
Abstract
Adult-derived mesenchymal stem cells (MSCs) can be used in therapies for the treatment of various diseases. The MSCs derived from aging tissues or long-term MSC cultures could have diminished therapeutic effects compared with MSCs derived from younger tissues, but the underlying mechanism has not been completely established. Dysfunction of energy metabolism is one of the main mechanisms underlying cell senescence. Although cyclic adenosine monophosphate (cAMP) is known to inhibit cell division and proliferation in vitro, its impact on MSC senescence has not been described. In this study, we used forskolin, an adenylate cyclase agonist and cAMP inducer, to disrupt metabolism in human adipose-derived MSCs and investigate the effects of metabolic dysfunction on MSC senescence. Treatment of human MSCs with forskolin resulted in senescence phenotypes, including reduced proliferation, cell-cycle arrest, and enhanced expression of the cell aging markers p16 and p21. Further, the senescent MSCs exhibited increased adipogenesis capacity and decreased osteogenesis capacity as well as a senescence-associated secretory phenotype characterized by increased expression of several inflammatory factors. Forskolin-associated MSC senescence was mainly caused by oxidative stress–induced disruption of mitochondrial metabolism, and the senescent MSCs had high levels of reactive oxygen species and reduced sirtuin gene expression. Lastly, we found that cAMP inhibitor SQ22536 protects MSCs from forskolin-induced senescence and senescence-related inflammatory phenotype. Our results indicate that forskolin can cause senescence of human MSCs through oxidative stress–induced mitochondrial metabolic dysfunction, and thus the results provide a basis for developing strategies for improving the quality and efficacy of cultured MSCs for clinical use.
Introduction
Adult mesenchymal stem cells (MSCs) have multiple applications for the treatment of various diseases, mainly owing to their powerful immunomodulatory and regenerative features, paracrine effects, multi-lineage differentiation potential, and migration ability. Because the human body has relatively few MSCs, MSCs are typically expanded in vitro to obtain a sufficient number of cells for clinical applications.
During culture in vitro, MSCs may undergo senescence owing to their limited capacity for continuous cell division [1 –5]. Aged MSCs generally have relatively reduced efficacy compared with younger MSCs in various disease models, and the donor's age and/or health status may limit the therapeutic potential of the harvested MSCs [6,7]. This can be partly attributable to the fact that senescent MSCs may produce inflammatory cytokines, chemokines, and/or extracellular matrix–remodeling factors that can disrupt tissue structure and function.
Cellular metabolic dysfunction may reflect cellular senescence, tissue aging, and several aging-related diseases such as Alzheimer's disease, Parkinson's disease, stroke, and osteoarthritis [8 –12]. The cyclic ADP-ribose hydrolase CD38 regulates metabolism during aging by modulating cellular NAD+ level and the activity of the histone deacetylase sirtuin 3 (SIRT3) [13]. Agonists of the nuclear receptor PPARγ have shown therapeutic potential for certain age-related brain disorders [14].
Cyclic adenosine monophosphate (cAMP) is a second-messenger molecule that plays important roles in various cellular physiological and pathological processes, including metabolism, cell differentiation, and inflammation [15]. Forskolin, a natural compound produced in the roots of the Indian plant Coleus forskohlii, directly activates adenylate cyclase, which generates cAMP from ATP, thereby increasing the intracellular concentration of cAMP [16].
As an adenylate cyclase agonist, forskolin inhibits cell proliferation, motility, colony formation, and migration, as well as promotes apoptosis and improves the sensitivity of various types of tumor cells to chemotherapeutic drugs; therefore, forskolin has applications for the treatment of several cancers [17 –21].
In this study, we carried out experiments to determine the effects of the cAMP agonist forskolin on MSC senescence and studied the underlying mechanism. We showed that forskolin treatment increased intracellular cAMP level, decreased ATP level, and induced senescence phenotypes of MSCs. Mechanistically, forskolin treatment disrupted mitochondrial metabolism of MSCs and the senescent MSCs exhibited reduced expression of the longevity-related sirtuin gene family.
The forskolin-induced senescence phenotypes could be rescued by a adenylate cyclase inhibitor SQ22536, but not an NF-κB inhibitor pyrrolidine dithiocarbamic acid (PDTC). Thus, our study not only revealed the effects and mechanisms of forskolin-induced MSC senescence but also provided a research model for future studies to further investigate how to prevent MSCs from senescence.
Materials and Methods
Preparation and treatment of MSCs
The MSCs were isolated from discarded adipose tissue removed from healthy patients undergoing liposuction surgery, as previously described [22]. The median age of patients was 25, and informed consent was obtained from all patients. The procedure was approved by the Ethics Committee of the Chinese Academy of Medical Sciences and Peking Union Medical College. The isolated cells were seeded in T75 flasks containing 12 mL culture medium and incubated under humidified conditions at 37°C and 5% CO2.
The MSCs were continuously cultured to the third passage and then used for experiments. Cells were either left untreated or treated for 24 h with forskolin (60 μM; Target Mol, Boston, MA). NF-κB inhibitor PDTC (100 μM; Beyotime, Shanghai, China) or adenylate cyclase inhibitor SQ22536 (30 μM; Abcam, Cambridge, United Kingdom) was added to study their effects on MSCs senescence.
MTS assay
An MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) assay kit (Sigma-Aldrich, St. Louis, MO) was used to assess cell viability. The MSCs were seeded into five 96-well plates at a density of 2 × 103 cells per well, with each plate being cultured for 1, 24, 48, 72, or 96 h. One hour before each time point, 20 μL MTS reagent was added into each well of the 96-well plate containing 100 μL culture medium. After incubation at 37°C for 1 h, absorbance at 490 nm was measured using a 96-well plate reader (Bio-Rad, Hercules, CA).
Assessment of senescence using β-galactosidase staining
A senescence β-Galactosidase staining kit (Beyotime) was used for staining senescent cells based on the upregulation of senescence-associated β-galactosidase activity during senescence. Culture medium was aspirated from six-well plates, and the wells were washed once with phosphate-buffered saline (PBS). The cells were then fixed at room temperature for 15 min with 1 mL β-galactosidase staining fixative solution per well and then washed three times with PBS. Finally, 1 mL of working solution of the stain was added to each well, and the plates were incubated overnight at 37°C in a CO2-free incubator. The cells were then observed and counted under an optical microscope.
