
Abstract
The ShcA adapter protein is necessary for early embryonic development. The role of ShcA in development is primarily attributed to its 52 and 46 kDa isoforms that transduce receptor tyrosine kinase signaling through the extracellular signal regulated kinase (ERK). During embryogenesis, ERK acts as the primary signaling effector, driving fate acquisition and germ layer specification. P66Shc, the largest of the ShcA isoforms, has been observed to antagonize ERK in several contexts; however, its role during embryonic development remains poorly understood. We hypothesized that p66Shc could act as a negative regulator of ERK activity during embryonic development, antagonizing early lineage commitment. To explore the role of p66Shc in stem cell self-renewal and differentiation, we created a p66Shc knockout murine embryonic stem cell (mESC) line. Deletion of p66Shc enhanced basal ERK activity, but surprisingly, instead of inducing mESC differentiation, loss of p66Shc enhanced the expression of core and naive pluripotency markers. Using pharmacologic inhibitors to interrogate potential signaling mechanisms, we discovered that p66Shc deletion permits the self-renewal of naive mESCs in the absence of conventional growth factors, by increasing their responsiveness to leukemia inhibitory factor (LIF). We discovered that loss of p66Shc enhanced not only increased ERK phosphorylation but also increased phosphorylation of Signal transducer and activator of transcription in mESCs, which may be acting to stabilize their naive-like identity, desensitizing them to ERK-mediated differentiation cues. These findings identify p66Shc as a regulator of both LIF-mediated ESC pluripotency and of signaling cascades that initiate postimplantation embryonic development and ESC commitment.
Introduction
Embryonic development and cellular differentiation rely on the accurate transduction of extrinsic cues to mediate transcriptional programs. These complex cellular events are characterized by just a handful of intracellular cascades that elicit a broad range of cellular responses through regulation of signal timing, magnitude, and localization [1]. Signaling is achieved, in part, through adaptor proteins that possess two or more distinct protein binding domains which facilitate interactions between proteins [2].
ShcA (Src homology domain containing transforming protein 1) belongs to a family of adaptor proteins that regulate pathways responsible for cell proliferation and differentiation. ShcA plays a key role in facilitating the rat sarcoma/mitogen activated protein kinase (RAS/MAPK) cascade [3 –6], which mediates many stages of embryonic development, particularly the early acquisition of cell identity [7,8]. The ShcA locus encodes three isoforms with molecular weights of 46, 52, and 66 kDa (p46Shc, p52Shc, and p66Shc, respectively), which are largely conserved across vertebrates [5,9,10].
Despite their structural similarity, the ShcA isoforms have functionally distinct roles in signal transduction. The p52/46 kDa isoforms predominantly transduce mitogenic signaling by binding to Grb2, facilitating its interaction with Son of Sevenless, which subsequently activates the MAPK cascade [10 –12]. In contrast, p66Shc antagonizes mitogenic signaling and promotes oxidative stress [13 –15]. Although p66Shc binds to Grb2 [16], this interaction does not necessarily lead to the activation of ERK1/2. Expression of p66Shc is correlated with the inhibition of ERK1/2 signaling in some cell lineages, including fibroblasts [16], T-cells [17], and CHOK-1 cells [18], while in other cell types, p66Shc expression is associated with the activation of ERK1/2 [19 –21]. These conflicting findings suggest that p66Shc regulates ERK1/2 signaling in a cell-context dependent manner.
Several studies have implicated p66Shc in the regulation of cell fate: kidney development in rats [22], morphogenesis of fetal mouse lungs [23], and B cell maturation [24]. We previously demonstrated that p66Shc transcript abundance alters the expression of lineage-associated transcription factors (TFs) and the timing of blastocyst cavitation [25,26]. Collectively, these studies implicate p66Shc in a wide variety of developmental roles; however, the extent and mechanism by which p66Shc affects cell fate have not been established.
To explore the involvement of p66Shc at the earliest stage of lineage commitment, we employed the use of mouse embryonic stem cells (mESCs). In vitro, mESCs can be maintained in a state transcriptionally similar to the inner-cell mass of the preimplantation blastocyst, referred to as naive-like pluripotency, and are capable of recapitulating the early events of epiblast maturation [27].
The propagation of naive-like mESCs can be achieved by the addition of growth factors and pathway inhibitors, to reinforce self-renewal while preventing differentiation. Current approaches commonly involve modulating key pathways: (1) blocking extracellular signal regulated kinase (ERK) activity, traditionally through the inhibition of mitogen-activated protein kinase (MEK), the kinase responsible for ERK1/2(T202/Y204) phosphorylation, (2) inhibition of glycogen synthase kinase-3 beta (GSK3β), the kinase responsible for the phosphorylation of β-catenin(S33/S37/T41), targeting β-catenin for proteasomal degradation, and (3) enhancing STAT3 activity, through supplementation with exogenous LIF which stimulates Janus kinase (JAK) the enzyme responsible for the phosphorylation of STAT3(Y705). When combined, the in vitro supplementation of LIF together with pharmacologic inhibitors of MEK and GSK3β is commonly referred to as 2i/LIF media [28].
The relevance of these pathways to the acquisition of cell fate is evident from the effect of their targeted disruption on mouse embryogenesis. Germline deletion of ERK2 results in developmental abnormalities before E5.5 and embryonic lethality by E8.5, with failure of trophoblast maturation and endoderm specification [29]. Deletion of β-catenin prevents mesoderm formation, causing embryonic lethality at the gastrulation stage E6.5-E9.5 [30]. Targeted disruption of STAT3 similarly prevented mesoderm formation, resulting in embryonic lethality by E7.0 [31].
In vitro, the imbalance of these signals can induce precocious differentiation, loss of ESC self-renewal, or impair lineage commitment. Notably, ERK and STAT3 have been shown to have opposing roles at this stage of naive-like identity. ERK is generally considered essential for early ESC differentiation, driving commitment toward primed-like identity, which is representative of postimplantation epiblast cells [8,32 –35]. STAT3 activity, conversely, opposes ESC differentiation and its forced activation can durably suppress ESC differentiation [36 –38].
Phosphorylation of STAT3 at Y705 is primarily mediated by JAK, which is critical for its nuclear translocation and transcriptional activity. However, STAT3 activity can also be modulated by phosphorylation at S727. Although the regulation of S727 phosphorylation and its contribution to STAT3 transcriptional activity remain controversial [39 –42], preventing STAT3(S727) phosphorylation, by replacement of serine residue with alanine, has been reported to reduce the expression of core pluripotency TF Nanog in mESCs [43].
Embryogenesis relies on coordinated timing and activity of these critical signal effectors, and consequently, disrupting their upstream regulators can also have dramatic effects across multiple stages of embryogenesis. The targeted disruption of the upstream activators of ERK can lead to delayed ESC differentiation and embryonic lethality early to mid-gestation [44 –47]. Similarly, germline deletion of ShcA in mice causes profound cardiovascular defects and results in embryonically lethal by E11.5 [12].
