
Abstract
Atherosclerosis (AS) is a chronic inflammatory disease associated with lipid deposition, which could be converted into acute clinical events by thrombosis or plaque rupture. Adipose-derived mesenchymal stem cell (ADSC)-encapsulated repair units could be an effective cure for the treatment of AS patients. In this study, we encapsulate human adipose-derived mesenchymal stem cells (hADSCs) in collagen microspheres to fabricate stem cell repair units. Besides, we show that encapsulation in collagen microspheres and cultured in vitro for 14 days maintain the viability and stemness of hADSCs. Moreover, we generate AS progression model and niche in vitro by combining hyperlipemia serum of AS patients with AS cell models. We further systematically demonstrate that hADSC-based microspheres could ameliorate AS progression by inhibiting oxidative stress injury, cell apoptosis, endothelial dysfunction, inflammation, and lipid accumulation. In addition, we perform transcriptomic analysis and functional studies to demonstrate how hADSCs (three dimensional cultured in microspheres) respond to AS niche compared with healthy microenvironment. These findings reveal a role for ADSC-based microspheres in the treatment of AS and provide new ideas for stem cell therapy in cardiovascular disease. The results may have implications for improving the efficiency of hADSC therapies by illuminating the mechanisms of hADSCs exposed in special pathological niche.
Introduction
Atherosclerosis (AS) is an aging-associated chronic inflammatory disease with lipid deposition in arterial intima, which is the major cause of cardiovascular diseases (CVD) [1]. Despite remarkable achievements being made and therapeutic advanced over past decades, AS remains the leading cause of mortality worldwide [2]. The pathophysiological mechanisms of AS are very complex, which probably involves dyslipidemia, lipoprotein accumulation, endothelial dysfunction, foam cell formation, and immune cell infiltration [2,3]. Increasing studies support the roles of anti-inflammation and anti-immunity therapies in mitigation of AS [3]. Therefore, stem cell-based therapy presents an innovative approach to the treatment of AS [4].
Mesenchymal stem cell (MSC)-based cell therapy has been tested in many clinical trials, which demonstrated favorable safety profiles, potent immunomodulatory effects, and anti-inflammatory action [5 –7]. Among the cell clusters, adipose-derived mesenchymal stem cells (ADSCs) are easy to obtain and expand and have lower donor site morbidity, which provided a promising future in the field of stem cell therapy and regenerative medicine [8]. Several clinical trials have tested ADSC-based treatment of inflammation-linked diseases [9,10]. Moreover, studies have demonstrated the remarkable aptitude of ADSCs as a treatment for AS by intravenous infusion into mammals [11,12]. Most of these studies have attributed the therapeutic efficacy of ADSCs to the paracrine release of anti-inflammatory and immunomodulatory effects. In addition, researchers have indicated that infusion with ADSCs could take part in the lipid metabolism by reducing serum lipid strikingly in type 2 diabetic models [13].
Studies have indicated that the behavior of ADSCs and their secreted exosomes and factors varies between two-dimensional (2D) and three-dimensional (3D) culture environments [14,15]. In addition, previous studies also demonstrated that encapsulation of donor cells improves therapeutic outcomes in kinds of disease models by delaying clearance and preventing host rejection [16]. In particular, encapsulation of therapeutic cells into microscale hydrogels forms a biofunctional microdomain or repair units, which could be used for in situ interventional therapy in many diseases. However, it remains unclear whether microencapsulation of ADSCs in hydrogel could maintain their activity and biological function, and whether ADSC-based repair units could be an effective cure for the treatment of AS.
In this study, we present an approach to fabricate human adipose-derived mesenchymal stem cell (hADSC)-based microspheres (ABMs) by combining hADSCs with collagen type I, which is the major constituent of extracellular matrix (ECM) and a natural material that is widely used for 3D cell culture [17]. In our study, we first confirmed the viability, proliferation, and stemness of hADSCs after encapsulation in ABMs and extraculturing in vitro for 14 days. We then established an AS progression and niche in vitro by combining hyperlipemia serum of AS patients with cell models. We further used a transwell system to indicate ABMs could ameliorate AS progression in vitro. In addition, we analyzed the transcriptome and function of hADSCs in microspheres, which exposed to AS niche compared with healthy microenvironment.
Materials and Methods
Ethics approval
Written informed consent was obtained from all participants in accordance with the Declaration of Helsinki. All collection of specimens and procedures were approved by the Ethics Committee of Sichuan University, West China Hospital. The ethical approval numbers are 2020-Audit-(269) and 2022-Audit-(1029).
Preparation of hADSCs
Human raw lipoaspirates from thighs of donors (n = 6) undergoing suction lipectomy were collected after obtaining informed consent from the donors as described previously [18]. The aspirate was extensively washed with saline containing 1% (v/v) penicillin/streptomycin to remove contaminating blood cells and local anesthetics. The ECM was digested with 0.1% collagenase I (Gibco) at 37°C for 1 h to release the cellular fractions. The cells were washed thrice using phosphate-buffered saline (PBS) and cultured in T-75 flasks using alpha-MEM (α-MEM; Gibco) containing 5% Helios (Gibco). HADSCs were subcultured to new flasks with fresh medium every 3 days.
Preparation of ABMs
According to the methods provided by the manufacturer (Revotek, CHN), ABMs were fabricated by loading hADSCs (0.5–1 × 108 cells) into 6 mg mL−1 collagen type I (5 mL), followed by a droplet microfluidic approach in a sterile environment (Fig. 1A). ABMs were collected and washed with solution B4 thrice in the kit. Then, ABMs were centrifuged and resuspended with complete α-MEM for further use. The sterilization methods for the instrument replaceable parts were ultraviolet irradiation and ethylene oxide treatment. For part detections, hADSCs were released and collected form ABMs after treatment with 0.1% collagenase I for 10 min at 37°C. For partial analysis, hADSCs were recollected from ABMs after preparation or extraculturing in vitro for 3, 7, and 14 days.