Quantification of cAMP
The intracellular concentration of cAMP was determined with a cAMP enzyme-linked immunosorbent assay (ELISA) kit (Elabscience, Wuhan, China), which uses a competitive ELISA method. Briefly, wells of a microtiter plate were coated with cAMP (the antigen), and the cAMP present in each cell extract or standard solution was allowed to compete with the immobilized cAMP for binding to a biotin-labeled monoclonal antibody against cAMP.
Unbound components were then washed away. Horseradish peroxidase (HRP) conjugated avidin was then added, resulting in the formation of an immune complex; the solution in each well was aspirated; and the plates were washed three times with PBS. The chromogenic substrate 3,3′ 5,5′-tetramethylbenzidine was then added, which turned the solution blue on catalysis by HRP and then yellow after the stop solution was added. Finally, absorbance at 450 nm was measured with a microplate reader, and the cAMP concentration in each sample was calculated based on a standard curve.
Cell-cycle analysis
Cell-cycle phase was determined by flow cytometry using propidium iodide staining. The MSCs were seeded into 6-well plates at a density of 2 × 105 cells per well and then treated with dimethyl sulfoxide (DMSO; vehicle control) or forskolin (60 μM final concentration) for 24 h. Cells were then harvested and fixed in 1 mL of 75% ethanol at 4°C for 24 h. The fixed cells were then washed twice and resuspended in 50 μL PBS, followed by addition of 5 μL RNase A (10 mg/mL) and incubation at 37°C for 30 min. Next, 450 μL PBS with 10 μL propidium iodide (1 mg/mL) was added, and the samples were incubated at 4°C in the dark for 30 min before being detected with a flow cytometer.
Transmission electron microscopy
The MSCs were treated with DMSO or forskolin and then fixed in 2% glutaraldehyde solution in 0.1 M cacodylate buffer for 24 h, post-fixed in 1% osmium tetroxide for 1 h, and dehydrated in sequential dilutions of acetone (25%, 50%, 75%, and 100% twice) before impregnation in resin of increasing concentration (25%, 50%, 75%, and 100% three times) in acetone over a 24-h period. Each sample was subjected to transmission electron microscopy, and mitochondrial morphology was assessed and imaged at magnifications of 8,000 × and 10,000 × .
Measurement of intracellular ATP content
Intracellular ATP content was measured with an ATP assay kit (Beyotime). Briefly, cells were lysed and then centrifuged at 12,000 g for 10 min at 4°C. The supernatant was removed and mixed with dilution buffer from the kit, after which the relative light units were measured using a luminometer. A standard curve was prepared for each round of assays to calculate ATP content.
Measurement of intracellular reactive oxygen species
Mitochondrial reactive oxygen species (ROS) formation was assayed using 2,7-dichlorofluorescein diacetate, a fluorescent probe, as part of an ROS assay kit (Beyotime) with a slight procedural modification. Briefly, MSCs were incubated with the probe (10 μM) dissolved in serum-free Dulbecco's modified Eagle medium at 37°C for 20 min. Fluorescence signal was measured with a flow cytometer.
Mitochondrial superoxide assay
Mitochondrial superoxide (MitoSOX) formation was assayed with the MitoSOX Red kit (Yesen, Shanghai, China). First, 13 μL DMSO and 50 μg MitoSOX Red (the probe) were mixed well to prepare a 5 mM MitoSOX Red working solution. Next, a 5 μM probe working solution was prepared by diluting the MitoSOX Red working solution with serum-free culture medium. The cells were washed twice with PBS and resuspended with 2 mL of the probe working solution. After incubation at 37°C for 10 min in the dark, the cells were washed three times with PBS. Fluorescence was measured with a flow cytometer.
Quantification of nitric oxide
Nitric oxide was detected with 3-amino,4-aminomethyl-2′,7′-difluorescein, diacetate (DAF-FM DA; Beyotime). DAF-FM DA was first diluted with the DAF-FM DA diluent provided in this kit to a final concentration of 5 μM. Then, the cells were collected and incubated with diluted DAF-FM DA for 20 min at 37°C. Cells were washed with PBS to remove excess probe before fluorescence signal was measured with a flow cytometer.
Oxygen consumption analysis
The Seahorse XF24 Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA) was used to measure oxygen consumption in MSCs. Forskolin-treated and control MSCs were plated onto 24 well XF cell culture microplates at a density of 50,000 cells/well in 250 μL culture medium on the day before analysis. XF assay medium was supplemented with 250 mM glucose and 2 mM sodium pyruvate. The sensor cartridge was hydrated in XF Calibrant at 37°C in a non-CO2 incubator overnight.
After basal respiration measurements, cells were treated sequentially with oligomycin (1 mM) to measure the non-phosphorylating oxygen consumption rate (OCR), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (0.5 mM) to determine maximal OCR, and rotenone and antimycin A mixture (0.5 mM) to measure the residual mitochondrial OCR, and changes in respiration were recorded. Results were analyzed using the default settings in Wave or XF software.
Wound-healing assay
Wound-healing assays were performed using Culture-Insert two-well inserts (Ibidi, Munich, Bavaria, Germany). Briefly, the inserts were placed in six-well plates before seeding the cells and were removed after the cells formed a confluent layer (12–24 h after seeding). The remaining medium was replaced with fresh serum-free medium, and images were acquired after another 24 h.
Alizarin red staining
Cells plated in 24-well plates were washed twice with PBS and fixed with 4% paraformaldehyde for 10 min. Then, the cells were washed with distilled water and stained with 1% Alizarin red (Leagene, Beijing, China) with pH 4.2 for 30 min. The cells were rinsed with distilled water again to remove unbound dye, then observed, and photographed under a light microscope.
