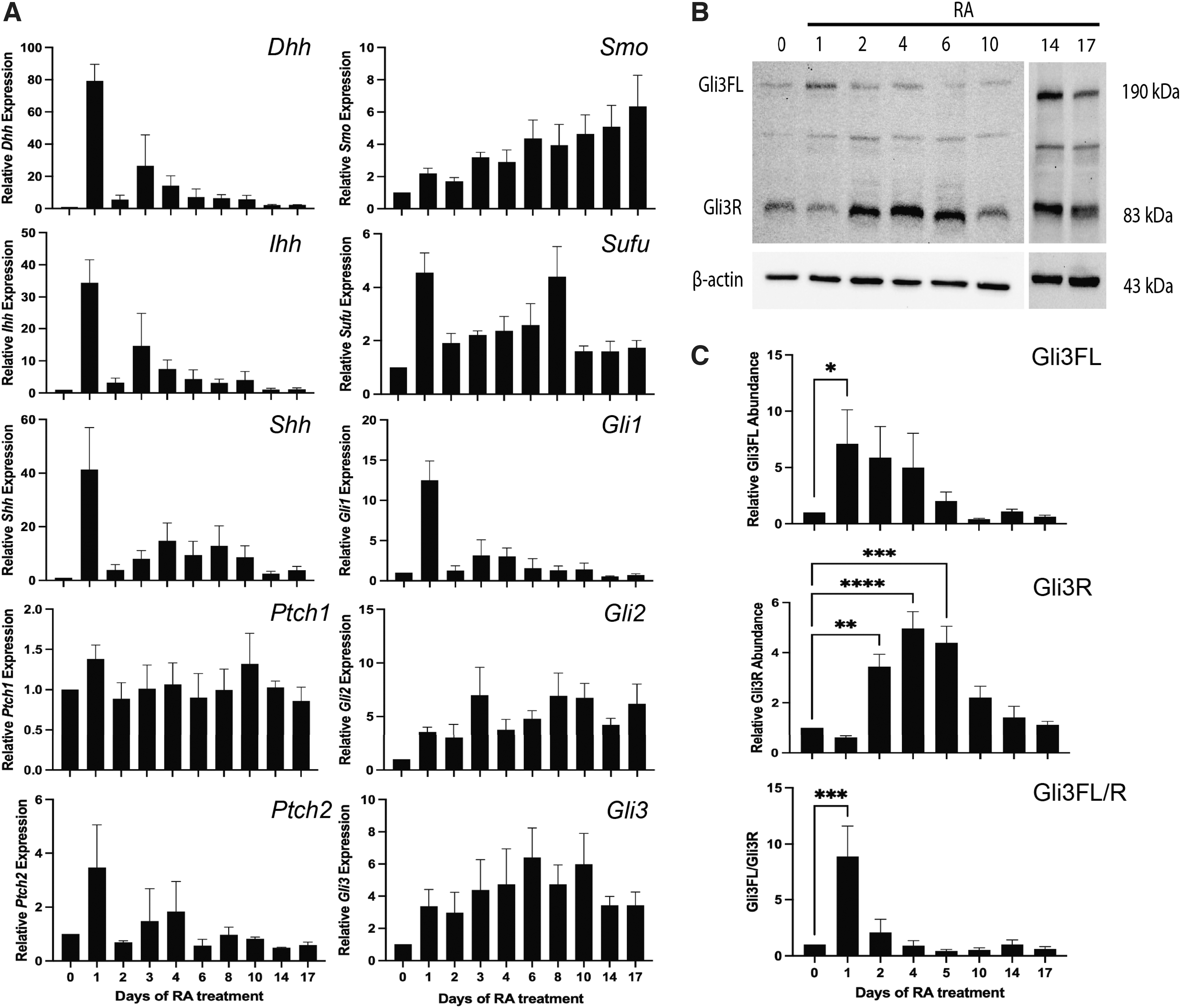
Hh signaling is activated early in RA-induced differentiation.
Full-length Gli3 (Gli3FL) was significantly increased 1 day after RA treatment (Fig. 2B, C), whereas Gli3 repressor (Gli3R) was increased on days 2, 4, and 6 of RA treatment. As the activation of Hh signaling is dependent on the ratio between Gli3FL and Gli3R, this ratio was examined, and results showed an increase in the Gli3FL to Gli3R ratio 1 day after RA treatment (Fig. 2C). Given these data, it would appear that Hh pathway activity is increased early in neural differentiation of P19 cells, where it subsequently returns to basal levels coinciding with neuron and astrocyte differentiation.
Early Hh signaling is required, but not sufficient for neural differentiation
To first address if activation of Hh signaling would affect differentiation, cells were treated with 0.5 μM RA (RA), 0.5 μM RA plus 10 nM SAG on day 0 (RA + SAG d0), 10 nM SAG alone on day 0 (SAG d0), or 10 nM SAG on day 0 with subsequent 0.5 μM RA treatment on day 4 (SAG d0 + RA d4). Immunoblots showed β-III-tubulin in RA, RA + SAG d0, and SAG d0 + RA d4 treatments, but not in SAG d0 treatment alone on day 10 (Fig. 3A). Densitometric analysis revealed an increase in β-III-tubulin intensity in the RA + SAG d0 treatment compared to RA alone, but no difference between RA alone and the SAG d0 + RA d4 treatment (Fig. 3B).