We hypothesized that p66Shc could act as a negative regulator of ERK activity in mESCs, to block lineage commitment and promote the retention of naive-like pluripotency. To explore this hypothesis, we examined the differentiation capacity of p66Shc knockout (KO) mESCs in vitro and the effect of p66Shc deletion on the critical signaling effectors involved in pluripotency. Consistent with our hypothesis, the deletion of p66Shc enhanced ERK phosphorylation but, surprisingly, also simultaneously reinforced mESC naive-like identity. We discovered that loss of p66Shc enhanced not only ERK phosphorylation but also STAT3(S727) phosphorylation. We propose that p66Shc intersects with both ERK1/2 and STAT3 pathways during mESC commitment, thereby sensitizing mESCs to differentiation by suppressing STAT3(S727) phosphorylation, despite simultaneously inhibiting ERK1/2.
Materials and Methods
Generating p66Shc KO mESCs
R1 wild type (WT) mESCs (provided by Janet Rossant, Sick Kids Hospital, Canada) were targeted with CRISPR/Cas9 to induce a knockout of p66Shc. Guide RNAs were designed to target the p66Shc-specific promoter and the 5′ region of exon 2, which encodes for the CH2 region unique to p66Shc. Guide RNA sequences, 5′-TCGGGGTCTACCCCTCCGG-3′ and 5′-ATATATCTGTAGGTCCGAG-3′, were synthesized by Life Technologies and were introduced into the pSpCas9(BB)-2A-Puro (PX459, No. 48139; Addgene [48]) plasmid following digestion with the restriction enzyme Bbs1. The plasmid was transfected into the R1 mESC line using Lipofectamine 3000 (Life Technologies) according to manufacturer's guidelines. Transfected cells were selected following exposure to puromycin, and single-cell clones were picked and expanded upon mouse embryonic fibroblasts (MEFs) and feeder-free conditions (2i/LIF).
Genomic DNA of clonal lines was isolated using GenElute™ Mammalian Genomic DNA Miniprep (Sigma), amplified using LA Taq DNA polymerase (Takara Bio, USA), and screened by polymerase chain reaction (PCR) product size using agarose gel electrophoresis. Genomic PCR products were extracted, purified, and sequenced at the London Regional Genomics Center (Robarts Research Institute, London, Canada). P66Shc deletion was further confirmed by immunoblotting with a ShcA specific antibody (610879; BD Biosciences), and densitometric analysis was performed to identify lines with loss of p66Shc expression but unmodified levels of p52/46Shc.
Cell culture
R1 WT and p66Shc KO mouse embryonic stem and p66Shc knockout clonal lines were maintained on a mouse embryonic fibroblast (MEF) feeder layer and in Dulbecco's modified Eagle's medium (DMEM) containing 15% ESC qualified fetal bovine serum (Life Technologies), 0.1% 2-Mercaptoethanol (Millipore Sigma), 1 × GlutaMAX (ThermoFisher), and 1000 U/mL mouse Leukemia Inhibitory Factor (mLIF; Millipore Sigma) in a 37°C, 5% carbon dioxide incubator. Cells were dissociated using 0.025% Trypsin-EDTA (ThermoFisher) at 37°C and passaged onto MEF-coated cell culture plates (Millipore Sigma).
For feeder-free conditions, R1 WT and p66Shc KO mESCs were plated on 0.2% wt/vol porcine gelatin coated plates (Millipore Sigma) and cultured with 2i/LIF media comprising: N2B27 media [KO 50% DMEM F12 medium (Life Technologies), 50% Neurobasal Medium (Life Technologies), 1 × GlutaMAX (ThermoFisher), 0.1% vol 2-Mercaptoethanol (Millipore Sigma), 0.5 × N2 Supplement (ThermoFisher), 0.5 × B27 Supplement without vitamin A (ThermoFisher)], 3 μM CHIR99021 (Reagents Direct), 1 μM PD0325901 (Reagents Direct), and 1000 U/mL mLIF (Millipore Sigma). Cells were passaged using StemPro™ Accutase™ Cell Dissociation Reagent (Life Technologies), according to the manufacturer's instructions, onto 0.2% wt/vol porcine gelatin coated plates (Millipore Sigma). Under all conditions, cells were passaged —three to four times post-thaw before experimentation.
The cytotoxicity of 2i was quantified using the trypan blue exclusion assay during the transition of mESCs to feeder-free conditions. In brief, mESCs maintained on MEFs were dissociated using 0.025% Trypsin-EDTA and separated from MEF feeder layer by allowing the MEFs to readhere to the culture plates. The mESCs were passaged onto 0.2% gelatin coated plates in N2B27 media containing LIF/2i and serum with a seeding density of 40,000 cells/cm2. The cells were then passaged every 48 h and plated with N2B27 media containing LIF/2i. For assessment of cytotoxicity, media was removed during passaging and sampled along with the dissociated cell suspension to calculate the net concentration of live and dead cells. Cells were counted in duplicate using a hemocytometer following the addition of 0.4% Trypan Blue (Gibco, ThermoFisher).
For colony forming assays, dissociated cells were seeded in 6-well plates, 600 cells/well on 0.2% gelatin, with a minimum of three technical and biological replicates. Colonies were counted after 4 days.
Differentiation of mESCs
Mouse (m)ESCs were differentiated using a monolayer differentiation protocol established for 2i culture systems, with minor modifications [28]. Briefly, mESCs were dissociated using StemPro Accutase Cell Dissociation Reagent (Life Technologies) and then seeded on gelatin-coated tissue culture plates at 104 cells/cm2 in N2B27 or N2B27 supplemented with LIF. JAK inhibitor treatments used a final concentration of 0.5 μM Ruxolitinib (Cayman Chemical Company Culture). Medium was changed daily.
Immunocytochemistry and microscopy
Cells were seeded in eight-well plastic chamber slides (Ibidi) coated with gelatin. Cells were fixed with 4% paraformaldehyde (Electron Microscopy Sciences) for 20 min and washed with cold phosphate-buffered saline (PBS). Cells were then permeabilized with 0.1% Triton X (Millipore Sigma) in PBS for 30 min then incubated with blocking solution containing 3wt/vol% bovine serum albumin (BSA; ThermoFisher) and 0.1% Tween 20 in PBS (PBS-T). Following blocking, cells were incubated with primary antibodies diluted in PBST containing 1% BSA overnight at 4°C. A list of the primary antibodies used is shown in Supplementary Table S1. Cells were washed with PBS, counterstained with NucBlue (ThermoFisher, Invitrogen) according to manufacturer instructions, and then mounted using ProLong Gold™ (ThermoFisher, Invitrogen).