Oil red O staining
Cells plated in 24-well plates were washed twice with PBS, fixed with 4% paraformaldehyde for 10 min, and stained with filtered oil red O solution (stock solution: 1 mg/mL in isopropanol; working solution: 60% oil red O stock solution mixed with 40% distilled water) at 37°C for 30 min. The cells were rinsed with distilled water to remove unbound dye and then observed and photographed under a light microscope.
RNA isolation and quantitative reverse transcription–polymerase chain reaction analysis
Total RNA was extracted from cultured cells with TRIzol reagent (Invitrogen, Carlsbad, CA), and the RNA was quantified with a NanoDrop ND1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). Reverse transcription was performed using a Reverse Transcription kit (Takara, Tokyo, Japan). Quantitative polymerase chain reaction (qPCR) was performed with SYBR premix Ex Taq (Takara) with the Step One Plus Real-Time PCR Detection System (Applied Biosystems, Foster City, CA). The relative expression of mRNA was evaluated by the 2−ΔΔCt method and normalized to the expression of glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Supplementary Table S1 lists the primers used in this study.
Western blotting
Cells were harvested in RIPA lysis buffer (Beyotime) supplemented with 1 mM phenylmethanesulfonyl fluoride. Total protein was quantified using a BCA Protein Assay kit (Beyotime). Cell extracts (20 μg protein) were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE; 10% acrylamide gels) and then transferred to a 0.45-μm polyvinylidene difluoride membrane (Millipore, Boston, MA).
After blocking with 5% fat-free milk in Tris-buffered saline Tween-20 for 1 h at room temperature, each membrane was incubated with a specific primary antibody at 4°C overnight. The next day, each membrane was incubated with an HRP-conjugated secondary antibody for 1 h at room temperature. Immunopositive bands were visualized with HRP substrate (Proteintech, Rosemont, IL) and quantified using an Image Quant LAS 4000 mini-imaging system (GE Healthcare, Chicago, IL). GAPDH was used as an internal control. Primary antibodies (rabbit IgG; from Proteintech) were used that targeted the following proteins: p16 (diluted 1:1,000), p21 (1:1,000), and lamin B1 (1:2,000).
Statistical analysis
Data were analyzed using Prism 8 software (GraphPad Software, Inc.). Unless annotated otherwhere, the Student's t-test was used for comparisons between groups. P values of <0.05 were considered to reflect statistically significant differences between data. Error bars indicate standard deviation.
Results
Forskolin increases cAMP level and reduces ATP level in MSCs
To investigate the effects of forskolin on MSC senescence, we first treated MSCs with an increasing concentration of forskolin for 24 h and then performed β-galactosidase staining as an indicator of senescence. Forskolin treatment induced senescence at a dose-dependent manner and reached a plateau at 60 μM (Fig. 1A). To further validate the effect of forskolin on MSC senescence that was dependent on its function as a cAMP inducer, we detected the cAMP and ATP productions.

Increase in cAMP level and decrease in ATP level in forskolin-treated MSCs.
The cAMP level in MSCs treated with forskolin was ∼3-fold greater than that in control MSCs after 24 h, whereas this difference subsided after 48 h (Fig. 1B). On the other hand, forskolin treatment significantly reduced intracellular ATP level after 24 h, and a similar difference was maintained till 48 h (Fig. 1C). Thus, the effect of 60 μM forskolin as a cAMP inducer reached the maximum at 24-h post-treatment.
Forskolin leads to a senescence phenotype in MSCs
Certain cAMP-elevating agents and cAMP analogs can inhibit cell proliferation [23]. We found that a 24-h incubation with forskolin significantly decreased MSC proliferation compared with control MSCs (Fig. 2A). Cell-cycle analysis of forskolin-treated MSCs revealed a significant arrest at the S and G2+M phases (Fig. 2B). The effects of forskolin on MSC senescence were further supported by the forskolin-induced upregulation of molecular markers associated with cell aging, namely p16 and p21, and downregulation of lamin B1 (Fig. 2C, D).
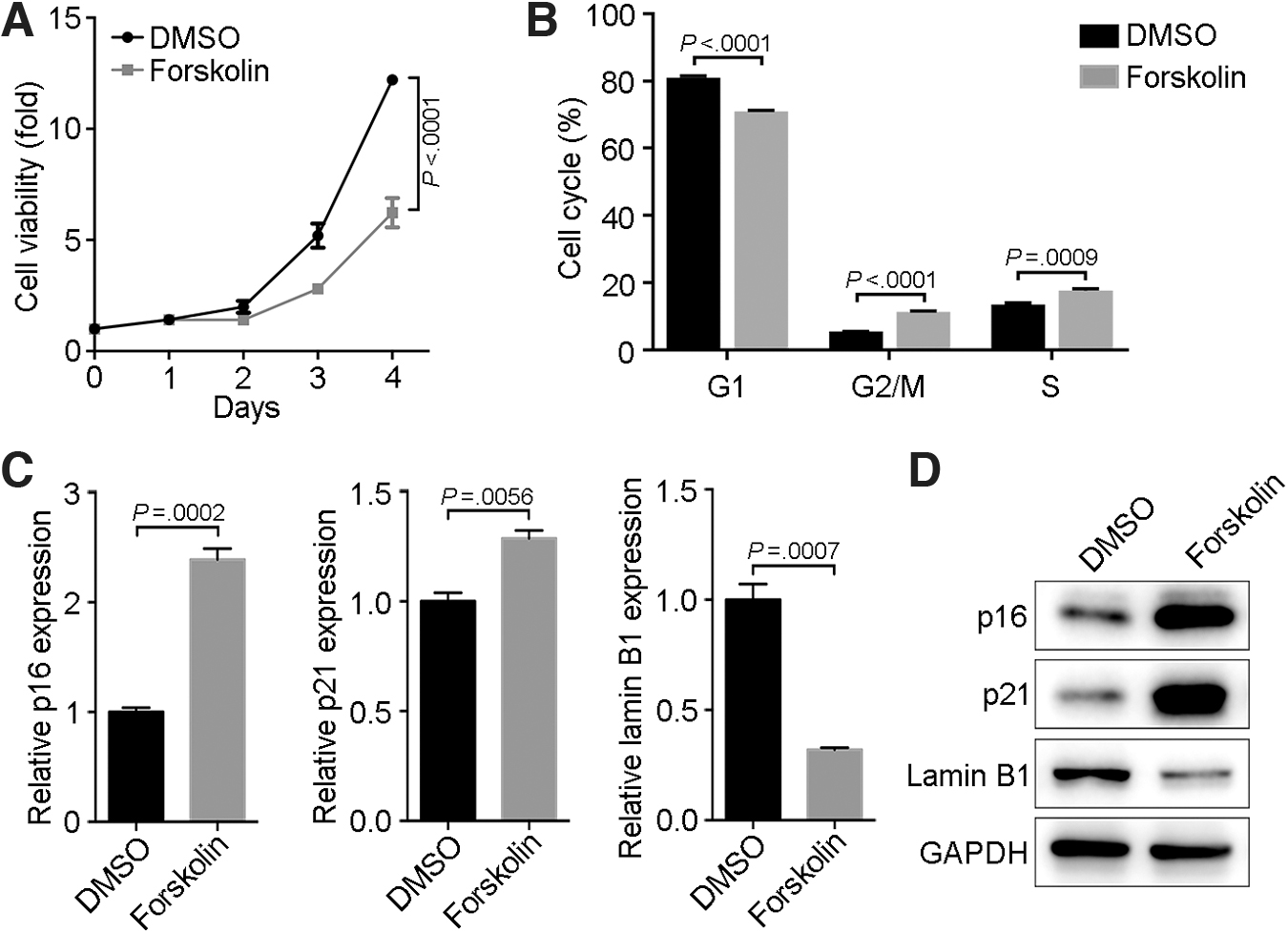
Forskolin causes a senescence phenotype in MSCs.