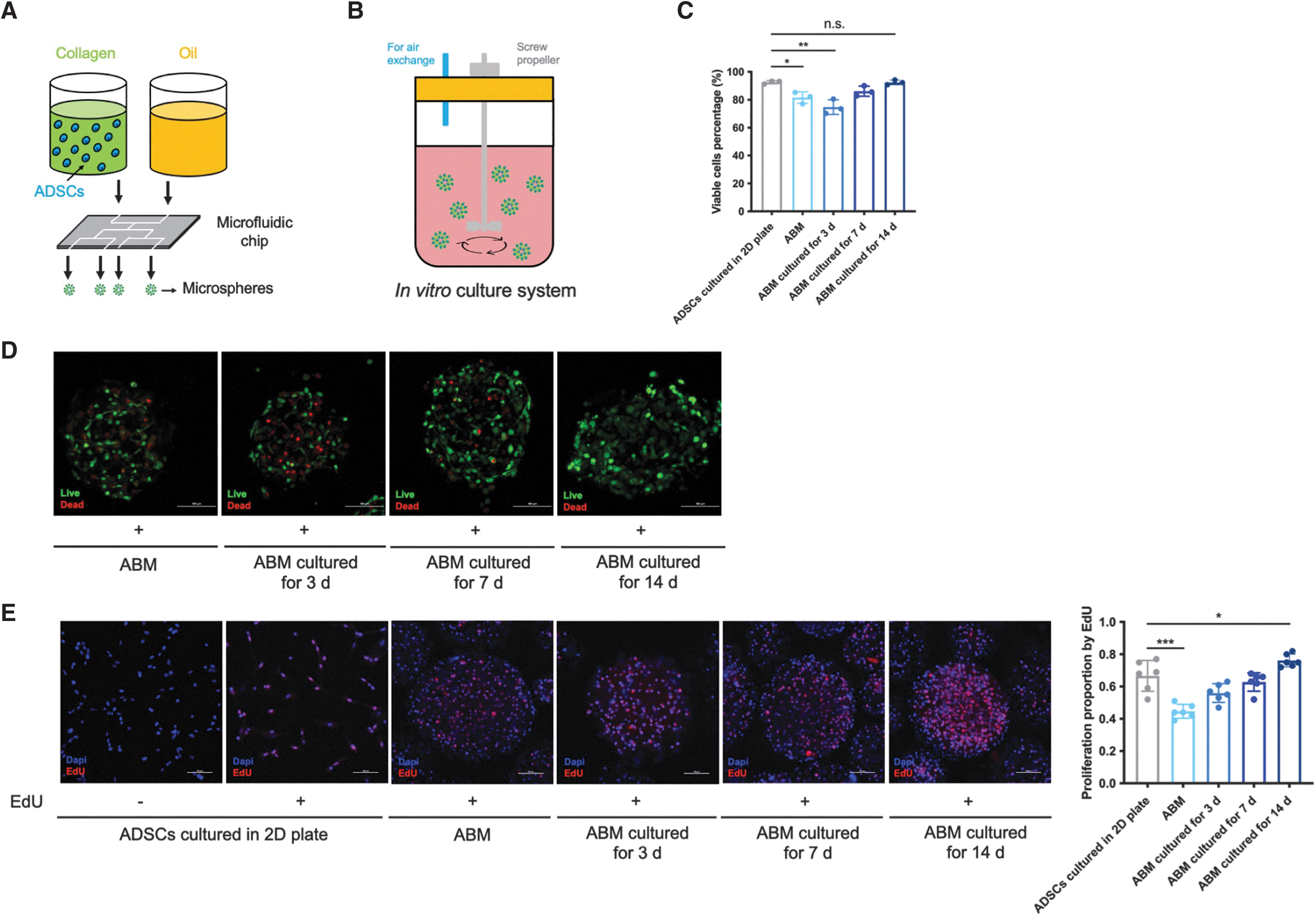
Encapsulation in microspheres maintained the viability and stemness of hADSCs.
Cell apoptosis analysis
Annexin V-FITC/PI Kit (Thermofisher) was used for tracking cell apoptosis percentage. Briefly, cells were washed with PBS and filtered through 0.22 μm membrane filters (Millipore) to remove the undigested collagen if necessary. After centrifugation at 400 g for 5 min, cells were resuspended in 100 μL of Annexin V-binding buffer. A mixture of 5 μL fluorescein isothiocyanate (FITC)-labeled Annexin V with 5 μL PI solution was added to incubate hADSCs for 10 min at room temperature in the dark before flow cytometry (FCM; Beckman Coulter) analysis.
Cell viability staining
The cells encapsulated in ABMs were stained using Calcein-AM/PI Kit (Invitrogen) as LIVE/DEAD staining to evaluate the viability percentage. ABMs were incubated for 1 h in a medium supplemented with Calcein-AM in a final concentration of 4 μM at 37°C, while PI was added to a final concentration of 5 μM. The cells in ABMs were imaged using a confocal laser-scanning microscope (Nikon, Japan).
Cell proliferation analysis
Proliferation of cells in ABMs was evaluated by 5-ethynyl-2′-deoxyuridine (EdU; Invitrogen) incorporation into DNA for 2 h. Briefly, according to the manufacturer's protocol, ABMs were incubated with α-MEM containing 10 mM EdU for 12 h before fixation, permeabilized, and stained according to the standard protocol. EdU-positive cells in ABMs were imaged using a confocal laser-scanning microscope.
Cell cycle analysis
Cell cycle assay (Invitrogen) was performed after hADSC collection from the microspheres. Briefly, detached cells were fixed with 70% ethanol at 4°C overnight. Cells were then stained with PI containing Cell Cycle Reagent. Analyses were performed by using FCM.
DNA content analysis
The DNA content in ABMs was measured by Picogreen assay (Yeasen, China) per manufacturer's instructions after treatment with 0.1% collagenase I for 10 min at 37°C. Relative DNA content of ABMs was measured by using spectrofluorophotometer (Thermofisher) at wavelength 480/520 nm.
Cell differentiation capacity analysis
For the analysis of adipogenic, osteogenic, and chondrogenic differentiation capacities, the details have been described before [18]. For the analysis of adipogenic differentiation capacity, 5 × 104 cells were plated in six-well plates. When reaching 100% confluence, the medium was changed to the adipogenic differentiation basal medium-A (ADBM-A; Cyagen) for 3 days. Following the ADBM-A was changed to the ADBM-B (Cyagen) for 1 day. ADBM-B-maintained cells until lipid droplets became larger after ADBM-A and ADBM-B were used alternately three to five times. Finally, hADSCs were fixed in 4% paraformaldehyde for 10 min, stained with fresh Oil Red-O (ORO) solution to stain lipid droplets, and then photographed. For the detection of osteogenic differentiation capacity, 5 × 104 cells were plated in six-well plates until 70% confluence.
The cells were then cultured in osteogenic differentiation basal medium (Cyagen) for 3 weeks before fixing in 4% paraformaldehyde and stained by Alizarin Red. For the study of chondrogenic differentiation capacity, 5 × 105 cells were centrifuged and collected in the bottom of 15 mL centrifuge tube. The culture medium was changed to the chondrogenic differentiation basal medium (Cyagen) for 4 weeks after cells gathered to a pellet. Alcian blue staining was used to evaluate the capacity of hADSC pellets to differentiate toward chondrocyte pellets.