Immunofluorescence (IF) images were obtained using a LSM800 laser-scanning confocal microscope (Zeiss) and Zen imaging software (Zeiss). Laser settings were unchanged when imaging the same primary antibodies across conditions. Each condition was examined with three biological replicates. Phase contrast images were acquired using Leica DMI 6000 microscope (Leica Microsystems) and Application Suite X software (Leica Microsystems).
Immunoblotting
Cells were lysed and scraped into radioimmunoprecipitation assay (RIPA) buffer containing 1 × phosphatase inhibitor cocktail 2 (Millipore Sigma) and protease inhibitor cocktail 1 (Millipore Sigma) and stored at −80°C until processing. Proteins were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) on 4%–12% gradient gels (NuPage™, Life Technologies). Membranes were treated with 0.1 wt/vol% Ponceau S for 15 min, washed in methanol, and then imaged using a ChemiDoc MP Imaging system (Bio-Rad). A list of the primary antibodies used for immunoblotting is shown in Supplementary Table S2. Protein bands were visualized using enhanced chemiluminescence (Forte ECL, Millipore Sigma) and imaged using a ChemiDoc MP Imaging System (Bio-Rad). Densitometric analysis of Western blots was performed using ImageLab (Bio-Rad).
RNA extraction and quantitative real time PCR
Cells were scraped and lysed in TRIzol reagent (ThermoFisher, Invitrogen), then stored at −20°C until RNA extraction. RNA was extracted into chloroform then isolated using the RNeasy Mini Kit (Qiagen), according to manufacturer's instructions. Genomic DNA was digested using the RNase-Free DNase Set (Qiagen), and cDNAs were generated using SuperScript III Reverse Transcriptase (ThermoFisher, Invitrogen). Quantitative real time PCR (RT-qPCR) was performed using a SensiFast™ SYBR No-ROX Kit (FroggaBio), and amplification was assessed using a real-time CFX384 thermal cycler (Bio-Rad). Relative transcript abundance was determined by the
Teratomas
Experimental animals
Male and female immunodeficient mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl /SzJ, Stock No.: 005557) were ordered from The Jackson Laboratory (Bar Harbor, ME, USA) and kept in the Robarts Research Institute's mouse barrier facility (University of Western Ontario, London, Ontario, Canada). Mice were housed in Thoren vent rack cages (Thoren caging systems, Inc., PA, USA), in rooms kept at 23–26°C with a 35%–60% relative humidity, 12-h light/12-h dark cycle, and provided with constant food (Teklad 2920X irradiated soy protein-free diet) and water access. Cages were changed once per week in a biosafety cabinet, and mice were checked daily for overall health. All mice were handled according to the Western University Animal Care Committee guidelines approved by the Canadian Council on Animal Care (UWO animal utilization protocol 2022-072).
Stem cell injections
WT and p66Shc KO mESCs were cultured and passaged as described above. Cells were counted using a hemocytometer, and one million cells were collected, spun down, resuspended in 50 μL aliquots of 100 × Geltrex (Life Technologies catalog No. A1413302), and placed on ice. Mice were anesthetized using isoflurane (5% induction, 2% maintenance) through inhalation and injected with the 50 μL cell/geltrex mixture subcutaneously in the dorsal region.
Teratoma collection and characterization
All mice were euthanized using isoflurane and cervical dislocation once WT teratomas reached 1.5 cm in length (∼21 days postinjection). Teratomas were frozen on dry ice, then placed in a cryo-block, covered in optimal cutting temperature. Compound (Fisher catalog No. 23-730-571) and snap frozen in liquid nitrogen-chilled isopentane, and stored at −80°C for future cryo-sectioning and slide preparation at the Robarts Research Institute's Molecular Pathology Core Facility (University of Western Ontario). Excised tissue underwent immersion fixation for 24 h in formalin followed by paraffin embedding and sectioning using routine histological methods. Representative sections of each teratoma were stained with Hematoxylin & Eosin (H&E) for microscopic examination. Germ layers were identified from the combined study of individual cell morphology and cellular arrangement observed in the H&E-stained histological sections by histopathologist Dr. Patti Kaiser, without a priori knowledge of experimental conditions. For individual cell morphology, cell size, shape, and borders (indistinct vs. distinct) were considered. In addition, cytologic and nuclear cellular features were assessed to identify cell type. Cellular arrangements were assessed by cell density, pattern, and stroma. Microscopy was performed using an optical microscope (Olympus CX33) equipped with a camera (Lumenera INFINITY1-1M). Digitally recorded images' histologic measurements were conducted using Infinity Analyze software.
Chimera and embryo culture
To identify contribution of p66Shc KO mESCs to blastocysts, we genetically modified WT and p66Shc KO mESC lines with a mCherry fluorescent reporter and injected the cells into eight-cell mouse embryos. In brief, WT and p66Shc KO mESCs were transfected with H2B promoter-driven mCherry with a PiggyBac™ vector using Lipofectamine 3000 according to manufacturer's protocols. Transfected mESCs were selected by puromycin (2 μg/mL) over 72 h. Mouse embryos were harvested from oviducts 2 days after detection of mating plug and incubated in KSOMaa Evolve (Zenith Biotech) at 37°C. Noncompacted eight-cell embryos were selected and incubated for 30 min before injection. Using a micromanipulator-assisted injection pipette (CellTram, Eppendorf) and inverted microscope, ∼8–10 cells of either WT or p66Shc KO mCherry-mESCs were injected into the perivitelline space under the zona pellucida. A total of 10 embryos were injected with mESCs (5WT, 5KO) and then cultured in a hypoxic incubator for 24 h before fixation.
For extended culture, a similar approach was used, with modification, to remove the zona pellucida. The zona pellucida was removed at E3.5 by first washing each embryo in Acid Tyrodes droplets (30 μL, 1 min), transferring the embryo to a neutralizing M2 medium, then transferring them to M11-6 medium (Cell Guidance Systems) in eight-well culture slides (Ibidi, μ-plates). After 2 days the medium was changed to M12-6 (Cell Guidance Systems). Embryos were cultured for a total of 5 days in a hypoxic incubator at 37°C, before fixation with 4% paraformaldehyde.
Statistical analysis
Statistical tests were performed in Prism 8 (GraphPad Inc.) using Student's t-test (equal variance, unpaired, two-tailed) and one-way analysis of variance (ANOVA) followed by Tukey's honestly significant difference test to correct for multiple comparisons. Two-way ANOVAs were used for transcriptional analysis across multiple experimental conditions and cell types, and Dunnett's test was used when all comparisons were made to a single control. Experiments were performed with a minimum of three biological replicates.