Forskolin alters the differentiation capacity of MSCs
We next studied the immunophenotype of the MSCs after forskolin treatment. Both the forskolin-treated and control MSCs expressed CD44 and CD29 but not HLA-DR, CD45, and CD31 (Fig. 3A). Oil red O staining is commonly used to detect lipogenesis, and alizarin red staining is commonly used to detect osteogenesis. Forskolin-treated MSCs showed stronger Oil red O staining (Fig. 3B) yet weaker alizarin red staining (Fig. 3C).
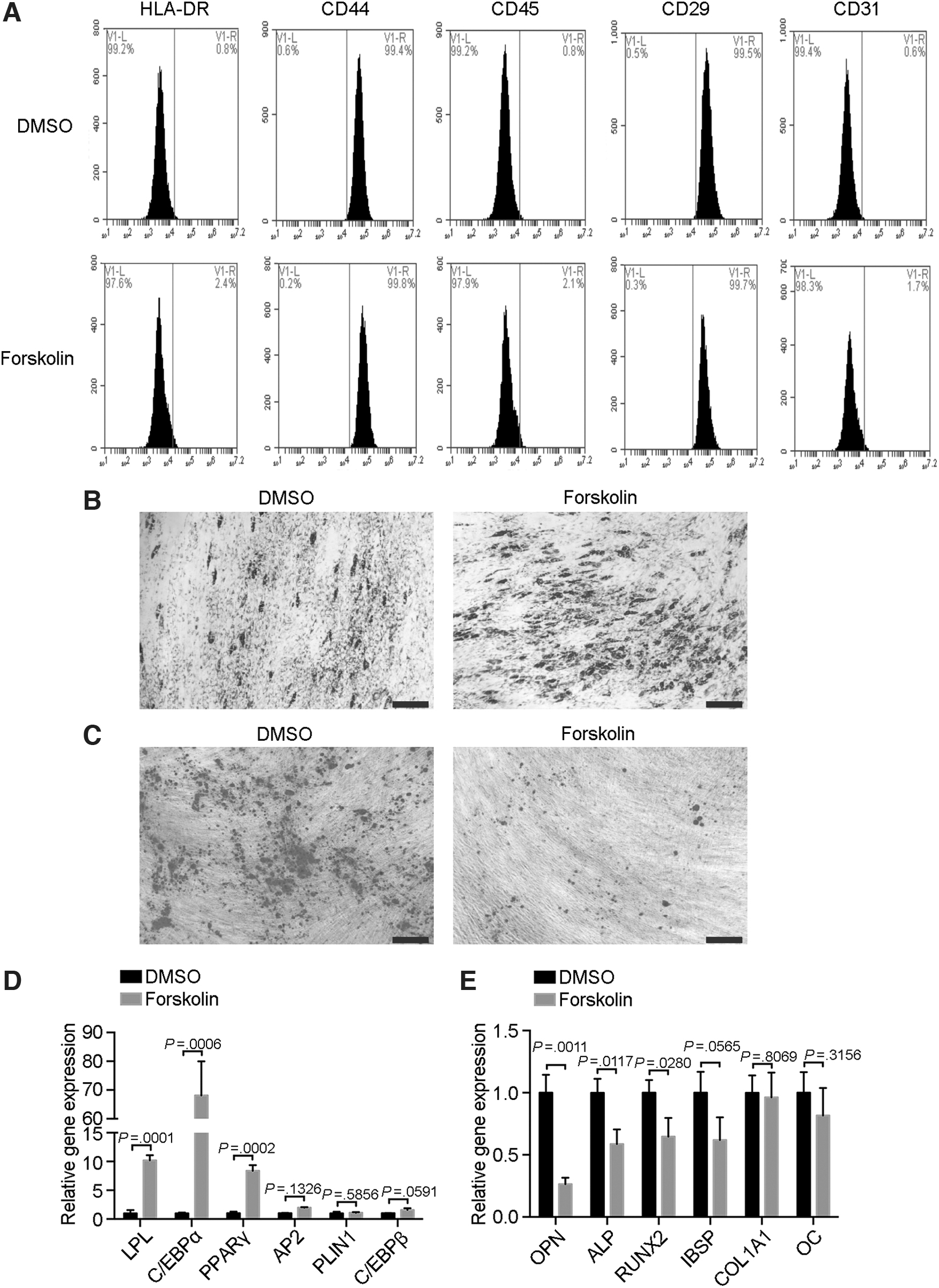
Immunophenotype and differentiation abilities of forskolin-treated senescent MSCs.