Surface markers analysis
Cells were incubated with fluorescent-labeled monoclonal antibodies CD45-FITC (1:10; BD), CD19-FITC (1:33; EBioscience), CD34-FITC (1:100; Abcam), HLA-RD-PC5.5 (1:10; BD), CD14-PC7 (1:50; BD), CD90-PE (1:33; BD), CD73-APC (1:50; BD), CD44-PE (1:50; BD), CD29-APC (1:10; BD), and CD105-PC7 (1:50; EBioscience) at 4°C for 30 min in the dark. After washing steps, the labeled cells were analyzed by using FCM.
Immunofluorescent staining
ABMs or cells were washed with PBS and fixed with 4% paraformaldehyde for 30 min. Samples were then blocked with 2% bovine serum albumin (BSA) in PBS for 1 h and incubated with the primary antibody Oct-4 (1:50; Abcam), Sox-2 (1:50; Abcam), CD31 (1:100; Abcam), and SM22α (1:50; Abcam) at 4°C overnight. Following incubation with FITC-conjugated goat anti-rabbit IgG (1:100; Abcam) for 1 h at room temperature, ABMs/cells were counterstained by incubation with DAPI. Images were obtained using a phase contrast fluorescence microscope.
Obtaining of human serum
Human hyperlipemia serum samples (n = 51) were obtained from patients undergoing AS or coronary heart disease. The inclusion criterion for patient selection was hyperlipidemia. The exclusion criteria were hyperglycemia, hypertension, or endocrinopathy. Patient characteristics are presented in Supplementary Table S1. Human healthy serum samples (n = 50) were obtained from the healthy donors going through medical examinations. The serum samples of each donor were collected and mixed homogeneously to minimize donor variability.
Cell culture
Human umbilical vein endothelial cells (HUVECs, cell line) were obtained from Regenerative Medicine Research Center and cultured in Dulbecco's modified eagle medium (DMEM) (Cyagen) containing 10% fetal bovine serum (FBS). Human monocytes (THP-1, cell line) were obtained from the Lab of Transplant Immunology. The cells were cultured in RPMI-1640 medium (Cyagen) supplemented with 10% FBS. THP-1 monocytes were incubated in a complete RPMI-1640 medium containing 100 ng mL−1 phorbol-12-phorbol myristate acetate (PMA; Sigma Aldrich) for 48 h to differentiated into THP-1 macrophages.
Generating of AS cell model
HUVECs and THP-1 macrophages were treated with DMEM or RPMI-1640 complete medium containing 20, 50, and 100 μg mL−1 ox-LDL for 48 h.
Cell viability analysis
Cells were incubated with CCK-8 (Beyotime, China) solution and media (1:9 dilution) at 37°C for 2 h after washing with PBS, and then absorbance was measured at 450 nm using a microplate reader (Thermofisher).
Determination malondialdehyde production and superoxide dismutase activities
Cells were harvested, resuspended in extracting solution, lysed by ultrasonic pyrolysis, and centrifuged at 8,000 g for 10 min at 4°C. Two hundred microliters supernatant was mixed with detection working fluid for malondialdehyde (MDA; Solarbio, China) at 37°C for 30 min or superoxide dismutase (SOD) (Solarbio) at 100°C for 60 min, respectively. A microplate reader was used to measure absorbance values of the reaction mixture at 450, 532, and 600 nm (MDA), and at 560 nm (SOD).
ORO staining
After washing with PBS, cells were fixed with 4% paraformaldehyde for 20 min. ORO solution was added before the cells were incubated for 30 min at 37°C. The ORO in the cell cultures was redissolved into isopropanol and analyzed at 490 nm by a microplate reader, respectively.
Generating of AS progression and niche in vitro
HUVECs and THP-1 macrophages at ratio of 1:1 were stimulated in RPMI-1640 complete medium with 50% human hyperlipemia serum pool (v/v) and 100 μg mL−1 ox-LDL for 48 h.
Lactate dehydrogenase content measurement
Cells were pretreated with various condition medium (CM) for 48 h. To collect lactate dehydrogenase (LDH) content, 2 mL of RPMI-1640 basal medium was added to replace CM. After 24 h, LDH content in the media was assessed using an LDH activity kit (Solarbio) and absorbance was measured at 450 nm using a microplate reader.
Reactive oxygen species production measurement
After pretreatment, cells were incubated with 10 μM dichlorofluorescin diacetate (DCFH-DA) solution and washed with serum-free RPMI-1640 medium. Intracellular reactive oxygen species (ROS) accumulation of cells was measured by using spectrofluorophotometer (Thermofisher) at wavelength 488/525 nm.
Western blot
Cells were washed with PBS and collected from well plates by scraping. Cells were lysed in radioimmunoprecipitation assay (RIPA; Beyotime) buffer supplemented with protease inhibitor cocktail and phosphatase inhibitors (Roche, SUI). Bicinchoninic acid (BCA) Protein Assay (Thermofisher) was used to determine protein concentration. Twenty micrograms of total protein was loaded for every sample. Samples were run on sodium dodecyl-sulfate polyacrylamide gel electrophoresis (4%–20% Tris Glycine gel; Yeasen) at 100 V and transferred at 300 A onto polyvinylidene fluoride membranes (Bio-Rad).
Membranes were blocked in 5% BSA for 1 h at room temperature and incubated overnight at 4°C with the following primary antibodies: goat anti-CD36 (1:2,000; Abcam), Bax (1:1,000; Abcam), Bcl-2 (1:1,000; Abcam), Caspase-3 (1:1,000; Abcam), CD31 (1:2,000; Abcam), SM22α (1:2,000; Abcam), and mouse anti-β-actin (1:2,000; Abcam). Membranes were washed in PBST (PBS with 0.1% Tween-20 detergent) buffer before incubation with secondary antibodies at room temperature for 1 h (goat anti-rabbit (1:3,000; Proteintech) and rabbit anti-mouse (1:5,000; Proteintech). Membranes were washed in PBST and imaged using densitometry with a computer image analyzing system (GE).
Determination of lipids and proinflammatory factors
Supernatant medium was collected after treatment. Liposome levels were measured using an automatic biochemical analyzer (Beckman Coulter). The levels of proinflammatory factors were measured using ELISA kits (EBioscience) according to the methods provided by the manufacturer.