Results
Deletion of p66Shc in mESCs increases basal ERK activity, Stat3(S727) phosphorylation, and enhances expression of pluripotency TF Nanog in serum and LIF containing media
ShcA has pleiotropic roles in RTK signal transduction, but is most notably involved in promoting ERK phosphorylation [5]. To explore the potential involvement of p66Shc in mESCs, we assessed the effect of p66Shc deletion on the core effectors of signaling pathways that govern ESC self-renewal and pluripotency: ERK, Stat3, and β-catenin.
p66Shc-null (p66Shc KO) mESCs were generated from the R1 mESC line using CRISPR-Cas9 nickase-mediated editing, targeting the p66Shc promoter and a part of p66Shc exon-specific region of the ShcA locus (Fig. 1A) [49]. Immunoblot analysis using a SHCA specific antibody revealed that the p66Shc isoform was absent but the 52 and 46 kDa isoforms were unaffected by genomic editing of the R1 mESC line (Fig. 1B).
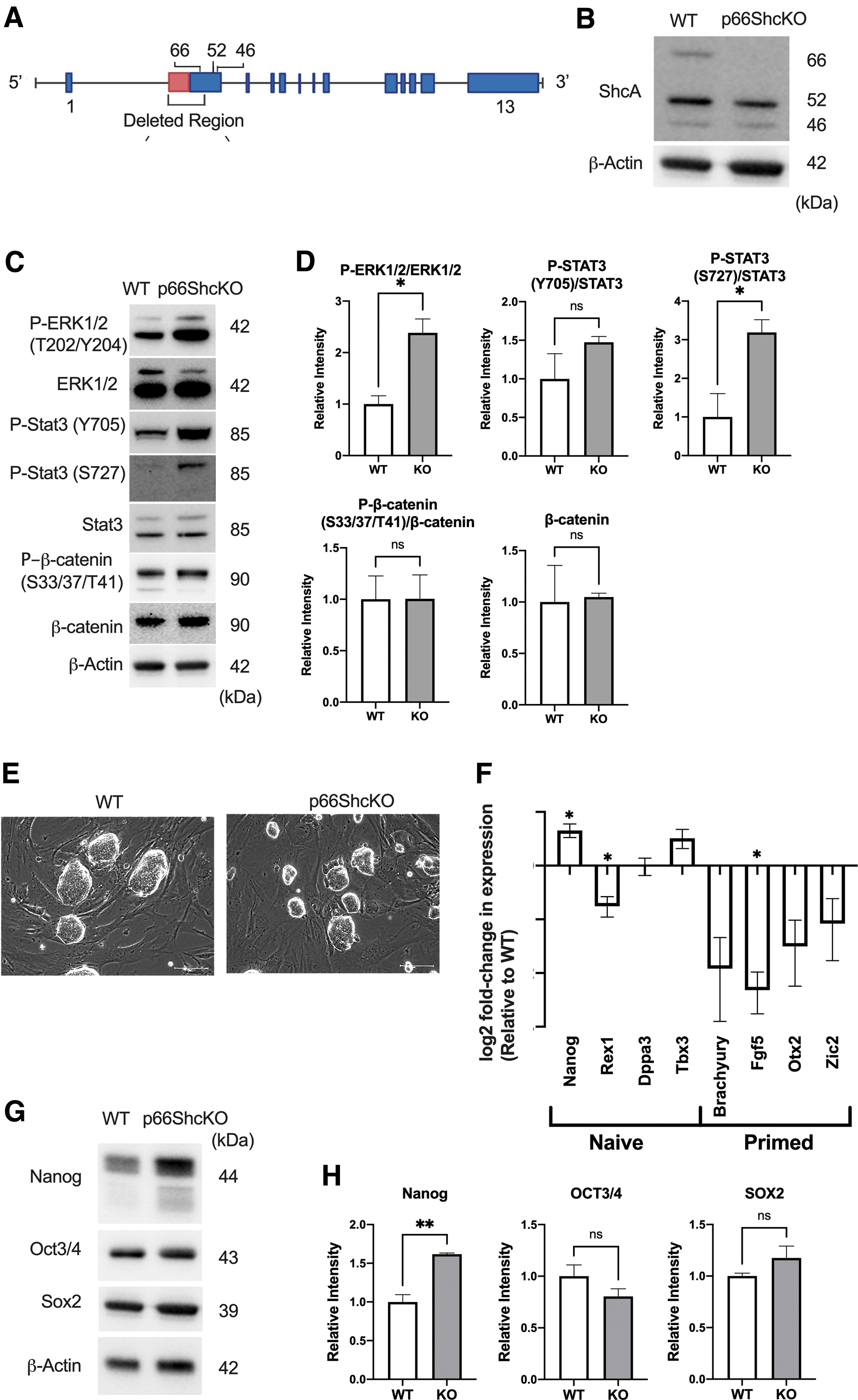
P66ShcKO mESCs have enhanced P-ERK and P-STAT3(S727) and have a more naive-like pluripotency state in serum/LIF-containing media.
Deletion of p66Shc significantly increased ERK1/2(T202/Y204) phosphorylation, without influencing levels of WNT pathway effector, β-catenin, or its GSK3β mediated phosphorylation (Fig. 1C, D). We also observed a significant increase in Stat3(S727) phosphorylation, while no significant alterations in Stat3(Y705) phosphorylation were observed (Fig. 1C).
Despite increased ERK phosphorylation, the p66Shc KO mESCs did not exhibit evidence of early commitment. P66Shc KO mESCs maintained tight colony boundaries and limited cell spreading (Fig. 1E), consistent with the morphology of naive mESCs. We observed alterations in naive-like marker expression, with a significant decrease in Rex1, but also a contradictory increase in Nanog. In addition, the p66Shc KO mESCs had reduced expression of the primed transcript FGF5 and a collective decrease across the remaining primed markers, although individually, the changes in each were not significant (Fig. 1F). We observed negligible changes in core pluripotency factors Oct3/4 and Sox2 and a significant increase in the levels of Nanog (Fig. 1G, H).
Collectively, these findings did not support our prediction that the loss of p66Shc would impair mESC self-renewal and pluripotency. Unexpectedly, p66Shc deletion appeared to stabilize naive TF circuitry, despite increasing ERK activity.
MEK and GSK3β inhibition abrogates differences in P-ERK and P-STAT3 signaling and Nanog expression between WT and p66Shc KO mESCs
Media supplemented with serum and MEFs contains a collection of cytokines that contribute to RTK signaling and that support mESC naive pluripotency, despite high levels of ERK activity. To eliminate the contributions of serum and MEFs to extrinsic signaling, we transitioned the WT and p66Shc KO mESCs to a defined media containing LIF, MEK inhibitor PD0325901, and GSK3β inhibitor CHIR99021, commonly referred to as 2i/LIF conditions. This media promotes naive-like pluripotency and Nanog expression by inhibiting ERK phosphorylation and the degradation of β-catenin [8,28,50]. These conditions would bypass upstream adaptor-control over ERK and WNT signaling, allowing us to better assess whether the loss of p66Shc was contributing to Nanog expression through alterations in canonical pluripotency signaling cascades.