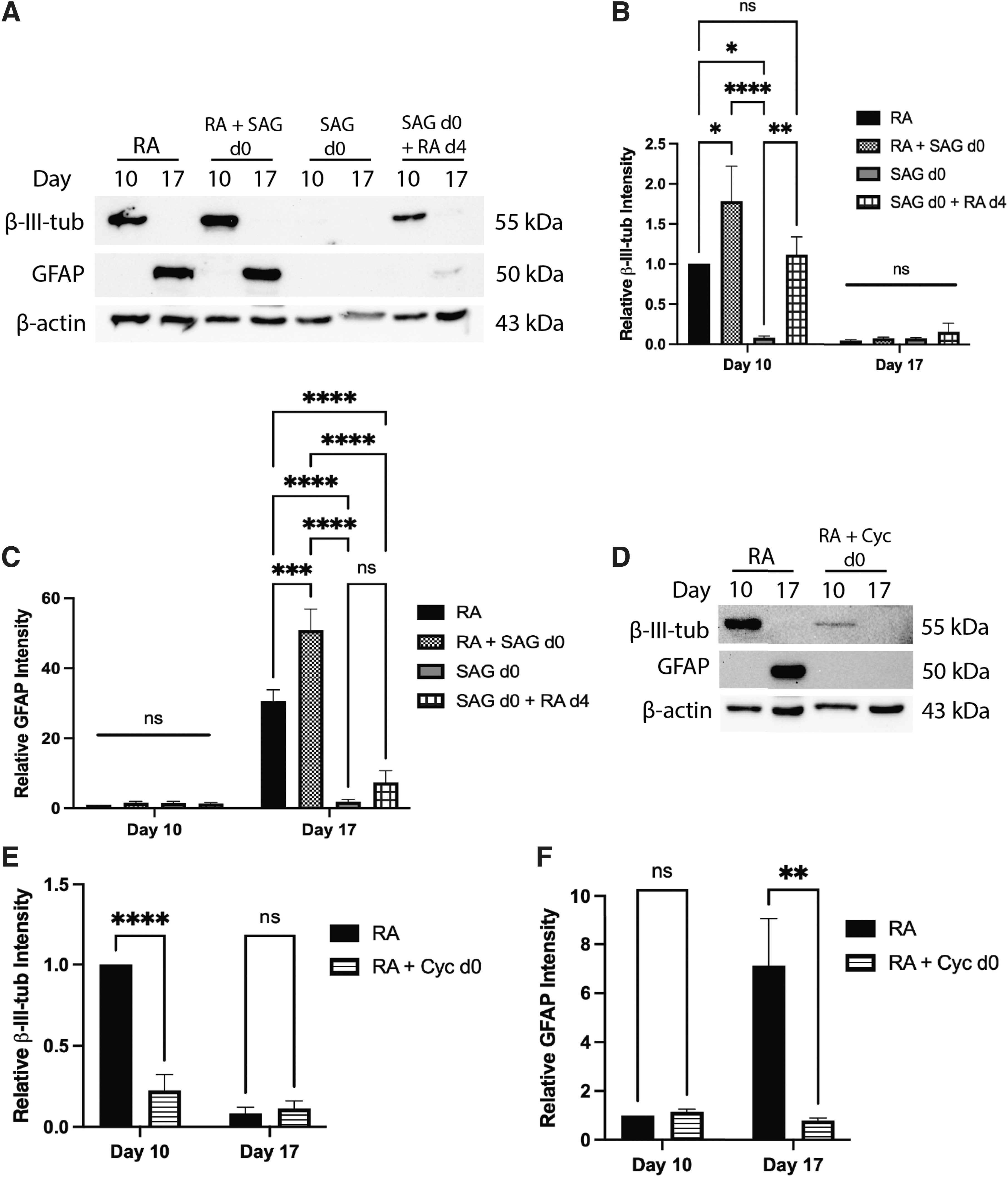
Early Hh pathway activation is required, but not sufficient to induce neural lineages.
Experiments allowing cell aggregation for 4 days in the absence of RA, and then subsequent 0.5 μM RA treatment on day 4 [dimethyl sulfoxide (DMSO) d0 + RA d4], also showed the β-III-tubulin signal on day 10, as did cells in the presence of DMSO only (DMSO d0 + DMSO d4); however, both treatments were significantly less than the RA treatment alone (Supplementary Fig. S2A, B). Therefore, SAG overactivation of Hh signaling, in the presence of RA, appeared to enhance the neuron cell fate, while SAG alone was not sufficient to induce neurons.
Immunoblot analysis of GFAP showed detection on day 17 in the RA and RA + SAG d0 treatments, but no signal was seen in the SAG d0 alone treatment (Fig. 3A). Furthermore, densitometric analysis showed increased GFAP intensity in the RA + SAG d0 treatment compared to RA treatment alone (Fig. 3C). A GFAP band was also seen in the SAG d0 + RA d4 treatment (Fig. 3A), but densitometric analysis revealed that this signal was not significantly different from the SAG d0 alone treatment (Fig. 3C). DMSO d0 + RA d4 cells also showed this low level of GFAP on day 17 (Supplementary Fig. S2A), indicating that RA treatment at this later time was sufficient to induce astrocytes; however, not to the extent observed with the initial RA treatment.
As the changes to neural cell fates in the presence of SAG were subtle, we confirmed SAG activation of Hh was occurring in these cells through a luciferase assay (Supplementary Fig. S2D). Thus, it appears from immunoblot data that supplementing the initial RA treatment with SAG increases astrocyte differentiation, but activation of Hh signaling alone using SAG was not sufficient to induce the astrocyte fate.