Quantitative real-time polymerase chain reaction
Total RNA from cultured cells was extracted using TRIzol reagent (Thermofisher) and reverse transcribed into cDNA using ABScript III RT Master Mix (Abclonal, CHN). Real-time polymerase chain reaction (PCR) was performed on the Applied Biosystems. The quantitative real-time PCR system contained 1 μL of cDNA sample solution, 5 μL of SYBR-Green PCR master mix, 0.2 μL of forward and reverse primers, and 3.8 μL of H2O. The data were analyzed using the ΔΔCt method, with a β-actin reference in the mRNA analysis. The primer sequences are in Supplementary Data S1.
Bulk RNA-seq
Total RNA was isolated from ABMs using TRIzol Reagent (Thermofisher) and following the manufacturer's protocol. The samples, diluted with RNAse-free water, were shipped to Applied Protein Technology (CHN) for RNA sequencing and data analysis. The details have been described in Supplementary Data S2.
Statistical analysis
All statistical analyses were performed using GraphPad Prism version 9.3.1. Data are presented as the mean ± standard deviation (SD). The results were analyzed by one-way analysis of variance or unpaired Student's t-tests when continuous variables were normally distributed. A two-tailed P value <0.05 was considered statistically significant. (* indicates P < 0.05, ** indicates P < 0.01, *** indicates P < 0.005, **** indicates P < 0.001, n.s. indicates nonsignificant difference, and n.d. indicates nondetected).
Results
Encapsulation in microspheres maintained the viability and stemness of hADSCs
We leveraged a droplet microfluidic approach to fabricate ABMs by encapsulating hADSCs with collagen type I (Fig. 1A) and an ABM culture system for in vitro culturing (Fig. 1B). This approach yielded collagen-encapsulated microspheres with a high proportion of viable hADSCs (Fig. 1C). The confocal images revealed that hADSCs were well distributed throughout the microspheres (Fig. 1D). However, collagen encapsulation decreased hADSC viability and kept small part of hADSCs from proliferating for the first 3 days of culturing in vitro (Fig. 1C–E). The viability and proliferative ability of hADSCs were significantly rescued after 14 days of culture (Fig. 1C–E).
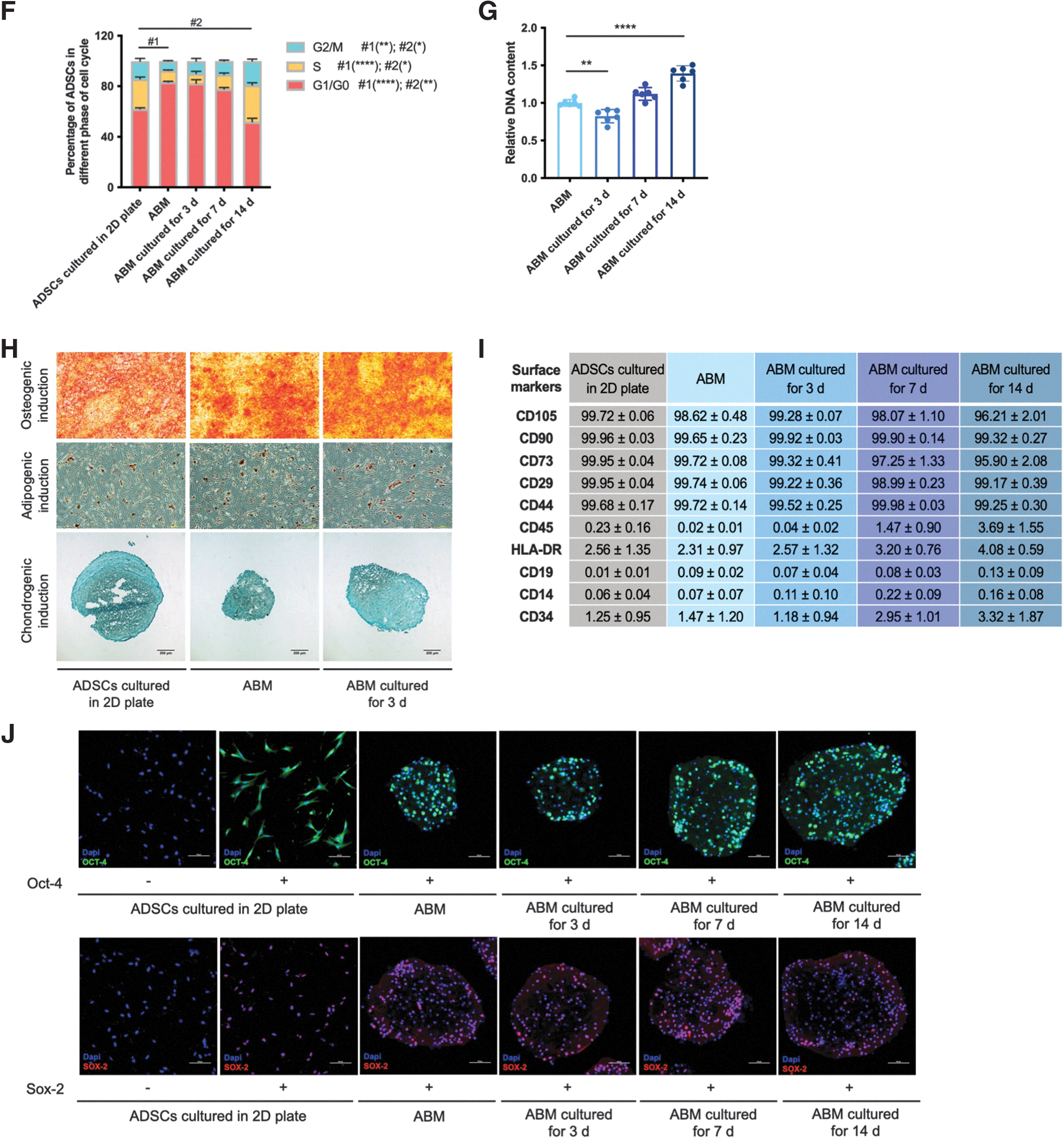
Notably, 3D encapsulation and culturing for 3 days decreased dividing cells as the percentage of G1/G0 phase increased, an effect that could be rescued by 14 days of culturing (Fig. 1F). In addition, the detection of DNA content in ABMs after 3D encapsulation and culturing for 3, 7, and 14 days verified the results above (Fig. 1G). Importantly, the stemness of hADSCs maintained for at least 14 days after encapsulation and culturing in vitro, which confirmed by three directional differentiation capacities (Fig. 1H), FCM analysis of 10 surface markers (Fig. 1I), and immunofluorescent staining of Oct-4 and Sox-2 (Fig. 1J).