Transitioning mESCs to 2i/LIF media required several passages before the mESCs adapted to the new conditions (Fig. 2A). The initial addition of 2i appeared to stress both WT and p66Shc KO mESCs, with significantly higher levels of cell death in WT mESCs (Fig. 2B). Following this transition, both WT and p66Shc KO mESCs could be maintained in tight naive-like colonies, without evidence of cell death or differentiation (Fig. 2C).
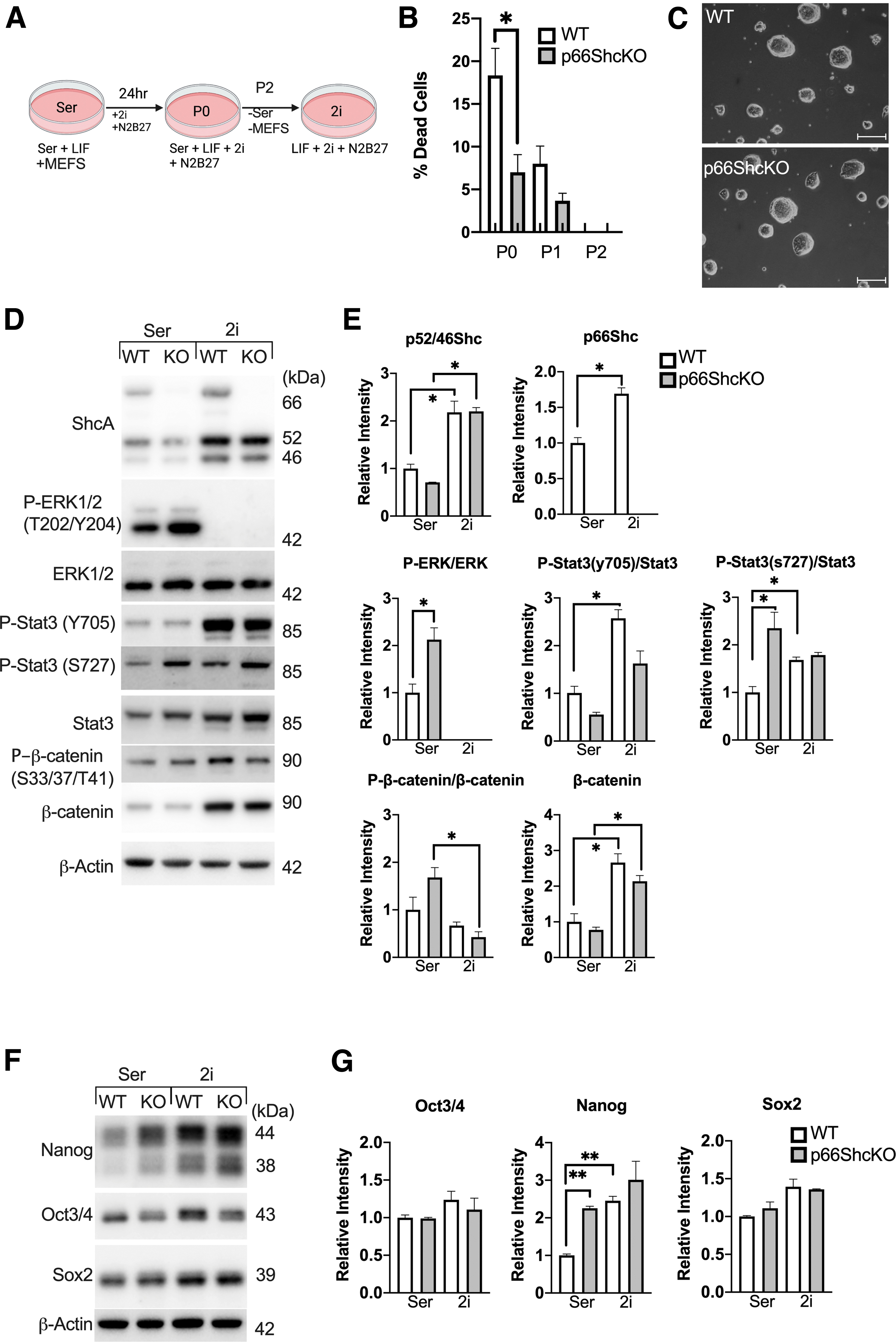
The addition of MEK and GSK3β inhibitors, PD0325901 and CHIR99021, abrogates P-ERK and P-STAT3(S727) differences between WT and p66ShcKO mESCs, and increases expression of Nanog in WT mESCs.
The presence of 2i abrogated all differences between WT and p66Shc KO phenotypes that we previously observed in serum/LIF media. The addition of 2i inhibited ERK activity and significantly enhanced levels of β-catenin in both WT and p66Shc KO mESCs (Fig. 2D, E). Notably, 2i also influenced STAT3 activity, abrogating the differences in STAT3(S727) phosphorylation, despite the inhibitors not being directly involved in canonical STAT3 signaling cascades. 2i also abrogated differences in Nanog expression by increasing levels of Nanog in the WT mESCs, without significantly altering levels of Nanog in the p66Shc KO mESCs (Fig. 2F, G).
Deletion of p66Shc permits the self-renewal of pluripotent mESCs in the absence of serum, 2i, or MEFs
2i appeared to abrogate intracellular signaling and pluripotency TF differences between WT and p66Shc mESCs; however, it remained unclear which aspect of the serum/LIF/MEF signaling environment was mediating these differences. It was also unclear whether p66Shc was directly driving these differences or whether they were due to indirect adaptations within the p66Shc KO mESCs compensating for the absence of p66Shc.
To clarify the conditions necessary to reassert these differences, we assessed early commitment of WT and p66Shc KO mESCs in response to withdrawal of one or more factors from the 2i/LIF media. Initial commitment, following the withdrawal of one or more components (MEKi, GSK3βi, and/or LIF) from the 2i/LIF media, was assessed by changes in mESC morphology over 2 days. Across most conditions, WT and p66Shc KO mESCs were morphologically indistinguishable (data not shown). Withdrawal of any single factor was insufficient to provoke noticeable morphological changes, while withdrawal of all three factors induced rapid cell separation and spreading in both the WT and p66Shc KO mESC lines. However, one condition, the withdrawal of 2i with maintenance of LIF provoked rapid morphological changes in the WT but not the p66Shc KO mESCs (Fig. 3A).

p66ShcKO mESCs maintain naive-like pluripotency in LIF containing media, despite withdrawal of 2i.
The effect of p66Shc deletion on mESC self-renewal was assessed by their capacity for colony formation. Although WT and p66Shc KO mESCs had similar colony formation efficiency in 2i/LIF, only the p66Shc KO mESCs maintained efficient colony formation in the absence of 2i (Fig. 3B). Moreover, colony formation efficiency decreased with repeat passaging of the WT mESCs in the absence of 2i. However, LIF alone was able to support p66Shc KO mESC self-renewal across multiple passages without loss of efficiency. The addition of higher doses of LIF (from 103 to 104 U/mL) also failed to support the colony formation in the WT mESCs (data not shown).