Since early Hh pathway activation enhances neural cell fates, would its attenuation affect neural differentiation? Cells were treated with either 0.5 μM RA (RA) or 0.5 μM RA plus 10 μM Cyc on day 0 (RA + Cyc d0), both with subsequent RA treatments on day 4. Immunoblot analysis of β-III-tubulin on day 10 showed signals in both RA and RA + Cyc treatments (Fig. 3D), but densitometry revealed the intensity was significantly reduced with Cyc treatment (Fig. 3E). Cyc also had dramatic effects on GFAP levels, eliminating the response induced by RA (Fig. 3D, F). Experiments with cells treated with RA plus the Cyc vehicle (RA + DMSO d0) showed no significant change in either marker (Supplementary Fig. S2C). These results suggest that inhibition of the Hh pathway attenuates neuron and astrocyte cell fates, and thus is required for proper differentiation of these cell types.
SUFU levels during neural differentiation
SUFU negatively regulates Gli activity and as reported by Humke et al. [7], promotes Gli3 conversion to a repressor. To determine its role in Hh signaling seen in P19 cells, Sufu gene expression was examined and found to be moderately increased on day 1, 8, and 10 of RA treatment (Fig. 2A); however, at the protein level, no change was seen across any time points of RA treatment (Supplementary Fig. S3). Since Sufu expression and protein levels remain largely unchanged, it is tempting to speculate that the early activation of the Hh pathway is controlled by the cellular localization or post-translational modification status of SUFU.
Sufu knockout activates Hh signaling
Since SUFU acts as an essential regulator of Gli3 [7] and Gli2 [4,7], its loss was expected to induce Hh signaling. To test this, CRISPR-Cas9 was used to target exon 6 of the Sufu gene (Fig. 4A), resulting in clones with an 81 bp deletion (KO no. 1) that spans the intron-exon boundary between intron 5 and exon 6 and another with a 1 bp insertion (KO no. 2). Sequence alignment, TIDE analysis [23], and in silico translation of clones revealed a premature stop codon shortly after the mutation (Supplementary Fig. S4).
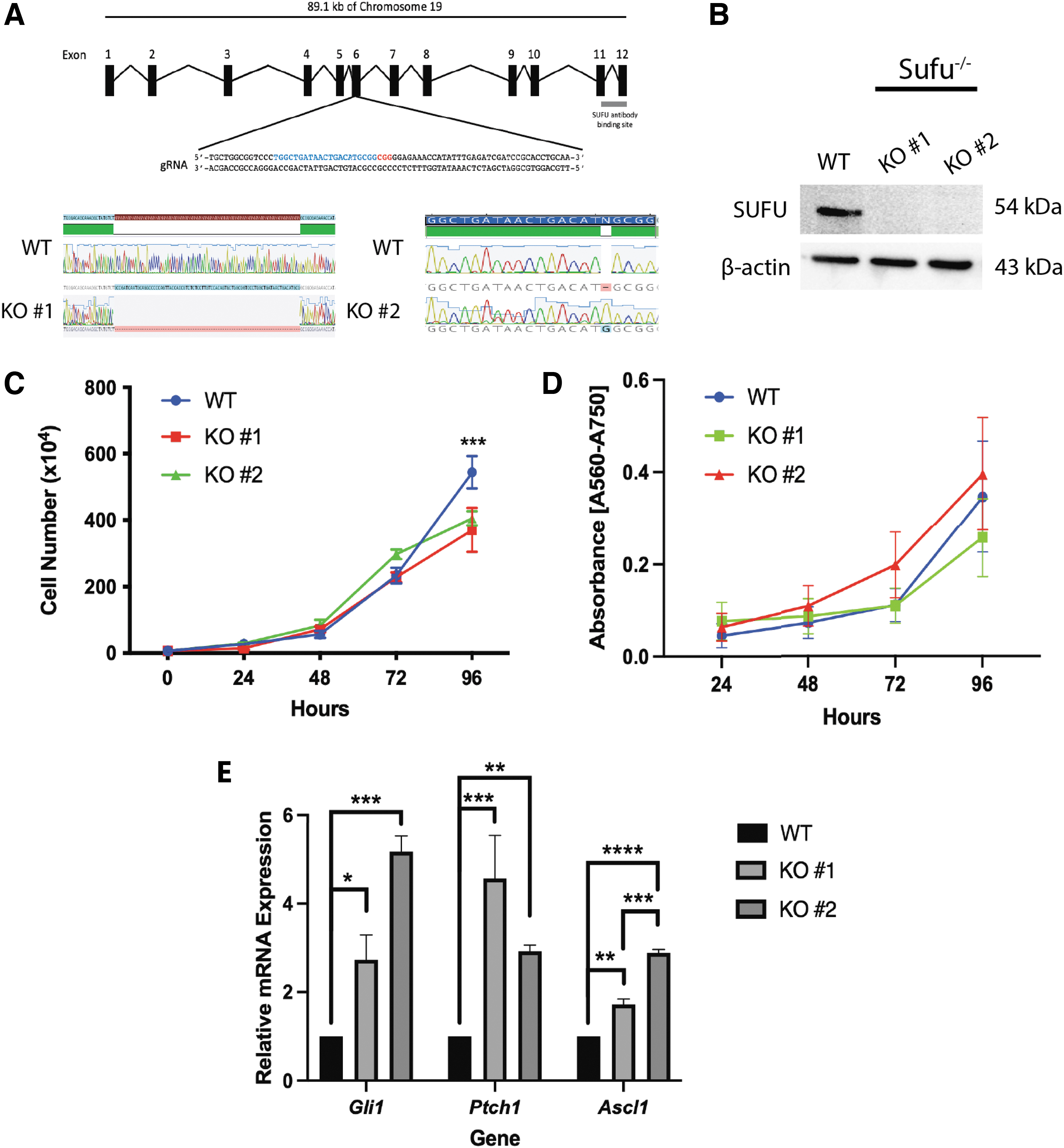
CRISPR-Cas9 knockout of Sufu activates Hh signaling in undifferentiated P19 cells.