AS progression and niche in vitro
For the generation of AS cell model in vitro, we used ox-LDLs to induce HUVECs to simulate endothelial injury and THP-1 macrophages to transform into foam cells. The cultured HUVECs displayed a reduction of cell viability dependent on various concentrations of ox-LDL (Fig. 2A). ox-LDL induced endothelia injury in HUVECs, as evidenced by remarkably increased MDA level and decreased SOD activity in all concentrations (Fig. 2A). Besides, ox-LDL significantly reduced activity of THP-1 macrophage (Fig. 2B). ox-LDL-induced THP-1 macrophages also had higher MDA release and lower SOD activity than the control group, which are involved in oxidative stress and progression of atherosclerotic plaque (Fig. 2B). In addition, the formation of foam cells was evaluated by ORO staining, and the results showed that ox-LDL treatment could induce accumulations of lipid droplets (Fig. 2C). Therefore, 100 μg mL−1 ox-LDL-induced HUVECs and THP-1 macrophages were used to produce AS cell model.
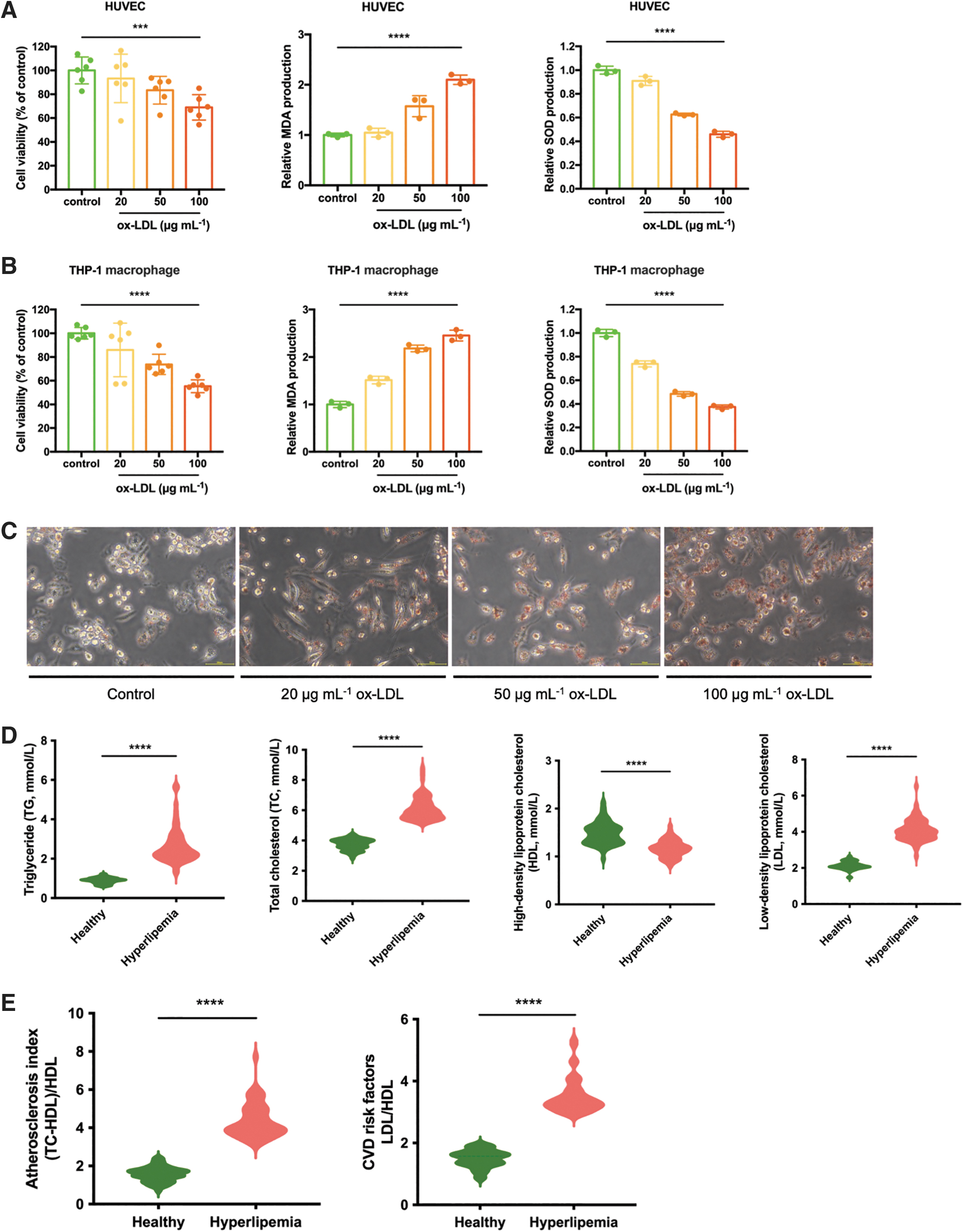
AS progression and niche in vitro.
We established AS progression and niche in vitro by combining hyperlipemia serum of AS patients with the generation AS cell model. The average age of the AS patients completing the study was 59.1 years (±12.4, SD, n = 51), and of healthy people was 22.8 years (±7.7, SD, n = 50). There was a significantly higher level of total cholesterol (TC), triglyceride (TG), and low-density lipoprotein (LDL) and lower level of high-density lipoprotein (HDL) in AS patients compared with healthy ones (Fig. 2D).
The significant increase of AS index indicated a higher risk of AS (Fig. 2E). Meanwhile, the high LDL/HDL ratio, a risk factor for cardiovascular disease, was also higher in AS patients (Fig. 2E). These results are consistent with the characteristics of AS and hyperlipidemia patients, which indicated that these blood samples with ox-LDL (100 μg mL−1)-induced cells could be effective in mimicking the AS progression and niche in vitro.
ABMs inhibit AS progression in vitro
We used a transwell system to examine whether ABMs could inhibit AS progression in vitro (Fig. 3A). The oxidative stress was first evaluated. ABMs rescued the high level of MDA (Fig. 3B) and increased the SOD activity (Fig. 3C) in cells of AS progression. In addition, the high level of LDH release in AS progression was attenuated by the treatment of ABMs (Fig. 3D). Besides, the augmented production of intracellular ROS in group AS progression was attenuated by ABMs, which was observed by detecting DCFH-DA (Fig. 3E).
