
Abstract
Intracerebral hemorrhage (ICH) is a common subtype of stroke with a very high mortality rate, but there is still no effective cure. Increasing evidence suggests that heme accumulation and neuronal ferroptosis play an important role in secondary injury after ICH. Neural stem cells (NSCs), as seed cells of the central nervous system, have received much attention due to their abundant paracrine product components and low immunogenicity. In this study, we focused on the protective mechanism of neural stem cell secretome (NSC-S) against neuronal ferroptosis in an ICH mouse model using hemin-induced in vitro models and collagenase type IV-induced in vivo models. The results showed that NSC-S could ameliorate neurological deficits and reduce neuronal injury in ICH model mice. In addition, NSC-S reduced heme uptake and ferroptosis in hemin-treated N2a cells in vitro. NSC-S induced the activation of Nrf-2 signaling pathway. However, these effects of NSC-S were abolished by the Nrf-2 inhibitor ML385. Notably, HSPE1 in NSC-S may be associated with the protection of NSC-S against hemin-injured neurons via the Nrf-2 signaling pathway. In summary, NSC-S protects against secondary neuronal injury in ICH via the Nrf-2 signaling pathway. Also, this functionality may be implemented by HSPE1.
Introduction
Intracerebral hemorrhage (ICH) is a subtype of stroke with a high mortality rate and morbidity, which affects more than 1 million people worldwide each year [1,2]. In the acute phase of ICH, a large amount of hemoglobin and heme are released after the destruction of erythrocytes, and the content of heme can reach 10 mM in the center of the hematoma. In addition, there are a large amount of ferrous ions in the extracellular space, which induces the formation of reactive oxygen species (ROS) and lipid peroxidation, and ultimately leads to neuronal ferroptosis and cerebral edema [3 –6]. Ferroptosis is a newly discovered programmed cell death caused by the accumulation of iron-dependent lipid ROS, which is closely related to the oxidative damage of cells [7,8].
Recent studies have shown that heme accumulation and neuron ferroptosis after ICH play an important role in secondary injury in ICH [9,10]. Therefore, targeting heme accumulation and ferroptosis in ICH may be a potential strategy for the treatment of ICH.
Hematoma clearance is still the primary treatment for ICH. However, it has many limitations and can lead to secondary injuries [11]. In addition, there is a lack of effective pharmacological treatments approved for the treatment of ICH. Therefore, it is urgent to find new strategies to promote neuron survival, remove hematoma, and accelerate the recovery of nerve function. Stem cell therapy has been a hot topic in recent years because of its ability of self-renewal and differentiation into lineage-specific cells (eg, neurons, oligodendrocytes), and neural stem cells (NSCs) become the best choice for the treatment of central nervous system (CNS) diseases [12 –14]. However, NSC transplantation has some problems such as insufficient sources and immune rejection, which leads to the new focus of paracrine therapy of NSCs. Notably, neural stem cell secretome (NSC-S) has been shown to play an important protective role in the CNS diseases [15 –19].
Our previous studies have found that NSC microvesicles and secretome (NSC-S) have the function of promoting nerve repair in vitro and in vivo [20 –23], especially for CNS diseases such as traumatic brain injury and Parkinson's disease, and NSC-S is superior to NSC microvesicles in the ability of comprehensive repair of nerve injury. Based on the above background, we hypothesized the following: Could NSC-S significantly inhibit the pathological damage caused by secondary injury after ICH? Is it by reducing the accumulation of heme and then inhibiting neuron ferroptosis?
To verify the scientific hypothesis, this study intends to investigate the effect of NSC-S in the treatment of ICH model mice, and to further explore the effect of NSC-S on heme metabolism accumulation and its protective effect on neuron ferroptosis and the underlying mechanism.
Materials and Methods
Cell culture
Cell cultures were performed according to our previous study [24]. NSCs derived from human embryonic stem cells were treated with 1% penicillin–streptomycin, 20 ng/mL of basic fibroblast growth factor (PeproTech, USA), and 20 ng/mL of epidermal growth factor (PeproTech) in Dulbecco's modified Eagle's medium (DMEM)/F12 (Gibco, USA). The differentiated human NSCs morphologically have a typical rosette-like structure (Supplementary Fig. S1) and express multiple NSC surface markers that can undergo adherent-sphere conversion; after being cultured on low adhesion plate for 48–72 h, neurospheres were formed. BV2 and N2a cells were purchased from the Cell Resource Center, Peking Union Medical College (Headquarter of National Infrastructure of Cell Line Resource, NSTI), and were cultured in DMEM (Gibco) with 10% fetal bovine serum (Gibco).