A SUFU antibody targeted to the C-terminus of polypeptide, a region required for GLI binding [4,26], was used for immunoblotting analyses and detected a signal in untreated wildtype (WT) cells, but not in either mutant clone (Fig. 4B). SUFU levels were also unchanged in Cas9 vector transfected cells without SUFU-specific gRNA (labeled PX459), in addition to vector transfected cells showing no change in differentiation marker abundance compared to WT cells (Supplementary Fig. S5).
To determine other effects, Sufu−/− alleles caused a decrease in cell numbers compared to WT cells cultured to 96 h (Fig. 4C); however, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) analysis at the same time points showed no change in MTT absorbance (Fig. 4D). As predicted, KOs showed increased expression of Hh targets Gli1, Ptch1, and Ascl1 in undifferentiated cultures (Fig. 4E). Thus, data show the loss of SUFU does not greatly affect the proliferation of undifferentiated cells, contrary to that reported in mouse neural stem cells of the dentate gyrus [27], and instead, it induced Hh target gene expression in P19 cells.
Sufu loss activates Hh signaling through loss of Gli3
RT-qPCR was used to determine if the observed activation of Hh signaling in the KO lines was maintained throughout RA-induced differentiation. Sufu−/− cells treated with RA maintained higher Gli1 (Fig. 5A and Supplementary Fig. S6A) and Ptch1 (Fig. 5B and Supplementary Fig. S6B) expression compared to that in WT cells. Since previous work also showed the connection between SUFU and Gli3 processing [7], we were particularly interested in Gli3 expression and protein levels.

Sufu−
/− maintains increased Hh signaling and causes the loss of Gli3. Expression of
Results show no significant difference in Gli3 expression between Sufu−/− and WT cells in KO no. 2 (Fig. 5C); however, significantly decreased Gli3 levels were observed on days 10, 14, and 17 in KO no. 1 (Supplementary Fig. S6C). At the protein level, neither the Gli3 repressor nor full-length Gli3 was detected in KO no. 2 (Fig. 5D), and this was consistent with KO no. 1 having decreased or absent Gli3 levels (Supplementary Fig. S6D). Thus, the loss of SUFU, specifically the region of the polypeptide required for Gli3 processing, likely contributes to post-translational perturbations to Gli3, causing its loss and the observed higher expression of Hh target genes.
Sufu knockout delays and decreases the astrocyte cell fate