To assess whether the observed morphological changes might be rationalized by changes in mESC identity, we assessed levels of pluripotency associated transcripts over 6 days. Withdrawal of 2i decreased the expression of core and naive pluripotency associated transcripts in the WT mESCs (Fig. 3C, D), concomitant with a depletion of Nanog and SOX2 protein levels (Fig. 3E, F). However, p66Shc KO mESCs were insensitive to the loss of 2i and maintained expression of both core and naive pluripotency transcripts after 6 days. Nanog protein levels decreased in the absence of 2i but remained significantly elevated relative to the WT mESCs. Levels of pluripotency markers in the p66Shc KO mESCs were retained even after repeat passaging in the absence of 2i, for a minimum of five passages (p5).
Immunofluorescence microscopy analysis corroborated these results, revealing decreased Nanog immunostaining within the WT mESCs and a loss of E-cadherin at the intercellular boundaries following withdrawal of 2i (Fig. 3G). Conversely, the p66Shc KO mESCs maintained stable Nanog levels within nuclei and E-cadherin at the cellular interfaces within colonies.
P66Shc regulates the timing and levels of ERK and STAT3 phosphorylation during mESC commitment in LIF-containing media
To examine the underlying signaling mechanism mediating the retention of naive pluripotency of the p66Shc KO mESCs, we examined the phosphorylation of STAT3 and ERK following the withdrawal of 2i, during the early stages of naive mESC commitment.
Over 6 days of differentiation, we observed a significant increase in levels of p66Shc in the WT mESCs (Fig. 4A, B), concomitant with a decrease in STAT3(Y705) and STAT3(S727) phosphorylation. The p66Shc KO mESCs, however, retained both P-STAT3(Y705) and P-STAT3(S727) across the period of 2i withdrawal.

p66ShcKO mESCs exhibit elevated ERK activity relative to WT mESCs, in response to LIF-JAK extrinsic signaling. Effect of 2i withdrawal on activity of pluripotency-associated signaling effectors in WT and p66ShcKO mESCs over a 6-day period, and after multiple passages (p5) in p66ShcKO mESCs:
The absence of p66Shc altered the timing of ERK phosphorylation following 2i withdrawal. Compared to the WT mESCs, which showed peak ERK phosphorylation at 2 days, ERK activity in the p66Shc KO mESCs was highest after 6 days. In contrast with STAT3 and ERK, the absence of p66Shc did not significantly modulate levels of β-catenin.
These findings indicate that p66Shc KO mESCs have altered patterns of ERK and STAT3 activity concomitant with the retention of their naive pluripotency in the presence of LIF. To explore the role of p66Shc in LIF signal transduction, the mESCs were starved of LIF for 4 h, then stimulated with LIF in conjunction with the targeted inhibition of either JAK1/2, using Ruxolitinib, ERK, using PD, or in the presence of serum for 1 h (Fig. 4C, D).
LIF appeared to enhance the P-STAT3(S727) phosphorylation of both WT and p66Shc KO mESCs. However, in contrast with STAT3(Y705) phosphorylation, JAK inhibition only partially inhibited the STAT3(S727) phosphorylation. This suggests that Stat3(S727) is, in part, mediated by non-LIF and non-JAK dependent cascades. Deletion of p66Shc did not significantly affect STAT3(Y705) phosphorylation; however, it significantly enhanced STAT3(S727) phosphorylation in response to LIF stimulation. Notably, the difference in STAT3(S727) phosphorylation could be abrogated by MEK inhibition and the addition of serum, but not by JAK inhibition. The deletion of p66Shc significantly enhanced ERK phosphorylation, both in response to LIF and its absence. Again, this difference could be abrogated by MEK inhibition or by the addition of serum, but not by JAK inhibition.
Collectively, these findings suggest that p66Shc antagonizes ERK and STAT3(S727) phosphorylation in naive mESCs. Once the constraints on signaling imposed by 2i were removed, the p66Shc KO mESCs responded by phosphorylating ERK and STAT3(S727), which could be durably sustained in LIF containing media. Thus, the regulatory effect of p66Shc on STAT3 phosphorylation appears to be ERK dependent and JAK independent.
P66Shc KO mESCs incorporate into the inner cell mass of chimeric embryos and contribute to all somatic germ layers but form small and immature teratomas
Elevated ERK activity has been shown to preferentially select for “primed” ERK-dependent epiblast-like ESC populations in LIF/serum, while STAT3 is necessary for the maintenance of naive inner cell mass (ICM)-like lineages [32,37,51,52]. Unlike naive mESCs, primed ESCs are incapable of contributing to the ICM when injected into preimplantation embryos [53,54]. Having observed both elevated ERK phosphorylation, but also increased levels of naive-associated transcripts in the p66Shc KO, we were interested in how these contradictory phenotypes would influence the ability of p66Shc KO mESCs to contribute to preimplantation embryos.
To assess the effect of p66Shc deletion on preimplantation development, we generated mCherry-reporter WT and p66Shc KO mESC lines. Between 8 and 10 labeled cells were injected into each mouse embryo at the 8-cell stage. After 1 day in vitro, both WT and p66Shc KO mESC chimeric embryos developed into the mid-blastocyst stage, similar to E3.5. Like WT chimeras, the donor p66Shc KO mESCs were restricted to the ICM and dispersed in a mosaic among the host's ICM cells (Fig. 5A). The WT and p66Shc KO cells predominantly expressed NANOG, a marker indicative of epiblast identity. We did not observe expression of GATA4, a primitive endoderm marker, in either the WT or p66Shc KO donor cells.

p66ShcKO mESCs contribute to the inner cell mass of blastocysts and contribute to all germ layers in teratomas, but teratoma-derived tissues remain immature.
The WT and p66Shc KO chimeras were also grown in extended embryo culture conditions to allow development to the egg-cylinder stage (Supplementary Fig. S1). The cells were stained with SOX17 and OCT3/4 to distinguish between their contribution to visceral endoderm and epiblast lineages, respectively. At this stage of development, we did not observe any distinct differences in their potential. Both WT and KO donor cells expressed OCT3/4, but not SOX17, suggesting that neither WT nor p66Shc KO donor cells contributed to visceral endoderm.
To explore the differentiation potential of the p66Shc KO mESCs, we used the teratoma assay [55]. Transplanted tumors were observed in nearly all mice, aside from one mouse injected with the p66Shc KO mESCs. WT derived tumors were all encapsulated masses with prominent solid components, while the p66Shc KO derived tumors were poorly defined soft tissue clumps, with significantly smaller mass than the WT derived tumors (Fig. 5B, C).
Histological analysis identified each of the three germ layers (ectoderm, mesoderm, and endoderm) in both the WT and p66Shc KO derived teratomas (Fig. 5D); however, these structures appeared less defined in the p66Shc KO tumors. WT teratomas consisted primarily of well-defined structures; however, the p66Shc KO teratomas were composed almost entirely of undifferentiated neoplasm (Fig. 5E, F).