Moreover, the early adipocyte genes LPL, C/EBPα, and PPARγ were significantly upregulated compared with the control groups (Fig. 3D), and the osteogenesis genes OPN, ALP, and RUNX2 were downregulated (Fig. 3E). Thus, forskolin-treated MSCs had a normal immunophenotype but greater adipogenic capacity and lesser osteogenic capacity than the control MSCs.
Forskolin induces senescence-related transcriptional reprogramming
To determine the mechanism underlying the effects of forskolin on MSCs, we exposed MSCs to 60 μM forskolin for 24 h and identified 3,154 upregulated and 2,731 downregulated genes, indicating that forskolin triggered global transcriptional reprogramming in MSCs (Fig. 4A). Gene Ontology enrichment of differentially expressed genes revealed cAMP catabolic processes and cAMP-mediated signaling as two of the top 4 activated processes (Fig. 4B), and thus forskolin, indeed, targeted the expected pathways in our experiment.
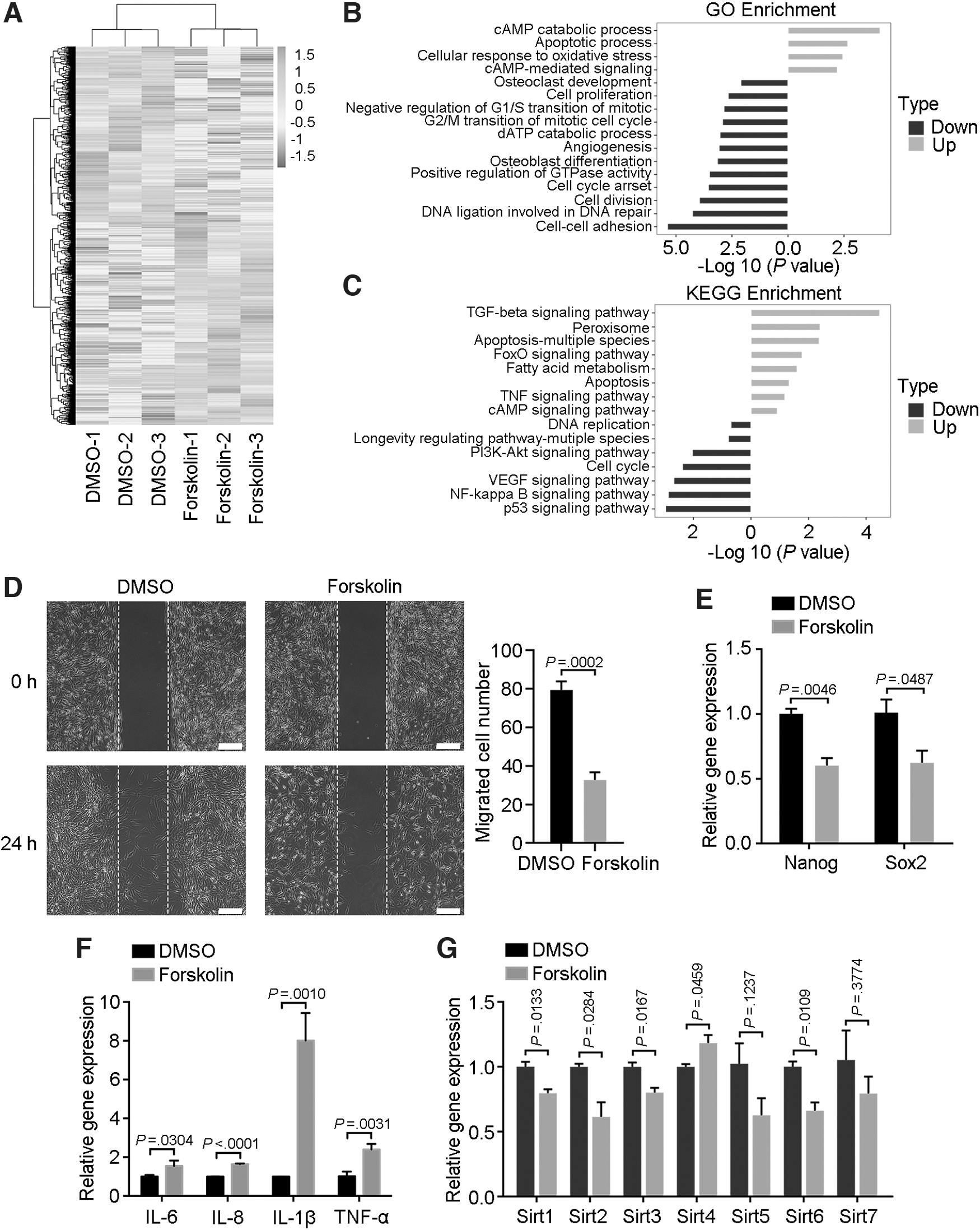
Cellular functions of senescent MSCs.
Two other activated processes, namely apoptosis and oxidative stress response, also supported our result that forskolin triggered MSC senescence (Fig. 4B). Several senescence- and differentiation-related processes also differed between forskolin-treated and control MSCs, such as osteoblast differentiation, cell proliferation, and cell-cycle processes (Fig. 4B). Moreover, several of the top 15 enriched KEGG pathways were related to senescence (Fig. 4C). Interestingly, certain inflammation-related pathways differed significantly between the two groups of cells, such as the NF-κB signaling and TNF signaling pathways (Fig. 4C).
Because cell–cell adhesion was the most significantly downregulated Gene Ontology process (Fig. 4B), we analyzed the effect of forskolin on MSC migration. In wound-healing assays, the number of migrated cells in the forskolin group was significantly less than that of control MSCs (Fig. 4D). Senescent cells typically exhibit characteristics of producing inflammatory factors, and thus they are often referred to as having a senescence-related secretory phenotype (SASP) [24,25].