As previously described [24], when the confluence of NSCs was about 90%, the P7-P15 NSC was washed four times with serum-free DMEM and maintained in DMEM for 6 h. The conditioned medium was collected and centrifuged at 2,000 rpm for 20 min. The collected supernatant was filtered with a 0.22 μm membrane to produce NSC-S and stored at −80°C. For in vivo experiments, NSC-S was concentrated 10-fold using a lyophilizer (Labconco FreeZone, England) and stored at −80°C.
ICH mouse model establishment and NSC-S treatment
Six- to eight-week-old male C57BL/6 mice (20–25 g) were purchased from the Laboratory Animal Center of Jiangsu University (Zhenjiang, China). The animals were raised in plastic cages with a 12-h light/dark cycle and free food and water intake. Take an animal as an experimental unit. All animal experiments were approved by the Ethics Review Committee of Jiangsu University (Zhenjiang, China) (Permit No. SCXK 2018-0012).
The ICH model was established by stereotactic brain injection of collagenase IV (Gibco; 17104-019) using the method described previously [25]. At first, the animals were anesthetized intraperitoneally with chloral hydrate (36 mg/kg) and fixed on a stereotaxometer (RWD, Shenzhen, China). A 0.5-mm cranial burr hole was drilled 0.2 mm anterior to the bregma and 2.0 mm lateral to the right of the midline by a 10 μL-sized microinjector (RWD). The ICH model group (n = 38) was injected with collagenase IV (0.045 U/1 μL PBS), and the sham group (n = 19) was injected with the same volume of PBS at the same site. The injection site was at a depth of 3.5 mm (related to bregma: AP = +0.2 mm, ML = −2 mm, DV = −3.5 mm). The injection rate was 0.2 μL/min. The needle was kept in place for 5 min after each injection to avoid backflow and pulled out slowly.
After 24 h, all animals were divided into three groups. Among them, the sham operation group (n = 19) was injected with concentrated 10 × DMEM and named the sham group. The successfully modeled mice in the ICH model group were divided into two groups according to the single blind method: ICH +10 × DMEM group (n = 19) and ICH +10 × NSC-S group (n = 19). The drug 10 × DMEM and 10 × NSC-S were similar in appearance and were both liquids. After 24 h of injection of bacterial collagenase IV, the ICH +10 × DMEM group received 4 μL of 10 × DMEM, while the ICH +10 × NSC-S group was injected with the same volume of 10 × NSC-S. The injection site of the two drugs was the same, near the edge of ICH, specifically AP = +0.2 mm, ML = −2 mm, DV = −3.0 mm [26]. Behavior evaluation was performed on the 0, 1, 3, 7, and 14 days after NSC-S injection.
Behavior evaluation
Animal behavior test was performed by a trained observer (other than the original researcher) who remained blind to the allocation group to which the mice belonged, throughout the entire study. After the behavioral test, the data were analyzed by the original researcher.
Modified Neurologic Severity Score
The researchers observed the degree of nerve defects and nerve damage in mice according to the 18-point Modified Neurologic Severity Score (mNSS) scoring standard [27]. mNSS contains sensory tests, motor tests, reflex tests, and beam balance tests, with a maximum score of 18. The higher the score, the more severe the behavioral deficits caused by nerve injury. Scores of 1–6 are mild injuries, scores of 7–12 are moderate injuries, and scores of 13–18 are severe injuries. Mice with mild and moderate injuries are selected as the experimental model animals.
Beam-walking test
For the beam walking test, mice were placed in the laboratory for at least 30 min each day before the start of the test. A beam with a 1.1-m length, 1.27-cm width, and 0.5-m height was used as previously described. All mice were trained three times per day before the ICH surgery. The performance score of the mice was based on a seven-point scale as previously described [28].
Adhesive removal test
Adhesion removal test is a sensitive method for monitoring sensorimotor defects after CNS injury [29]. For the adhesion removal test, mice were placed in the laboratory for at least 30 min each day before the start of the test. A 0.5-cm-length and 0.5-cm-width tape was used, and the tape strip was placed on the left hairless part of the mouse forepaw. The mice were then placed in a test cage and the time to remove the strip by any action was recorded. The test was repeated three times. Before each experiment, the experimental animals were placed in the laboratory cage and acclimated for 2 min.
Open field test
The open-field test (OFT) is often used for locomotor activity and anxiety-like emotion in ICH models [27]. The mice were placed in a relatively quiet, low-light chamber. Before the test, clean the bottom area with ethanol to avoid the influence of previous tests. Each mouse was observed for 30 min. Tracks and distances were recorded with the EthoVision 8.5 system (Noldus, The Netherlands).