If the Sufu knockout induced Hh target genes, what effect would this have on neural differentiation? To determine if KO cells could differentiate in the absence of RA, these cells were aggregated and replated in the absence or presence of RA. Both KO lines treated with RA showed β-III-tubulin signals on day 10 and GFAP signals on days 14 and 17; neither marker was detected in the absence of RA (Fig. 6A). To determine if the timing or degree of differentiation was affected by the loss of SUFU, the expression of these markers was compared in each knockout line with that in WT cells on days 10, 14, and 17 in the presence of RA (Fig. 6B). Densitometric analyses confirmed β-III-tubulin levels did not change between WT and KO cells (Fig. 6C).
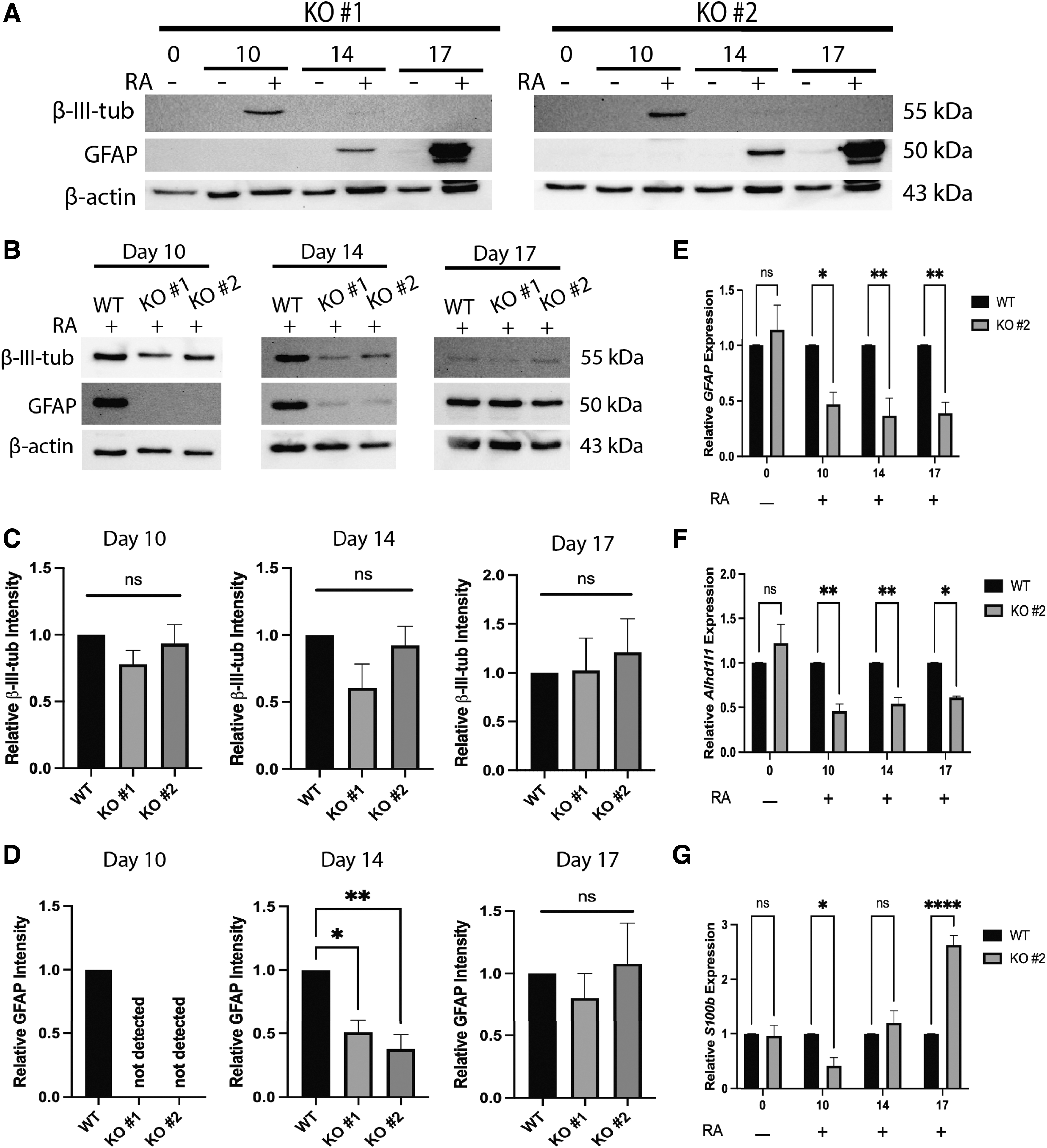
Sufu−/−
delays astrocyte formation without affecting neuron differentiation.
Significantly, astrocyte differentiation was affected and GFAP signals on immunoblots were absent on day 10 and reduced on day 14 of RA treatment in Sufu−/− cells compared to WT (Fig. 6B, D). The absence of GFAP on day 10 was interpreted cautiously, as in WT cells, we have seen inconsistencies in GFAP levels at this time point. Nevertheless, GFAP levels are consistently high in WT cells on day 14 (Figs. 1C, 8A) and it appeared removing SUFU delayed astrocyte differentiation when RA was present. To further confirm this result, RT-qPCR was used to measure expression of astrocyte markers GFAP, aldehyde dehydrogenase 1 family member L1 (Aldh1l1), and S100 calcium-binding protein B (S100b) (Fig. 6E–G, respectively).
Consistent with immunoblotting results, GFAP expression was reduced on days 10 and 14, and unlike the immunoblotting data, was also reduced on day 17 of RA treatment (Fig. 6E). Aldh1l1 expression, like GFAP, was also reduced in KO cells compared to WT on days 10, 14, and 17 of RA treatment (Fig. 6F). Interestingly, S100b expression was reduced on day 10 of RA treatment in KO compared to WT cells; however, it was not different from WT on day 14 and was increased compared to WT on day 17 (Fig. 6G). As S100b is also expressed in oligodendrocytes [28], it is possible the observed increase in expression on day 17 is due to an increase in oligodendrocyte or oligodendrocyte precursor populations; however, this result was not further explored.