Quantitative reverse transcription–polymerase chain reaction (RT-qPCR) was, thus, performed to assess whether forskolin treatment resulted in an SASP. The MSCs treated with forskolin expressed lower levels of the pluripotent genes encoding Nanog and Sox2 (Fig. 4E) but much higher levels of several proinflammatory factors, including IL-6, IL-8, IL-1β, and TNF-α (Fig. 4F), than did the control MSCs. Further, the longevity genes encoding SIRT1, SIRT2, SIRT3, and SIRT6 were downregulated in forskolin-treated cells, confirming cellular aging (Fig. 4G). Together, these results suggested that forskolin caused global transcriptional reprogramming in MSCs that contributed to senescence-related cellular functions.
Forskolin disrupts mitochondrial metabolism in MSCs
We next studied mitochondrial function, which is key for regulating cell viability, in forskolin-treated MSCs. Forskolin-treated MSCs had atypical mitochondria that were untethered, swollen, yet small, compared with mitochondria in control MSCs (Fig. 5A). Mitochondrial metabolism–associated functions were downregulated in forskolin-treated MSCs, including the ATP production, basal and maximal respiration, and the proton gradient (Fig. 5B–D).
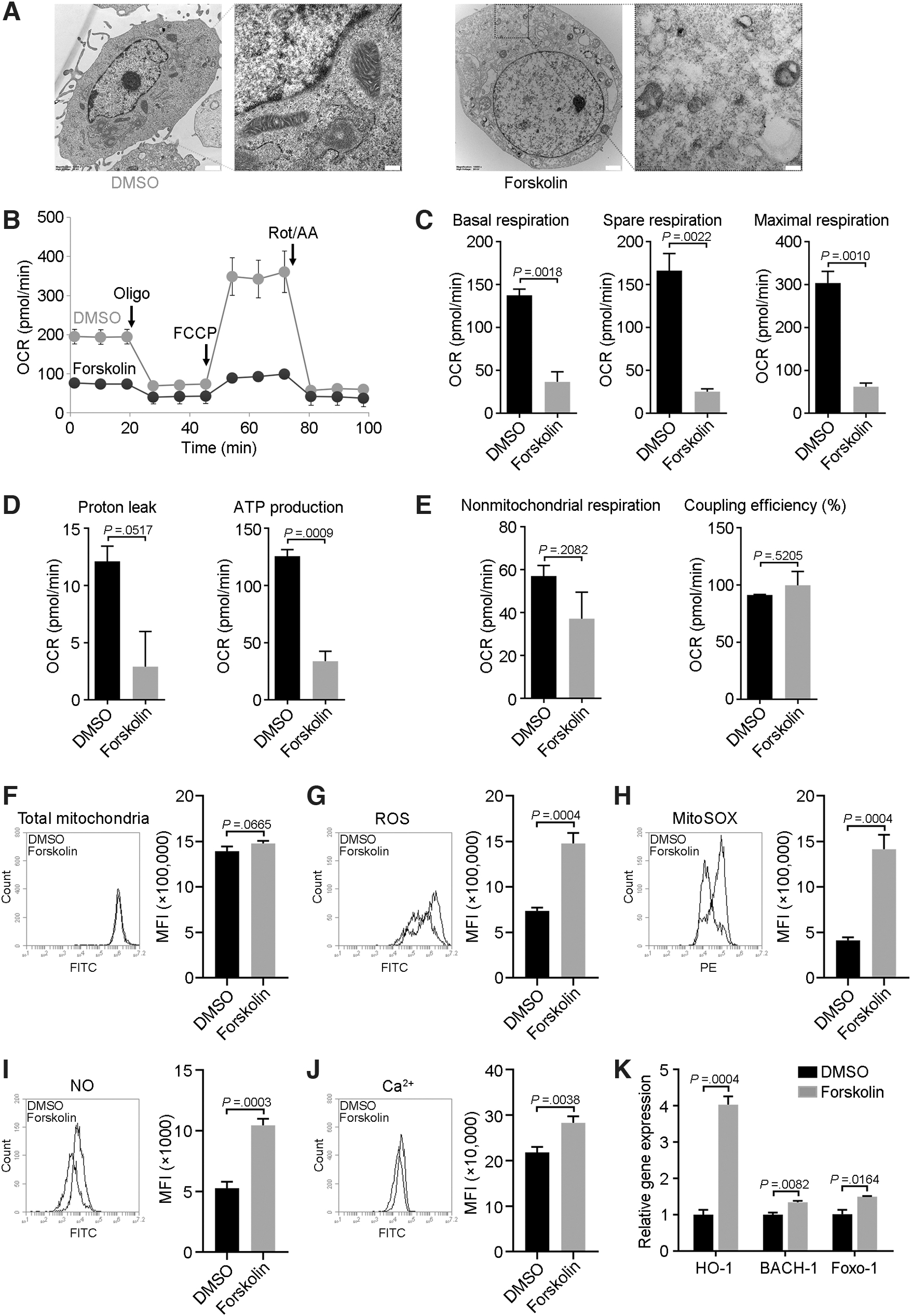
Analysis of mitochondrial metabolism in senescent MSCs.
However, there was no statistically significant difference in coupling efficiency and non-mitochondrial oxygen consumption between the two groups (Fig. 5E). Forskolin did not affect the total number of mitochondria (Fig. 5F) but increased the production of ROS and SOX (Fig. 5G, H), NO (Fig. 5I), and calcium flux (Fig. 5J). Moreover, forskolin-treated MSCs exhibited upregulated expression of the NO-induced genes HO-1 and FOXO-1 (Fig. 5K), with the latter being an ROS-regulated gene expressed at higher levels in older muscle than in younger muscle [26].
The expression of BACH-1, an inhibitor of HO-1, was also activated in forskolin-treated MSCs (Fig. 5K). These results suggested that mitochondria of forskolin-treated senescent MSCs were impaired with respect to both morphology and metabolism.
An adenylate cyclase inhibitor blocks forskolin-induced MSC senescence
The NF-κB pathway is essential for SASPs [27], and thus we treated MSCs with the NF-κB inhibitor PDTC to determine whether NF-κB inhibition could rescue forskolin-induced senescence. To our surprise, the cAMP content increased after 24-h of PDTC treatment (Fig. 6A). Although the levels of IL-6 and IL-8 mRNAs decreased, IL-1β expression was increased (Fig. 6B). Moreover, the senescence-related gene p16 was downregulated whereas p21 was upregulated (Fig. 6B).