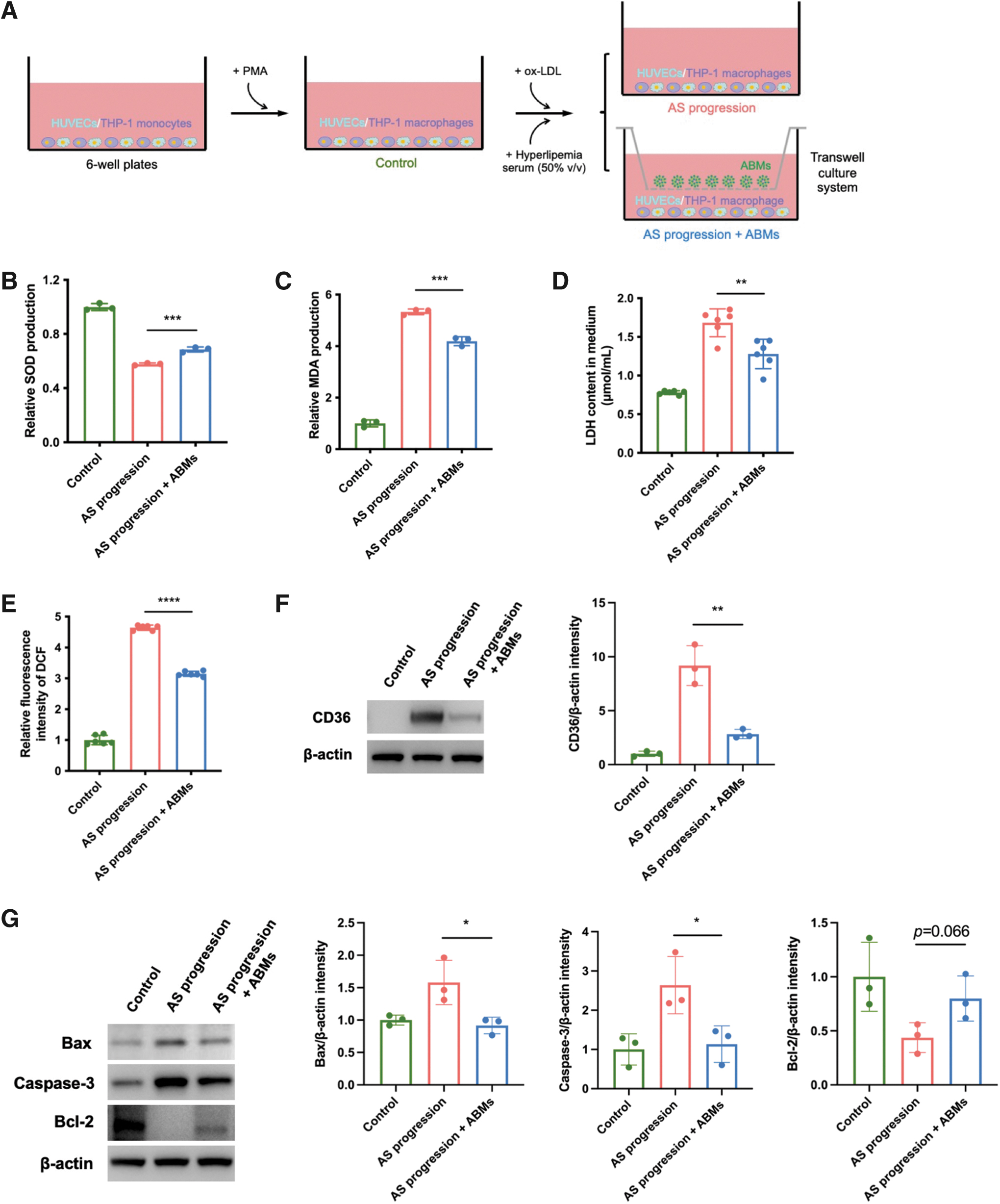
ABMs inhibit AS progression in vitro.

Moreover, the high expression of CD36 indicated the oxidative damage of macrophages in AS progression, which also decreased by the treatment of ABMs (Fig. 3F). The effect of ABMs on cell apoptosis in AS progression was examined. The proapoptotic proteins such as BAX and Caspase-3 were downregulated, although with slight upregulation (but no significant difference) in antiapoptotic protein (Bcl-2) (Fig. 3G); the result was verified by using FCM analysis (Fig. 3H).
To examine whether ABMs could facilitate lipid metabolism in AS progression, we preformed biochemical analysis to quantify the level of liposomes in the culture supernatant. Compared to the AS progression group, the levels of TG and TC decreased when treated with ABMs, despite no difference in HDL and LDL levels (Fig. 4A). Furthermore, we preformed ELISA test to know whether ABMs could reduce inflammation in AS progression. The levels of 3/4 tested proinflammatory cytokines, IL-1β, IL-6, and TNF-α (except interferon gamma), in culture supernatant significantly decreased in the presence of ABMs (Fig. 4B). We next examined whether ABMs could decrease endothelial dysfunction in AS progression.

ABMs inhibit AS progression in vitro.
The low expression of the endothelial marker CD31 and high expression of the mesenchymal marker SM22α indicated the endothelial-mesenchymal transition (EndMT) in AS progression. As we expected, these effects were mitigated by ABMs (Fig. 4C). Besides, the mRNA levels of AS-related genes such as VEGF, IL-6, MCP-1, and ICAM-1 were obviously upregulated in AS progression cells, while ABM treatment could reverse these aberrant changes (Fig. 4D). Also, of note, the treatment with ABMs had significantly lower THP-1 macrophage foaming, which was evaluated by ORO staining (Fig. 4E). These results together indicated that ABMs could be beneficial for the amelioration of AS progression in vitro.
ADSCs in microspheres respond to AS niche
To gain an understanding of the mechanism of hADSCs in microsphere response to AS niche, we used a transwell system to examine the transcriptome (Fig. 5A). To characterize early changes in gene expression of hADSCs, we performed bulk RNA-seq on ABMs treated with AS niche, healthy CM, or basal medium (BM, baseline control). We performed the principal component analysis, which indicated that AS niche and BM groups were clearly clustered into two separate groups (Fig. 5B). However, the distribution of healthy CM group was overlapped and could not be separated adequately with the other two groups (Fig. 5B).