Together, these data indicate that the p66Shc KO mESCs are pluripotent, with a naive-like identity, capable of differentiating to each germ layer; however, they have an attenuated maturation rate relative to WT mESCs.
The withdrawal of LIF or inhibition of JAK signaling restores p66Shc KO mESC differentiation competency
Although we had observed significant differences in intracellular activity of P-ERK and P-STAT3(S727) in p66Shc KO mESCs, these signaling differences did not appear directly mediated by canonical LIF-LIFR-JAK extrinsic cascades.
The reduced tissue complexity and size of the p66Shc KO derived teratomas appeared to suggest developmental impairments of the p66Shc KO mESCs, but it remained unclear whether these impairments were restricted to naive-commitment and whether LIF signaling was necessary for their enhanced naive-like self-renewal.
To assess early lineage commitment in the absence of LIF, we withdrew both 2i and LIF and monitored changes in expression of naive-associated genes. The withdrawal of 2i and LIF caused rapid morphological changes (Fig. 6A), concomitant with a loss of naive marker expression in both WT and p66Shc KO mESCs (Fig. 6B). Differences between WT and p66Shc KO were negligible across many of the markers assessed, including Nanog.
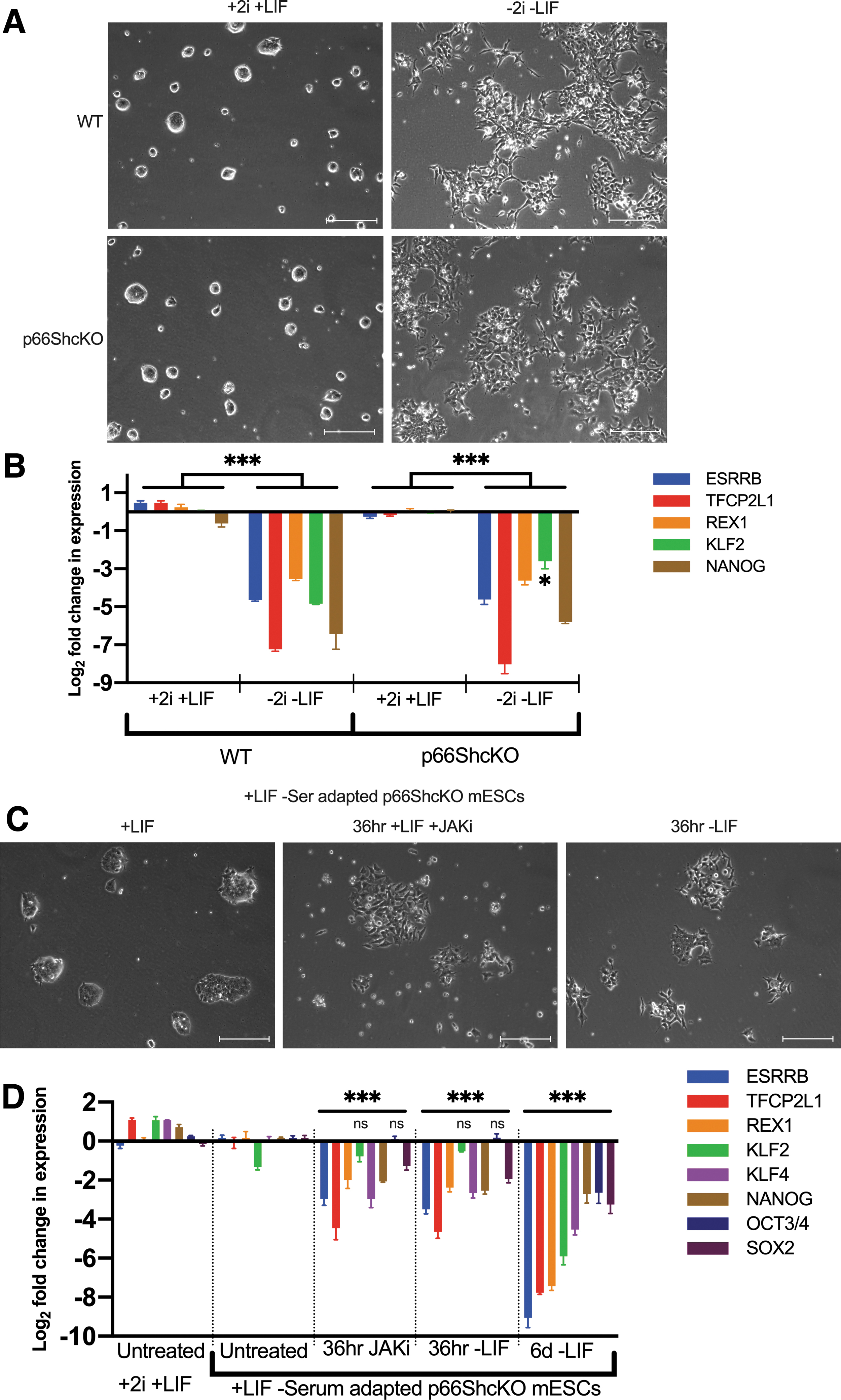
LIF-JAK signaling is necessary for enhanced naive-like pluripotency in p66ShcKO mESCs.
To explore whether canonical LIF-JAK-STAT signaling was mediating enhanced LIF permissiveness in p66Shc KO mESCs, we attempted to induce differentiation of p66Shc KO mESCs that we had adapted to LIF-only media, by the targeted inhibition of components of the JAK-STAT, MAPK, Phosphoinositide-3-kinase (PI3K), and BMP-SMAD pathways (data not shown). Notably, only the withdrawal of LIF or the presence of JAK/STAT3 inhibitors could recapitulate the morphological changes of mESC commitment (Fig. 6C). Concomitant with changes in morphology, LIF withdrawal induced the loss of both core and naive pluripotency markers from the LIF-only p66Shc KO mESCs (Fig. 6D). Moreover, persistent JAK1/2 inhibition could forcibly induce loss of naive marker expression and partially recapitulate the effect of LIF withdrawal (Fig. 6D).
Discussion
In this study, we show that the deletion of p66Shc from mESCs is sufficient to permit the maintenance of their naive pluripotency in minimal media containing LIF, without serum, supplemental growth factors, or pharmacologic inhibitors. Surprisingly, this effect was associated with a loss of sensitivity to P-ERK mediated differentiation cues in the p66Shc KO mESCs. This is the first study to identify the involvement of p66Shc in the negative regulation of ERK activity in ESCs and to implicate p66Shc in the negative regulation of LIF-permissive naive-like pluripotency.