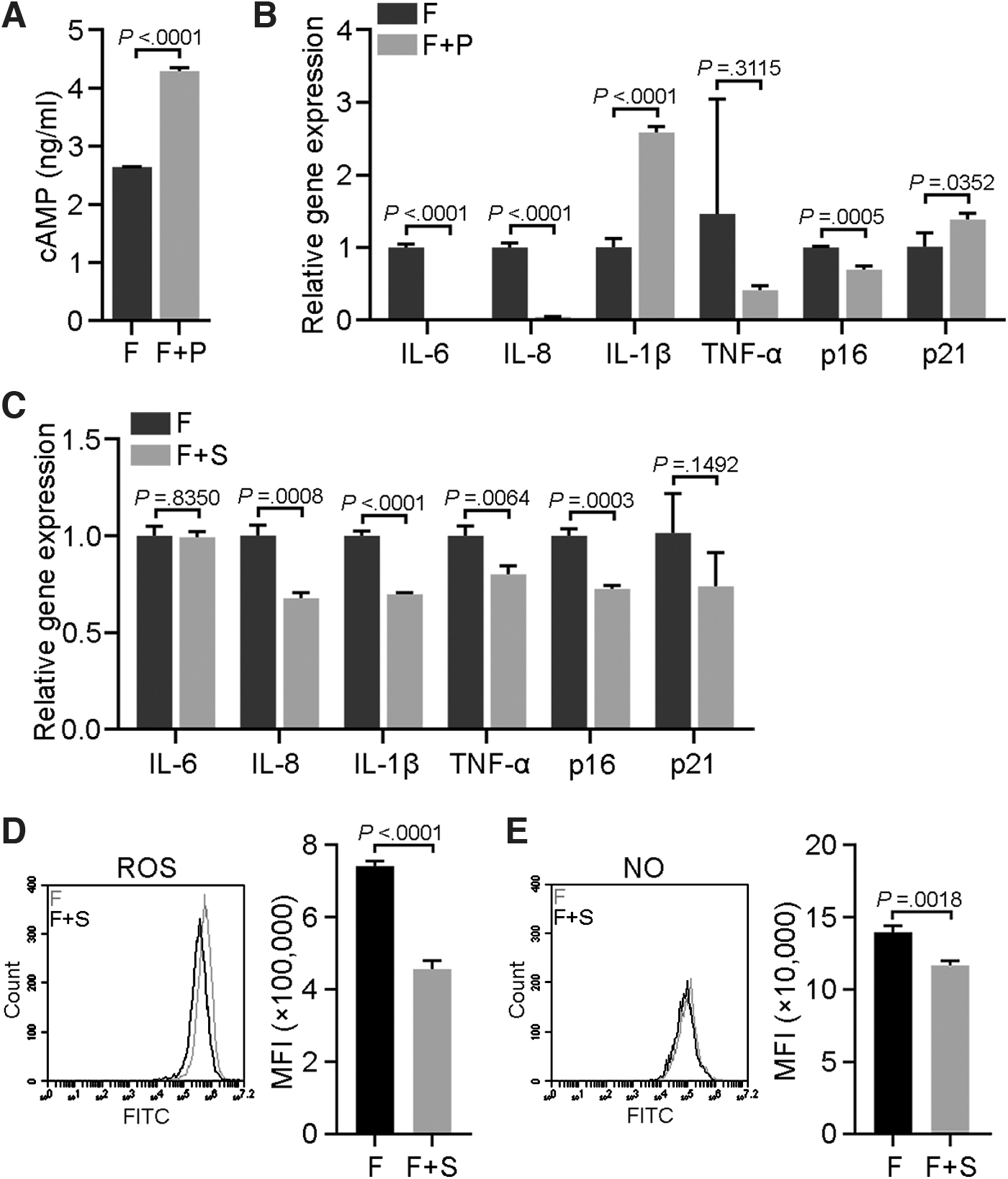
Effects of an NF-κB inhibitor and a cAMP inhibitor on forskolin-induced senescence in MSCs.
On the other hand, the adenylate cyclase inhibitor SQ22536 strikingly reversed the senescent state of forskolin-treated MSCs, exemplified by the decreased expression of p16, p21, and the inflammatory factors IL-8, IL-1β, and TNF-α (Fig. 6C). Moreover, SQ22536 treatment reduced ROS and NO levels in forskolin-treated MSCs (Fig. 6D, E). Thus, these results suggested that the adenylate cyclase inhibitor SQ22536, but not PDTC, could effectively reverse forskolin-induced senescence.
Discussion
The MSCs have shown potential in clinical trials for the treatment of several human diseases. However, preventing MSC senescence during in vitro culture remains a challenge for clinical applications of MSCs. Thus, deciphering the critical factors that contribute to MSC senescence is of great importance for developing effective strategies to prolong the viability of MSCs destined for clinical use.
In this study, we treated MSCs with forskolin in vitro and demonstrated that forskolin significantly induces MSC senescence and abnormal differentiation. Forskolin triggered global transcriptional reprogramming of MSCs, thereby reducing their viability, disrupting mitochondrial metabolism, and producing other senescence-related phenotypes.
An increasing body of evidence has shown that metabolic changes within individual cells affect cell-fate decisions, including entry into an apoptosis or senescence program [28 –32]. cAMP is generated in both the mitochondria and cytoplasm by adenylate cyclase in response to metabolically generated carbon dioxide [33], and cAMP signaling is important for regulating mitochondrial biogenesis. The natural cAMP-elevating compound forskolin raises intracellular cAMP levels by activating adenylate cyclase, which generates cAMP from ATP.
Because forskolin alters cellular energy metabolism, it has been proposed as an effective anticancer agent for several cancer types [34]. However, the role of forskolin in MSC senescence remains unknown. We found that forskolin-induced senescence of MSCs resulted in abnormal mitochondrial morphology and impaired metabolism-associated functions, particularly increased production of both ROS and NO and increased Ca2+ flux [35]. Surprisingly, although both RNA-seq and RT-qPCR data revealed that forskolin-treated MSCs had an SASP, the NF-κB inhibitor PDTC did not prevent forskolin-induced MSC senescence (rather, it only partially reduced the expression of genes encoding proinflammatory cytokines).