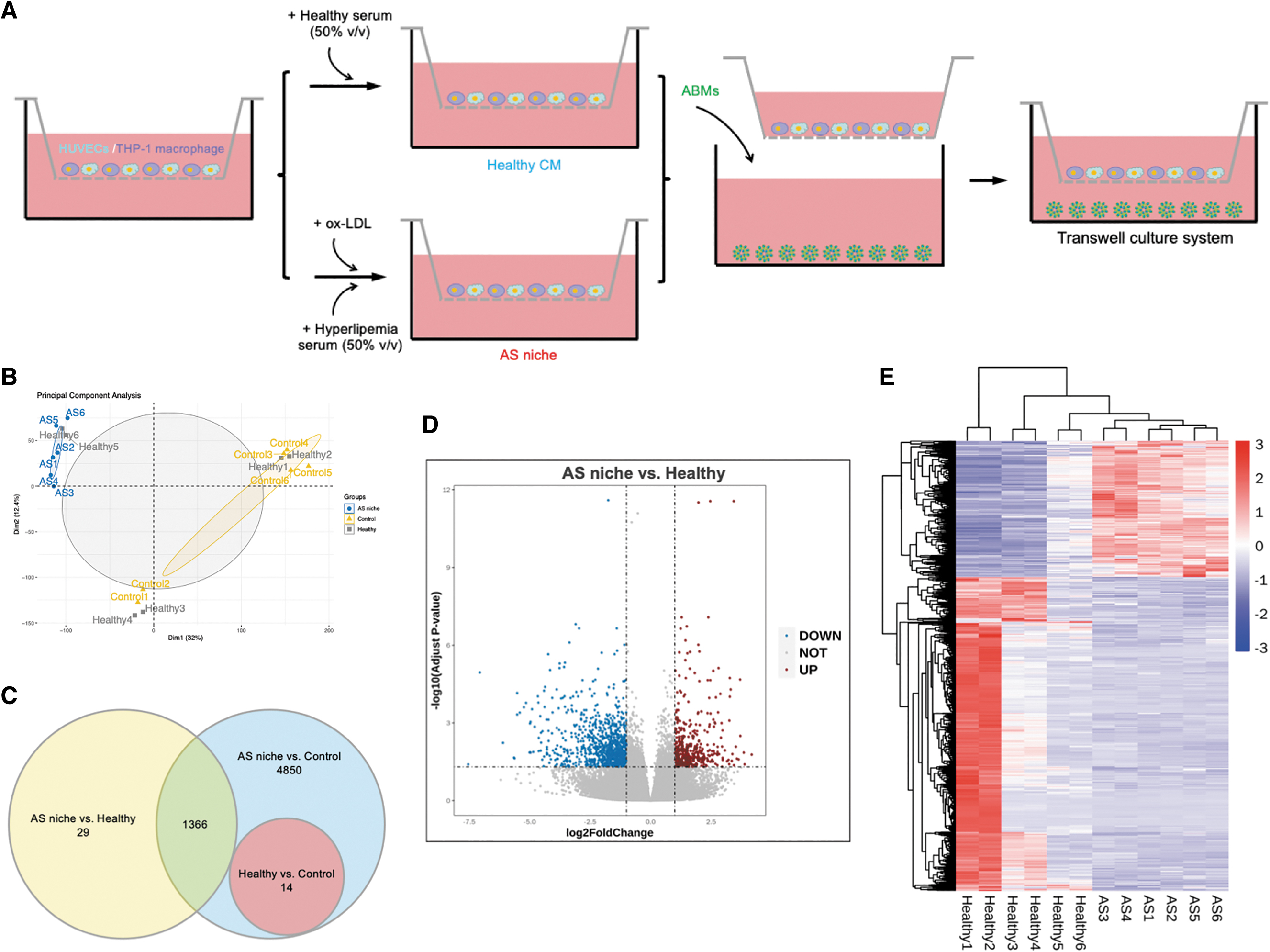
ADSCs in microspheres respond to AS niche.

A three-way Venn diagram analysis was performed to evaluate the number of differentially expressed genes (DEGs) among groups. Venn diagram analysis revealed 1395 DEGs between AS niche and healthy groups and 6216 DEGs with AS niche relative to baseline control (Fig. 5C). Besides, consistent with our expectation, few genes (14 DEGs) were differentially expressed upon comparing healthy group and BM (Fig. 5C). As a result, further comparison and analysis were made between group AS niche and healthy CM. We identified 422 upregulated and 973 downregulated genes upon AS niche treatment compared with healthy one in Volcano Plot and Heat Map (Fig. 5D-E).
To investigate the functions of the DEGs, we identified the enriched gene ontology (GO) terms, which covered a wide range of cellular components, molecular functions, and biological processes, and top 30 important upregulated/downregulated enriched GO terms are presented (Fig. 5F). Among them, 941 upregulated DEGs for AS niche treatment compared with healthy one were enriched for GO terms involved in vasculature development (GO:0001944), blood vessel development (GO:0001568), oxidative stress response (GO:0006979), ECM (GO:0031012), and angiogenesis processes (GO:0001525), which are fundamental to the development and progression of
The enrichment of biological pathways by our DEG dataset was assessed using Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis; the top 20 upregulated/downregulated pathways are as shown and including axon guidance, kinds of cardiomyopathy and vascular smooth muscle contraction (Fig. 5G and Supplementary Table S3). Taken together, these analyses provide evidence of a likely key role and mechanism of ADSCs (or ABMs) in ameliorating
Discussion
The identification of resident stem cells in the vessel wall has led to a significant investigation of MSCs into their therapeutic potential and contributions to the treatment of CVD [11]. Hence, MSCs have been widely studied in both human clinical trials and animal experiments, which have indicated a modulatory role in vascular physiology and disease [11]. Compelling evidence has been presented on bone marrow-derived MSCs (BMSCs) as a treatment for AS by inhibiting endothelial dysfunction, reducing hyperlipidemia, decreasing inflammation, and stabilizing existing atherosclerotic plaques [19]. However, ADSCs, another cluster of MSCs, are ubiquitous and can be easily harvested from adult, which their therapeutic effect on AS remains unclear for clinical study [20]. Our work systematically demonstrated similar roles of hADSCs on the progression of AS compared with BMSCs.
Most previous studies tested therapeutic effects of MSCs on disease by intravenous transfusion or injection [21]. However, it has been reported that about a quarter of intravenously infused MSCs survived after 1 day, and only 1% of MSCs persisted more than a week [22,23]. Besides, a large proportion of injected MSCs trapped in the lung microvasculature instead of the target tissues [24]. It is true that one of the important therapeutic approaches of MSCs is the infusion or perfusion therapy. However, MSCs also could be used to build 3D scaffolds for the reconstruction of damaged tissues. For instance, in the works by Ae-Kyeong Kim have revealed that fibrin matrix containing HUC-MSCs cooperate with balloon could be a potential and effective solution for the treatment of intimal hyperplasia [25].
Based on this approach, our study aims to fabricate ABMs, which could be further combined utilization with hydrogel for 3D bioprinting and in-situ remediation for atherosclerotic damaged vascular tissue. We believe our further study could provide another effective option for the treatment of AS. We encapsulate hADSCs in collagen microspheres to form cell therapy units, which not only provided a 3D culture environment, extended cell survival, and residence but also may expand its application range. More broadly, 3D bioprinting of hydrogels containing MSC microencapsulation units could be applied as dressing to promote wound healing, or even as organoid for local tissue regeneration [26].