Initially, we hypothesized that p66Shc would be essential to suppress epiblast commitment by negatively regulating ERK activity. Consequently, we predicted that p66Shc deletion from mESCs would increase ERK phosphorylation and drive differentiation. Contrary to our expectation, we discovered that p66Shc is neither essential for the self-renewal of naive mESCs nor their incorporation into the ICM of preimplantation embryos. Instead, we observed that the deletion of p66Shc increased the expression of naive-associated pluripotency genes and promoted the retention of Nanog. This was a surprising finding, considering that ERK is a negative regulator of Nanog and an established driver of epiblast and mESC commitment [28,33,34,56].
Notably, aberrant retention of naive pluripotency in p66Shc KO mESCs depended on the presence of LIF. LIF is conventionally added to mESCs in vitro to reinforce self-renewal and naive pluripotency through the downstream phosphorylation of STAT3(Y705) [36,51,57]. It has been previously shown that P-ERK mediated differentiation cues are blocked by JAK/STAT3 activity [38]. In this study, we suspect that the deletion of p66Shc is conferring P-ERK insensitivity due to elevated P-STAT3(S727). P-STAT3(S727) has been previously shown to enhance the transcriptional activity of P-STAT3(Y705) [39,40] and facilitate the maintenance of mESC naive pluripotency [43]. This interpretation is supported by enhanced expression of STAT3 target genes, klf4 and tfcp2l1, in the p66Shc KO mESCs and their sensitivity to both JAK inhibition and LIF withdrawal.
How might p66Shc inhibit ERK and STAT3(S727) phosphorylation in mESCs? Canonically, SHC adaptor proteins transduce RTK signaling and promote ERK phosphorylation by first binding to the intracellular motifs of activated RTKs, then binding to GRB2 to facilitate GRB2-RAS interactions [58 –60]. We observed that deletion of p66Shc enhanced ERK phosphorylation in the presence of LIF, consequently, one possibility is that p66Shc is antagonizing LIFR/gp130 signaling cascade. This is supported by previous findings suggesting that p66Shc can directly interact with gp130 and JAK2, in response to IL6 stimulation [61]. Alternatively, p66Shc could be directly binding to ERK to inhibit its activity, similar to what has been reported in cancer cell lines in the absence of EGF [62]. Given that SHC adaptors lack affinity for traditional LIFR/gp130 binding motifs [63], it is unlikely that p66Shc is directly participating in the regulation of LIFR/gp130 signaling.
We speculate that it is more likely that p66Shc is indirectly influencing ERK and STAT3 phosphorylation by competitively inhibiting other adaptors, most notably, SHP2 which directly participates in LIF-ERK cascades. In contrast with the other SHC isoforms, p66Shc may bind to RTKs and form stable complexes with GRB2, without activating ERK [17,18,64,65]. Analogous findings have been reported for the closely related SHCD adaptor, which has been observed to suppress ERK signaling in response to neurotrophic RTK activation by binding to and competitively inhibiting GRB2 [66]. The binding of p66Shc to a limited pool of GRB2 could competitively inhibit a wide range of GRB2 interactions and prevent ERK phosphorylation, both in response to LIF and in the absence of LIF (Fig. 7).

Proposed model for p66Shc's involvement in ERK and STAT3 signaling in mESCs in LIF-media. 1. P66Shc attenuates ERK activity by directly binding to the intracellular domains of RTKs with SHC binding motifs (EGFR, FGFR, IGFR, TRKA/Bs, IL-2/3, OCMR), competitively inhibiting the ERK-promoting SHC proteins: p52/46ShcA and the ShcB, ShcC, and ShcD families. 2. Binding of LIF to the LIFR/GP130 heterodimer activates both the JAK-STAT and SHP2-MAPK cascades. P66Shc competitively inhibits SHP2-ERK in LIF signaling by binding to and sequestering GRB2. P66Shc's inhibition of ERK impairs ERK-dependent phosphorylation of P-STAT3(S727), inhibiting STAT3 activity without influencing STAT3(Y705) phosphorylation. P66Shc may also participate in the negative regulation of P-STAT3(S727) in response to other kinases (PKC
Several studies have suggested that STAT3(S727) phosphorylation is mediated by ERK [67]. Consistent with this, enhanced P-STAT3(S727) levels in p66Shc KO mESCs coincided with elevated P-ERK. However, we also observed that ERK inhibition simultaneously promoted STAT3(S727) phosphorylation in the WT mESCs. To date, there is limited information surrounding the regulation of P-STAT3(S727); however, our findings suggest that p66Shc may antagonize STAT3 phosphorylation in mESCs in a P-ERK dependent manner. These findings warrant further investigation, which may offer insight into regulatory crosstalk between STAT3 and MAPK signaling cascades and the contradictory reports regarding P-STAT3(S727) regulation [68].
Given the dynamic nature of differentiation, and the limited number of timepoints assessed in this study, it's possible that the observed alterations in ERK and STAT3 activity are not directly responsible for the naive-like retention in the p66Shc KO mESCs. Future approaches could use phospho-proteomic screens with high temporal resolution, to closely establish the causal links between each phosphorylation event and the acquisition of cell fate. In addition, we did not assess how the deletion of p66Shc influenced mitochondrial function. P66Shc may be contributing to differentiation through its putative role in the production of reactive oxygen species, which may influence commitment through a mitochondrial-specific mechanism.
Derivation of naive-like mESCs, using conventional serum and LIF media, is restricted to certain mouse strains. Similarly, the ESCs derived from many other species, including humans, typically lack naive-like features and more closely resemble the developmentally primed epiblast stem cells of postimplantation embryos [53]. A central feature that distinguishes naive and primed ESCs is their responsiveness to LIF. Previous studies have shown that reinforcement of STAT3 activity in non-LIF-permissive mESCs, and even primed hESCs, reprograms these cells to a LIF-dependent naive-like state [69,70]. Based on these findings, studies have theorized that the function of LIF to maintain a naive-like pluripotency is conserved across species and that LIF permissiveness in vitro is determined by intracellular regulators that modulate the relative contribution of ERK and STAT3 activity [57].
Our findings implicate p66Shc's participation in the signaling cascades that mediate mESC commitment and may offer additional insight into the molecular basis of LIF permissiveness and the regulatory mechanisms contributing to preimplantation pluripotency status.
Footnotes
Acknowledgments
The authors thank the members of the Cumming and Betts labs for their valuable contributions to ideas and analysis, Jonathan Teichroeb and Courtney Brooks for their assistance in generating the p66Shc KO mESCs. The authors also acknowledge funding from the Western Strategic Support for CIHR Success Program and the Natural Sciences and Engineering Research Council of Canada (NSERC). A.P acknowledges support from the Ontario Graduate Scholarship.
Data Availability
Data available on request.
The data underlying this article will be shared on reasonable request to the corresponding author.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This research was funded by Discovery Grants from the Natural Sciences and Engineering Research Council (NSERC) of Canada, Seed and Bridge grants from Western Strategic Support and an internal research grant from the Children's Health Research Institute (CHRI). AMP was supported by an Ontario Graduate Scholarship (OGS).
Supplementary Material
Supplementary Figure S1
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.