These results indicate that the NF-κB pathway is not the primary mediator of forskolin-induced MSC senescence and that the observed SASP is a consequence of events occurring downstream of senescence. One the other hand, SQ22536, an adenylyl cyclase inhibitor that inhibits forskolin-induced cAMP elevation [36], remarkably reversed forskolin-induced MSC senescence.
Research has shown that forskolin at 10 μM promotes mitochondrial biogenesis and at 4 μM upregulates fatty-acid oxidation [35]. In contrast, use of a higher dose (60 μM) in our experiments revealed that forskolin alters mitochondrial morphology and function in MSCs and triggers their senescence (Fig. 7). This phenomenon suggests that forskolin may have contrasting effects in different contexts or at different doses.
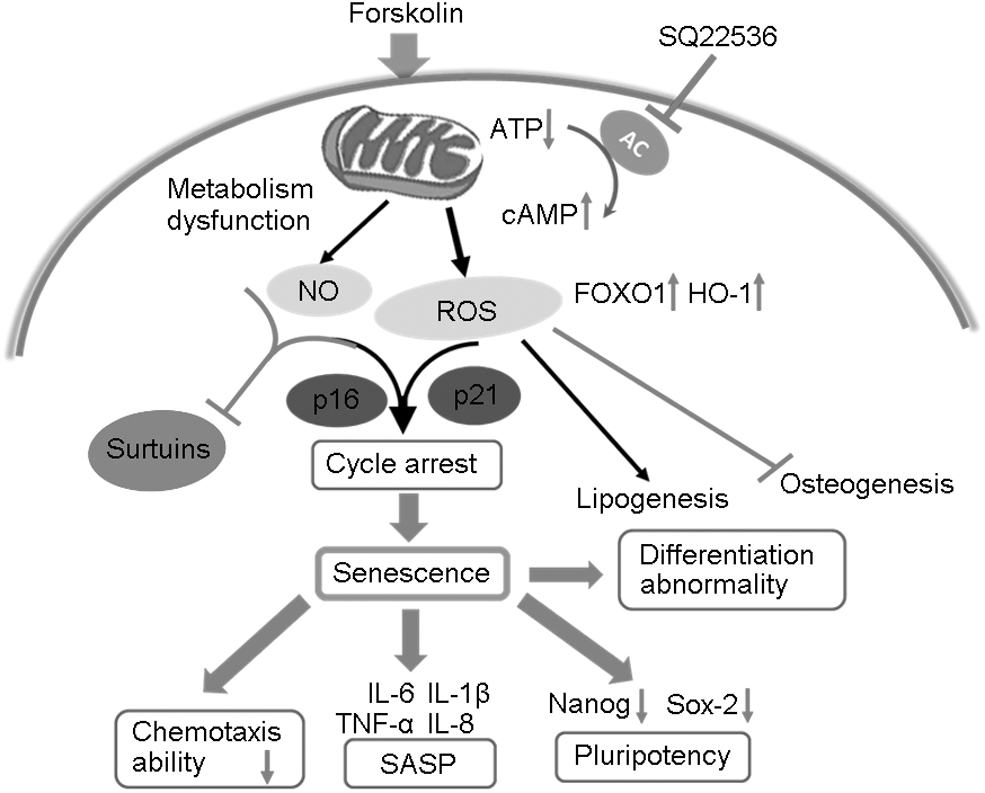
Schematic diagram of the mechanism by which forskolin induces senescence in MSCs. Forskolin induces an increase in cAMP level and a decrease in ATP level, leading to dysfunction of mitochondrial metabolism in MSCs and an increase in the levels of ROS, NO, FOXO1, and HO-1. In addition, expression of longevity genes sirtuins is suppressed. These changes lead to a senescence phenotype in MSCs characterized by cell-cycle arrest, SASP, reduced chemotaxis, and abnormalities in pluripotency and differentiation. SASP, senescence-related secretory phenotype.
For example, eye drops containing forskolin have passed all clinical trial phases, and 10 mg/day of forskolin could prevent asthma in children and adults with mild/moderate persistent asthma via an increase in FEV1 and a decrease in frequency in asthmatic attacks [37 –39]. Moreover, our study may provide probable references of forskolin for clinical use, such as cumulative dose repeatedly and drug resistance mechanism. In addition, forskolin has also been proposed for anticancer treatments. Thus, our results that forskolin can induce MSC senescence improves our knowledge of the role that forskolin plays in cellular energy metabolism yet raises caution regarding its clinical applications.
Preventing senescence has been a long-standing goal of developmental biologists, and MSCs have proved promising for anti-aging strategies countering the aging process. Our study reveals that forskolin can potently induce MSC senescence. Moreover, an adenylyl cyclase inhibitor could potentially be an effective means of countering forskolin-induced MSC senescence, thus providing a basis for further research toward the goal of optimizing cAMP signaling to minimize the impact of senescence on MSCs. However, our study has certain limitations that require further investigation.
For example, although our results provide solid evidence that forskolin induces MSC senescence via transcriptional reprogramming and disrupting mitochondrial metabolism, any potential interaction(s) between these two aspects remains unclear. Because mitochondrial metabolism is key for controlling cell fate, further studies to reveal subtle changes in both the nuclear and mitochondrial transcriptomes may suggest practical means for preventing forskolin-induced senescence in vitro.
Further, the opposing effects of low-dose and high-dose forskolin—as demonstrated in our current study and previous studies—raise the necessity to perform in vivo studies to further investigate the dose-related effects of forskolin on mitochondrial metabolism and senescence.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by the National Key Research and Development Program of China (2018YFA0109800, 2016YFA0101000, 2016YFA0101003), CAMS Innovation Fund for Medical Sciences (2017-I2M-3-007), the 111 Project (B18007), and the National Natural Science Foundation of China (81672313, 81700782, 81971324, 81972523, 81771349).
Supplementary Material
Supplementary Table S1
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.