Indeed, many studies have indicated that biological performances of MSCs such as anti-inflammatory ability, stemness maintain, cell survival after transplantation, angiogenic and tissue reparative effects, and paracrine function were enhanced by in 3D microenvironments compared with 2D plate [15,26,27]. In addition, in our work, the viscosity of the collagen solution is higher than the traditional hydrogel precursors like HAMA, GelMA, or AlgMA. This requires strong mechanical stirring to achieve satisfactory ADSC encapsulation.
However, the mixing process might result in undesired mechanical damage to the encapsulated ADSCs, which we hypothesis is the main reason for the unsatisfactory biological performance of ADSCs in viability, proliferation, and cell cycle at the early stages after cell encapsulation. However, after long-time culture in vitro, the cell viability and proliferation ability of hADSCs in ABMs significantly increased, which showed more advantages of hADSCs cultured in 2D plates. Moreover, encapsulation and long-time culture in vitro could maintain the stemness of hADSCs, which was beyond our expectations.
Dysfunction of endothelial cells, recruitment of macrophage, development of plaques, hyperlipidemia, and chronic inflammatory microenvironment are all involved in AS progression. Ox-LDL contributes to the formation and progression of atherosclerotic plaquehas been reported by many studies, which has been wildly used in the establishment of AS cell models [28]. Hyperlipidemia is considered to be a risk factor leading to AS plaques and always accompanies AS patients lifelong [29]. In addition, previous studies have suggested that serum of AS patients could be used in the construction of oxidative (morbid) environment and has the ability to induce cholesterol deposition in cultured VSMCs or blood-derived monocytes or macrophages [30]. Therefore, we believe the AS cell models added with hyperlipemia serum of AS patients in our experiment is better in mimicking the AS progression and niche in vivo compared with single ox-LDL-induced cell model.
For the initial progression of AS, oxidative stress plays central roles by acting on multiple cells through ox-LDL and downstream LOX-1 [28]. Oxidative stress activates pathways of apoptosis, increases ROS, causes endothelial dysfunction in endothelial cells, inhibits migration, and stimulates foam cell formation in macrophages [28]. Among the biomarkers, SOD family, one of the main antioxidant systems, plays an important role in oxidative stress modulation [31]; MDA is the most frequently measured in oxidative stress and lipid peroxidation [32]; LDH produces in normal cells and releases into the circulation when cells suffered damage such as oxidative stress injure [33]; Scavenger receptor CD36 is a multifunctional membrane protein that promotes thrombosis in conditions of oxidative stress and promotes ROS generation [34].
In addition, oxidative stress can also trigger and enhance apoptosis and programmed cell death, which is the characteristic of atherosclerotic lesions [35]. Our work suggests that ABMs could suppress oxidative stress injury and cell apoptosis when co-cultured with AS niche.
Accumulating evidence demonstrates that ADSC transplantation could decrease liposome level by facilitating lipid metabolism in several diseases, including non-alcoholic fatty liver disease [36]. A recent rat study has suggested ADSC transplantation could alleviate pathological symptoms of aortic AS by decreasing the serum levels of TC, TG, and LDL and increasing HDL [37]. Our result indicates a difference that ABMs reduced only TG and TC level, but not LDL, which probably is due to different metabolic mechanisms between in vivo and in vitro.
Limitations of this work include the relatively short time of ABM treatment, the AS progression model in vitro, which could not fully simulate the AS pathological process in vivo. This study aims to explore the effect and mechanism of ABMs on the treatment of AS preliminarily. Further experiments in vivo are going to be carried out. Studies also suggest ADSCs, and their secreted factors may attenuate inflammation in animals with AS, type 2 diabetes, or obesity [13,37,38]. Our work confirms this phenomenon in vitro by using ABMs in co-culture system.
For endothelial dysfunction, studies have indicated another role for the endothelium in AS development and progression -EndMT-, which linking to the vascular remodeling [39]. Our work demonstrates that EndMT can be reversed by the treatment of ABMs as evidence by increasing the expression of CD31 and lowering the expression of SM22α, which represent endothelial cells and mesenchymal transition cells, respectively.
Endothelial dysfunction also leads to the expression of chemokines and cytokines (IL6 and MCP-1) and adhesion molecules (VCAM-1 and ICAM-1) that attract and facilitate immune cell extravasation [39]. Our results indicate the mRNA levels in AS model cells could be downregulated by the treatment of ABMs. Furthermore, lipid accumulation in macrophage decreases significantly in the presence of ABMs.
A recent review suggests that preactivation in pathological microenvironment may improve therapeutic efficacy of MSCs [40]. Therefore, it is necessary to comprehend the response mechanism of MSCs in the pathological microenvironment. Our work aims to investigate the response of hADSCs in microspheres following exposure to the healthy or AS niche microenvironment by providing insight into transcriptome and function alterations. Our results indicate that hADSCs in microspheres elicit few DEGs when exposed to a healthy microenvironment compared with those in baseline control. The result is coincident with the expectation that there is no need for tissue repair and regeneration as in the absence of pathological conditions.
However, hADSCs elicit thousands of DEGs in AS niche group versus healthy or baseline control groups. The transcriptome of ADSCs dramatically alters following AS niche stimulus, potentially caused by participating in damage repair and regeneration. Of relevance, some DEGs are already known to play major roles in AS and CVD, which include SAA1 [41], ACTA2 [42], APOD [43], CSRNP3 [44], PDGFD [45], and PENK [46]. Function and pathway analysis were further identified based on the DEGs between groups. Some upregulation enrichment for GO terms are of central relevance in vascular disease, such as vasculature and blood vessel development, oxidative stress response, ECM, and angiogenesis processes.
Conclusion
Our study indicated that encapsulation in collagen microspheres and cultured in vitro for 14 days maintained the viability, proliferation, and stemness of hADSCs. Besides, the study established AS progression and niche in vitro by combining hyperlipemia serum of AS patients with cell model of AS. In addition, our results systematically demonstrated that ABMs could inhibit AS progression by suppressing oxidative stress injury, restraining apoptosis, inhibiting endothelial dysfunction and inflammation, and decreasing lipid accumulation. Furthermore, our study identified and validated the mechanism of ADSC response to AS niche, and the potential mechanism of ABMs in how to inhibit AS progression.
Footnotes
Acknowledgments
We thank to Prof. Kang, Y. James from Regenerative Medicine Research Center for his financial support. We thank members of the Regenerative Medicine Research Center for their support, expertise, and use of facilities.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This study was supported by grants from Sichuan University West China Hospital (ZY2016202).
Supplementary Material
Supplementary Data S1
Supplementary Data S2
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.