To resolve the discrepancy between immunoblotting and RT-qPCR results of GFAP, immunofluorescence analysis with antibodies to β-III-tubulin and GFAP was used on WT and Sufu−/− cells. Given the immunoblotting results (Fig. 6B, D), we were expecting to see no difference in either marker between the two cell lines on day 17 of RA treatment. However, this analysis revealed a reduction in GFAP-positive staining in Sufu− /− cells (Fig. 7A, quantified in B). This observed reduction also occurred, despite the KO cells having increased cell viability (Fig. 7C) and increased total cells (Fig. 7D) as measured by trypan blue exclusion assay on both days 10 and 17 of RA treatment.
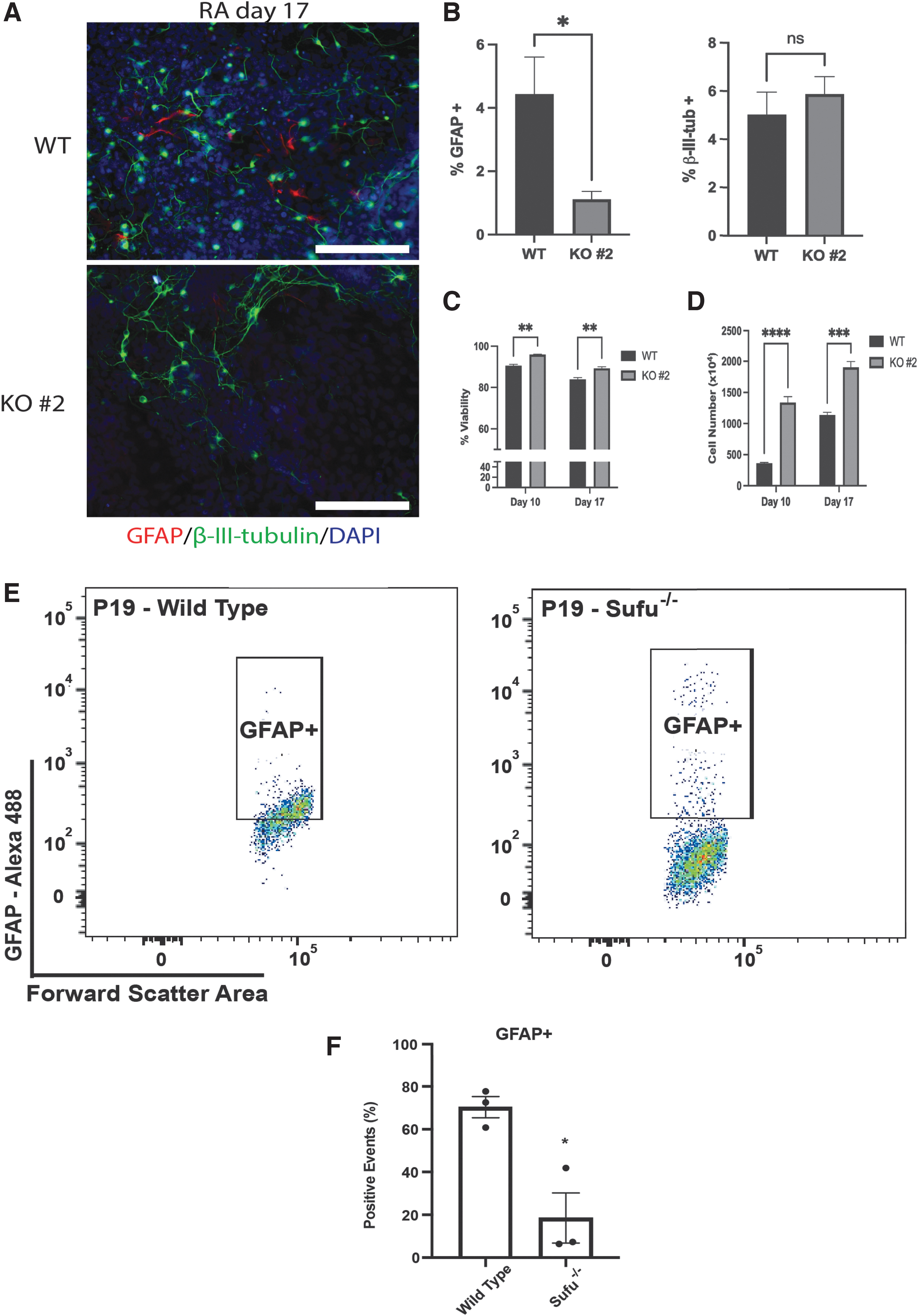
Sufu−/−
decreases astrocyte differentiation.
However, since these cells are highly heterogenous and exist in areas of high density, flow cytometry was used, and results confirmed this reduction showing Sufu−/− cells (∼6%–25%) had significantly less GFAP-positive staining compared to WT cells (∼60%–70%; Fig. 7E, F). It should also be noted that, although the immunoblot data on day 17 was inconsistent with the immunofluorescence and flow analysis, the flow cytometry had identified high GFAP levels (Fig. 7E) in some Sufu−/− cells, even though overall, there were fewer GFAP-positive cells present in the KO line (Fig. 7E). Nevertheless, these data support the idea that the loss of Sufu does not alter neuronal differentiation, but it does delay and decrease astrocyte differentiation.
Sufu or Gli3 overexpression partially rescues astrocyte phenotype
If the loss of Sufu resulted in delayed and decreased astrocyte differentiation (Figs. 6, 7) and this was likely due to the loss of Gli3 (Fig. 5D and Supplementary Fig. S6D), then overexpression of a CRISPR-insensitive chicken Sufu (pSUFU) or human GLI3 (pGli3) in Sufu knockout cells should rescue the phenotype and astrocyte levels should increase in these mouse cells. Overexpression was confirmed by RT-qPCR of chicken Sufu and immunoblotting of human Gli3 (Supplementary Fig. S7). To determine the effect this overexpression had on astrocyte differentiation, GFAP levels were explored following pSUFU or pGli3 stable transfection in knockout cells.
Results showed no GFAP detection on day 10, but there was an increase by day 14 of RA treatment, although not to the levels observed in WT cells (Fig. 8A, B). GFAP levels were maintained on day 17 in the human pGli3 stably transfected cells, but chicken pSUFU stably transfected cells showed significantly reduced levels compared to the WT or other transfected Sufu−/− cells (Fig. 8A, B). Despite these minor differences, these results show that chicken Sufu and human GLI3 overexpression partially rescue astrocyte differentiation and highlight not only the essential role of Gli3 in neural differentiation but also a major role for SUFU in regulating Gli3.