Hematoxylin and eosin staining
The coronal section of brain tissue was performed to obtain a brain section of 5 μm. The slices were dewaxed with xylene and gradient alcohol, and then hematoxylin and eosin (H&E) staining was performed. After xylene transparent treatment, the slices were sealed with neutral gum.
Hematoma volume measurement
According to previous standard procedures [30], whole-brain coronal sections of mouse brain tissue with a thickness of 1 mm were made, and brain sections were digitally analyzed and hematoma volumes were calculated using ImageJ.
Heme metabolism test
Hemin content detection
According to previous studies [31], a series of concentration heme standard solutions were first prepared with 0.25% sodium carbonate solution, and the solvent was adjusted to zero. The absorbance was detected at 385 nm in a 722 spectrophotometer, and the standard curve was drawn. For the determination of cell hemin content, the cell supernatant of each group was diluted with 0.25% sodium carbonate solution, and the normal control (NC) group was used for blank zeroing. Finally, the absorbance was measured at 385 nm.
For intracellular heme content determination, N2a cells and BV2 cells were inoculated and cultured for 24 h, respectively, and then, a coculture system was formed. At this time, according to the experimental design, hemin (HY-19424; MedChemExpress, USA) or NSC-S was added and treated for 24 h, and the concentration of hemin was 150 μM. Then the cells were washed twice with PBS, followed by addition of 1 mL of formic acid to solubilize the cell layer. The heme concentration in the formic acid solution was measured spectrophotometrically at 398 nm (ɛ = 1.56 × 105 M/cm). For brain heme content determination, brain tissue was dissolved with concentrated formic acid, then centrifuged at 15,000 rpm 4°C for 15 min, and the supernatant was taken to read heme absorption at 398 nm on a spectrophotometer.
Quantitative real-time polymerase chain reaction
Total RNA was isolated from brain tissue and N2a cells using the RNA-EASYTM isolation reagent (Vazyme, China) according to the manufacturer's instructions. For total RNA in N2a cells, N2a cells and BV2 cells were inoculated and cultured for 24 h, respectively, and then, a coculture system was formed. At this time, according to the experimental design, hemin or NSC-S was added and treated for 24 h, and the concentration of hemin was 150 μM. Then total RNA was isolated from N2a cells. cDNA was prepared using the Quantscript RT kit (Vazyme). The fivefold diluted cDNA was mixed with the SuperReal PreMix Plus kit (SYBR Green; Vazyme) for quantitative real-time polymerase chain reaction (qRT-PCR). The results were analyzed by the 2 − ΔΔCt method. The sequences of primer pairs used for gene amplification are listed in Table 1.
Primer Sequences Used for Quantitative Real-Time Polymerase Chain Reaction
CD91, cluster of differentiation 91; GPX4, glutathione peroxidase 4; HO-1, heme oxygenase-1; PTGS2, prostaglandin-endoperoxide synthase 2.
Western blot
Total protein was extracted from cells or tissues using RIPA buffer containing protease inhibitors (Beyotime, China). For the total cell protein, N2a cells and BV2 cells were inoculated and cultured for 24 h, respectively, and then, a coculture system was formed. At this time, according to the experimental design, hemin, NSC-S, or rhHSPE1 (HY-P76283; MedChemExpress) was added and treated for 24 h, and then prepared. The concentrations of hemin and rhHSPE1 were 150 μM and 50 ng/mL, respectively. The western blotting protocol was referenced as described previously [22]. The following primary antibodies were used: anti-β-actin (4970 T; Cell Signaling Technology, USA) (1:1,000), anti-histone 3 (A12477-2; BOSTER, China) (1:1,000), anti-glutathione peroxidase 4 (anti-GPX4; Ab125066; Abcam, USA) (1:1,000), anti-heme oxygenase (HO)-1(Ab52947; Abcam) (1:2,000), anti-Kelch-like ECH-associated protein 1 (anti-KEAP1; PB0813; BOSTER) (1:1,000), anti-NRF-2 (PB9290; BOSTER) (1:500), and anti-COX2 (BA0738; BOSTER) (1:1,000).
An HRP-conjugated goat anti-rabbit IgG (H+L) secondary antibody (RS0002; Ruiying Bio, China) (1:5,000) was used to detect the primary antibody. High-sensitivity enhanced chemiluminescence detection kit (Vazyme) was used to detect protein bands. After exposure, Lane 1 was used to analyze the gray level of immunoblotting.
Brain water content
Brains were quickly removed, and the brain samples were weighed on a precise electronic balance for wet weights and placed in an oven at 100°C for 48 h to weigh for dry weights. The water content was calculated according to the following formula: [(wet weight − dry weight)/wet weight] × 100% [32].