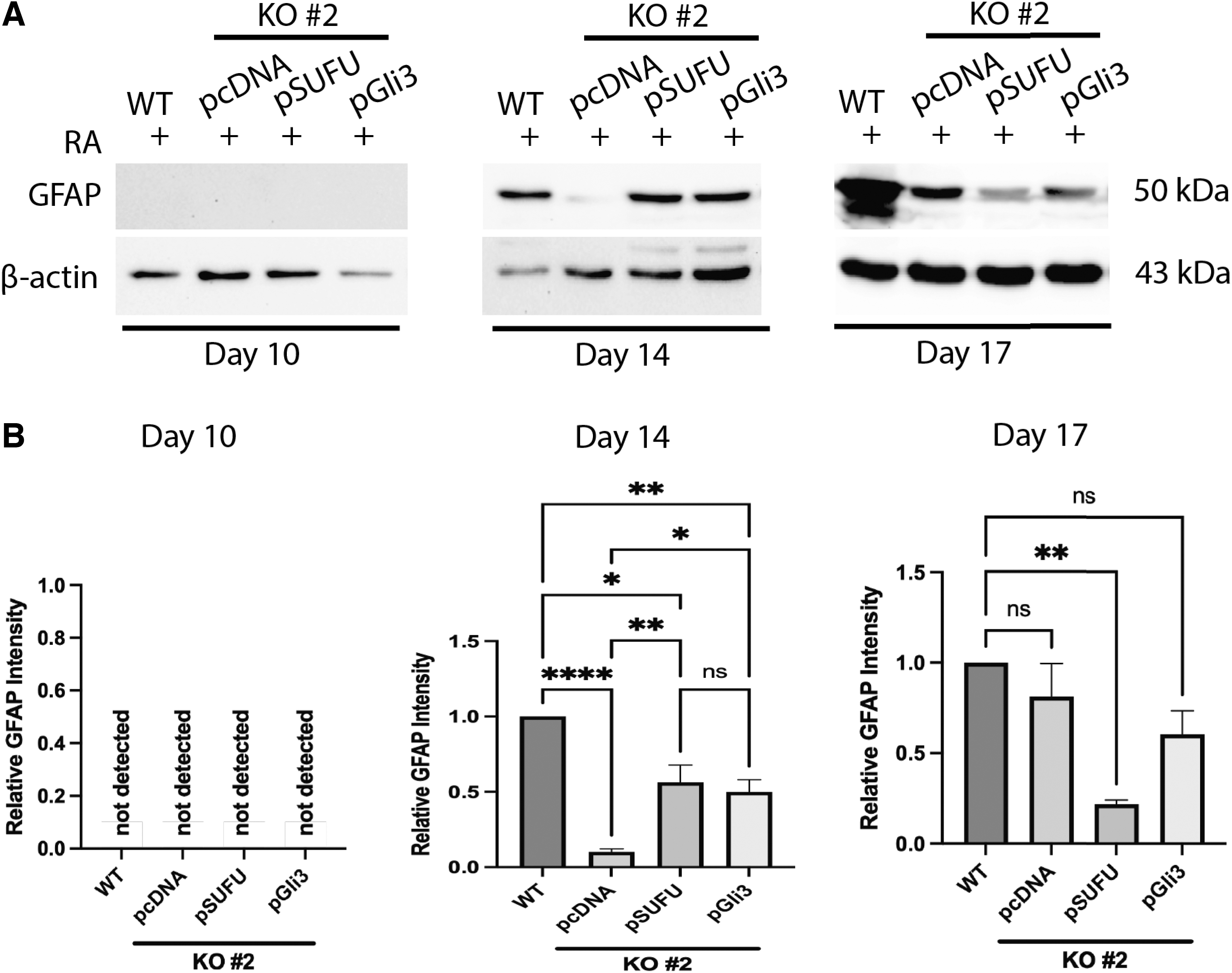
Sufu or Gli3 overexpression partially rescues the Sufu−/−
phenotype.
Discussion
Hh signaling is involved in neural tube closure and neural differentiation in vivo [1,3], where its activation results in transcription of target genes through Glis [3,7]. Negative regulators of the pathway include Patched and SUFU [3], the loss of which results in medulloblastoma and Nevoid Basal Cell Carcinoma Syndrome [13 –15,29]. Using the mouse P19 cell model, we report that Hh pathway activity is required for neural differentiation, but increasing Hh pathway activity alone is not sufficient to induce neural cell fates. Furthermore, SUFU was found to be essential for in vitro neural differentiation as its loss caused the loss of Gli3, the induced transcription of Hh target genes, and perturbed timing and proportion of astrocyte differentiation. Furthermore, human Gli3 overexpression was sufficient to rescue GFAP, the astrocyte marker, in these SUFU-deficient mouse cells, which indicates that Gli3 acts downstream or in parallel to the negative regulation imparted by SUFU.
RA-induced neural differentiation of P19 cells is well established [18,19,25,30]; however, few studies have explored Hh signaling in the model [20,21,31]. Voronova et al. [20] also explored expression of Gli transcription factors during RA-induced neurogenesis, and we have added to that work by reporting timeline expression of all components of the pathway. Specifically, we demonstrated that RA induced Shh expression (Fig. 2A), where RA likely acts in these cells through the transcription factor FOXA1, which promotes the early expression of Shh [31]; two other messages encoding Hh ligands (Dhh and Ihh) were also expressed (Fig. 2A).
IHH and DHH, like SHH, can activate the pathway as evident in long bone differentiation [32,33] and gonad development [34 –36]. Similar results showing transcriptional activation of all Hh ligands was reported with human embryonic stem cells (hESCs) induced to form neural tissues [37]. Thus, it is likely that SHH is responsible, but since transcripts encoding all ligands are induced by RA, it remains unclear which isoform triggers pathway activation in the P19 model. Although we, like others, have demonstrated the presence of Hh signaling in P19 differentiation, other signaling pathways, including those linked to Wnt, Fgf, and Yap are also involved [21,38 –40]. Considering our study, these other pathways require further investigation to determine if they too are temporally regulated to facilitate differentiation.
Utilization of RA to induce neural lineages in vitro goes beyond the P19 model, as RA is used for directed neural differentiation of human induced pluripotent stem cells and mouse embryonic stem cells (mESCs) [41,42]. Many neural differentiation protocols also supplement RA with exogenous SHH or SAG to enhance the differentiation of neurons [42 –44] and glial cell progenitors [45]. Like these previous results, co-treating P19 cells with RA and SAG on day zero in our protocol enhanced the differentiation of both neurons and astrocytes (Fig. 3), although treating with SAG alone was not sufficient to induce differentiation. We also investigated the effect of inhibiting the Hh pathway using Cyc, where Cyc treatment inhibited neuron and astrocyte differentiation (Fig. 3), a result consistent with previous work showing Cyc co-treatment with RA decreased expression of neural-related transcription factors in mESCs [44].
In vivo studies also highlight the requirement of Hh signaling in neural differentiation and development, where mutations reducing Hh activity cause reduced neuron differentiation in the embryonic midbrain [46], reduced neural progenitors in the developing cerebellum [47], reduced neurogenesis of the developing telencephalon [48,49], or large-scale alterations to the size of interneuron differentiation regions in the neural tube [50,51]. Thus, the early activation of Hh signaling in the differentiation of neural lineages is a requirement, and we next sought to understand the importance of later-stage reduction of the Hh signal by SUFU.
SUFU regulation of Hh signaling involves the ubiquitin-proteasome pathway following Hh pathway activation [52]. We showed Hh ligand expression is induced within 1 day of RA treatment (Fig. 2) and given previous reports on SUFU regulation, an early decrease in SUFU levels was expected. Our results, however, showed no significant change in SUFU levels during any stage of neurogenesis in P19 cells (Supplementary Fig. S3). Thus, it is likely that SUFU is being regulated through post-translational modifications to affect Hh signaling output, and although not explored in this study, it should be noted that differential phosphorylation and ubiquitination can both promote or inhibit SUFU stability and/or Gli interaction [53 –56].
CRISPR-Cas9, used to deplete SUFU from P19 cells, showed the loss of SUFU induces the ectopic transcription of Hh target genes (Figs. 4, 5) and while supported by studies using mice, mESCs and hESCs [15,37,57 –59], this is in contrast with a study showing reduced Hh target expression in adult neural stem cells [27] and in another showing loss of Sufu reducing maximal Hh pathway activity in the developing mouse spinal cord [60].
Given previous work showing Gli2 overexpression in P19 cells is sufficient to induce neuron differentiation in the absence of RA [20], we were surprised to observe SUFU-deficient P19 cells did not differentiate toward the neuron lineage in the absence of RA (Fig. 6A). That another study showed the knockdown of Gli2 in YAP overexpressing P19 cells causes an increase in neuron differentiation [21], would indicate that other Glis may be involved in the neural differentiation of P19 cells, specifically Gli3.
The connection between aberrant astrocyte differentiation and SUFU deficiency came from examining the transcriptional activation of Hh target genes (Figs. 4, 5). Gli1 and Ptch1 expression were higher when Sufu was knocked out, but not Gli3 (Fig. 5). Previous reports in mouse embryonic fibroblasts and mouse embryos show that SUFU is required for proper Gli3 processing [61] and its absence leads to reduced or absent Gli3 protein [9,57], which also occurs with conditional Sufu knockouts in the embryonic and post-natal mouse brain [16,17,62,63]. We reported similar results with Gli3 in P19 cells and showed that it affects neural differentiation. In support, conditional knockouts of Sufu at various stages of mouse brain development result in decreased neuron differentiation and disorganized glial cell arrangements in the cerebellum and forebrain [16,62].
Discrepancies, however, do exist even within Hh signaling in P19 cells [20,21], and although these differences can be explained, our work has shown the knockout of Sufu was accompanied by the consistent upregulation of Hh target genes, like that seen in mouse retinal progenitor cells [12], mESCs [58] and the developing mouse neocortex [62] lacking SUFU. This upregulation did not result in neuron differentiation without RA treatment (Fig. 6) and this result is supported by work in SUFU-deficient mESCs [58] and SHH-treated hESCs [37] maintaining their pluripotent state without additional differentiation cues.
Our results did show a delay and decrease in astrocyte differentiation with SUFU loss (Figs. 6, 7), which may explain the disorganization of glial cells observed in conditional Sufu knockouts described in mouse embryos [16,62]. However, previous work has shown the same reduction in astrocyte differentiation in P19 cells overexpressing a dominant negative form of Gli2, thus causing a reduction in Hh pathway activity [20]. This, combined with our work, shows that Hh signaling is induced by RA and this induction is required for differentiation.
The Hh pathway, however, must return to basal activity levels for proper astrocyte differentiation to occur. Given that glial cell populations are composed of astrocytes, oligodendrocytes, and microglia, further research must be done to explore the effect of SUFU loss on these other lineages. Initial work in the developing mouse forebrain suggests an essential role of SUFU in oligodendrocyte differentiation [64], where a reduction in Sufu levels promotes oligodendrocyte precursors [65]. We reported an increase in astrocyte and oligodendrocyte marker S100b [28] on day 17 in SUFU-deficient cells (Fig. 6); thus, future work with this model must explore the oligodendrocyte lineage.
In addition to the delay in astrocyte formation (Figs. 6, 7), there was a loss of Gli3 in SUFU-deficient cells (Fig. 5) and we have provided evidence that ectopic expression of Gli3 was sufficient to rescue the timing of glial cell differentiation (Fig. 8). That GLI3 was able to rescue the Sufu knockout phenotype is significant and supported by previous research showing Gli3 repressor overexpression in the SUFU-deficient embryonic mouse cerebellum was sufficient to rescue morphology and increase GFAP levels [17]. Together, our results show that SUFU imparts a negative regulation on the Hh pathway, likely by acting on Gli3, and is required during the specification of the astrocyte lineage.
Footnotes
Acknowledgments
The authors would like to thank members past and present of the Kelly laboratory for helpful discussions.
Author Disclosure Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
Funding Information
This research was funded by the Natural Sciences and Engineering Research Council (NSERC) of Canada, Discovery Grant R2615A02 to G.M.K. D.M.S. acknowledges support from the School of Graduate and Postdoctoral Studies, University of Western Ontario; the Collaborative Graduate Specialization in Developmental Biology, University of Western Ontario; the Children's Health Research Institute; and NSERC for a PGS D scholarship.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.