Glutathione assay
The level of glutathione (GSH) was measured by a GSH kit (Solarbio, Beijing). After preparation of brain tissue homogenate or cell homogenate, different solutions were added according to the instructions, and optical density (OD) values of each group were measured at 412 nm by a microplate reader. For cell homogenate, N2a cells and BV2 cells were inoculated and cultured for 24 h, respectively, and then, a coculture system was formed. At this time, according to the experimental design, hemin or NSC-S was added and treated for 24 h, and then, a cell homogenate was prepared. The concentration of hemin was 150 μM.
Prussian blue staining
After routine dehydration of brain sections, Prussian blue staining solution was added and incubated for 1 h at room temperature according to the instructions of the Prussian blue staining kit (Solarbio). Then, they were washed in double-distilled water for 5 min, stained with a nuclear red counter dye for 10 min, and sealed with neutral gum. Brain slices were observed by a scanning machine.
Ferrous iron colorimetric assay
The iron ion content in cells was detected by using the ferrous iron colorimetric assay kit (Elabscience, Wuhan). A series of iron standard solutions with a concentration gradient was first prepared, and then, the brain tissue homogenate or cell homogenate at the hematoma site was prepared, and different solutions were added according to the instructions. Finally, OD values of each group were measured at 593 nm of microplate reader. For cell homogenate, N2a cells and BV2 cells were inoculated and cultured for 24 h, respectively, and then, a coculture system was formed. At this time, according to the experimental design, hemin, NSC-S, or ML385 (HY-100523; MedChemExpress) was added and treated for 24 h, and then, a cell homogenate was prepared. The concentrations of hemin and ML385 were 150 and 25 μM, respectively.
Transwell coculture of BV2 and N2a
An indirect coculture model was adopted. BV2 microglia and N2a cells were cocultured in the Transwell-6 system or Transwell-12 system. BV2 cells were seeded in the upper inserts (2 × 105/well/Transwell-6 system or 105/well/Transwell-12 system) and N2a cells (2 × 105/well/Transwell-6 system or 105/well/Transwell-12 system) were seeded in the lower well. After 24 h, BV2 cells were transferred to 6-well or 12-well plates containing N2a neurons. The coculture system was treated with hemin in the presence or absence of NSC-S for 24 h for further experiments. Both BV2 microglia and N2a cells were cultured in DMEM (Gibco) supplemented with 10% FBS (Gibco) and 1% penicillin/streptomycin, and incubated at 37°C in an atmosphere of 5% carbon dioxide. Then BV2 microglia and N2a cells were separated with 0.05% and 0.25% Trypsin-ethylene diamine tetraacetic acid (Biosharp, China), respectively, after the cells reached 90% confluence for subculturing.
MTT and lactate dehydrogenase assay
The water-soluble tetrazolium salt 3-(4,5-dimethylthiazol)-2,5 diphenyl-tetrazolium bromide (MTT) and lactate dehydrogenase (LDH) kit (Nanjing Jiancheng, China) served to evaluate N2a cell viability and cytotoxicity.
N2a cells and BV2 cells were inoculated and cultured for 24 h, respectively, and then, a coculture system was formed. At this time, according to the experimental design, hemin, NSC-S, ML385, or rhHSPE1 was added and treated for 24 h, and then, MTT and LDH assays were performed following the manufacturer's instruction. The concentrations of hemin, ML385, and rhHSPE1 were 150 μM, 25 μM, and 50 ng/mL, respectively. The absorbance was determined by a microplate reader (Thermo Fisher Scientific, USA) at 490 and 450 nm, respectively.
Immunofluorescence staining
Immunofluorescence staining was performed as previously described [33]. N2a cells and BV2 cells were inoculated and cultured for 24 h, respectively, and then, a coculture system was formed. At this time, according to the experimental design, hemin or NSC-S was added and treated for 24 h, and the concentration of hemin was 150 μM. Then the cells were fixed with 4% paraformaldehyde for 30 min and permeated with phosphate buffer containing 0.1%Triton X-100 (PBS) for 20 min. For cluster of differentiation 91 (CD91), no permeation of the cell membrane was required, and cells were then blocked with 5% BSA for 1 h at room temperature.
Then they were incubated with immunofluorescent primary antibodies, including anti-CD91 (BM4098; BOSTER) (1:200), (67015–1-Ig; Proteintech, China) (1:200), and anti-NRF-2 (PB9290; BOSTER) (1:100) overnight at 4°C, and incubated with the CyTM3-conjugated AffiniPure goat anti-rabbit IgG (H+L) secondary antibody (111–165-003; Jackson, USA) (1:200) or Alexa Fluor® 488-FluoroNanogold-conjugated F(ab′)2-goat anti-mouse IgG (H+L) secondary antibody (A-24920; Thermo Fisher Scientific) (1:300) at 37°C for 1 h in the dark. The sections or cells were counterstained with 4, 6-diaminine-2-phenylindole for 5 min and sealed with an antifluorescence quench. Images were acquired using a fluorescence microscope (CKX53; Olympus Corporation, Japan) or confocal microscope (Carl Zeiss, Germany).
Reactive oxygen species
Cellular ROS were measured using the probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA; Nanjing Jiancheng). N2a cells and BV2 cells were inoculated and cultured for 24 h, respectively, and then, a coculture system was formed. At this time, according to the experimental design, hemin, NSC-S, or rhHSPE1 was added and treated for 24 h, and the concentrations of hemin and rhHSPE1 were 150 μM and 50 ng/mL, respectively. Then N2a cells were incubated with the 10 μM DCFH-DA probe at 37°C for 30 min in the dark and then washed with serum-free DMEM, and finally, the Zeiss fluorescence microscope was used for observation.
Transmission electron microscope
N2a cells and BV2 cells were inoculated and cultured for 24 h, respectively, and then, a coculture system was formed. At this time, according to the experimental design, hemin, NSC-S, or ML385 was added and treated for 24 h, and the concentrations of hemin and ML385 were 150 and 25 μM, respectively. Then 1 × 106 N2a cells were collected by a cell scraper. A 2.5% glutaraldehyde fixative solution precooled at 4°C was used to fix cells for 24 h at 4°C. The sample was rinsed with 0.1 M, pH 7.0 phosphate buffer three times; then the sample was fixed with 1% osmic acid solution for 1–2 h and rinsed with 0.1 M, pH 7.0 phosphate buffer three times. The sample was dehydrated with a gradient concentration of ethanol solution and finally treated with pure acetone for 20 min.
After the sample was treated with a mixture of embedding agent and acetone in different proportions, it was embedded and heated at 70°C overnight. The embedded sample was cut into slides of 70–90 nm in the LEICA EM UC7 ultrathin microtome. The slides were stained with lead citrate solution and uranyl acetate 50% ethanol saturated solution for 5–10 min, respectively. The slides were observed in a transmission electron microscope (TEM) after being dried.
NanoLiquid chromatography tandem mass spectrometry analysis
The concentrated NSC-S was subjected to vacuum freeze–drying, concentration detection, protein resolution, reduction, alkylation, trypsin digestion, extraction of coprecipitated peptide, and peptide desalting. Each sample was then separated and analyzed with a nano-ultra performance liquid chromatography (EASY-nLC1200) coupled to a Q Exactive HFX Orbitrap instrument (Thermo Fisher Scientific) with a nanoelectrospray ion source. Data-dependent acquisition mode was used for mass spectrometry analysis. Raw MS files were processed using Proteome Discoverer software (version 2.4.0.305) and the built-in Sequest HT search engine.
Statistical analysis
All data were processed by the SPSS 26.0 statistical software. First, all continuous variables were tested for normality, and Shapiro–Wilk tests (adaptation to small sample data) were used to test the normality of the distribution variables and assess the distribution of these variables. Parametric variables were presented as mean and standard error of mean. The comparison made between the two groups was analyzed using the t-test. The comparison made among multiple groups was first analyzed by test of homogeneity of variances, and the data that met the homogeneity of variance were analyzed by one-way analysis of variance, and then the post hoc Tukey honest test was carried out afterward; otherwise, the Brown–Forsythe test was carried out. For nonparametric variables, median and interquartile ranges were utilized with the nonparametric test used to compare such variables. A P value <0.05 was considered statistically significant.
Results
NSC-S improved neurological defects
An ICH in vivo model was established by injecting bacterial collagenase IV into the striatum of mice, and the flowchart of animal experiments is shown in Fig. 1A. After injection of bacterial collagenase IV for 24 h, NSC-S was injected. The mNSS, beam walking score, and the time to tape removal from the contralateral forepaw were significantly increased in the ICH +10 × DMEM group compared with the sham group, but significantly decreased after 10 × NSC-S treatment. After 3 days of NSC-S injection, there was no statistical difference between the sham group and ICH +10 × NSC-S group (Fig. 1B–D and Supplementary Fig. S2). Also in the OFT performed on mice, the ICH +10 × NSC-S group walked longer in the total distance (Fig. 1E, F) than the ICH +10 × DMEM group.
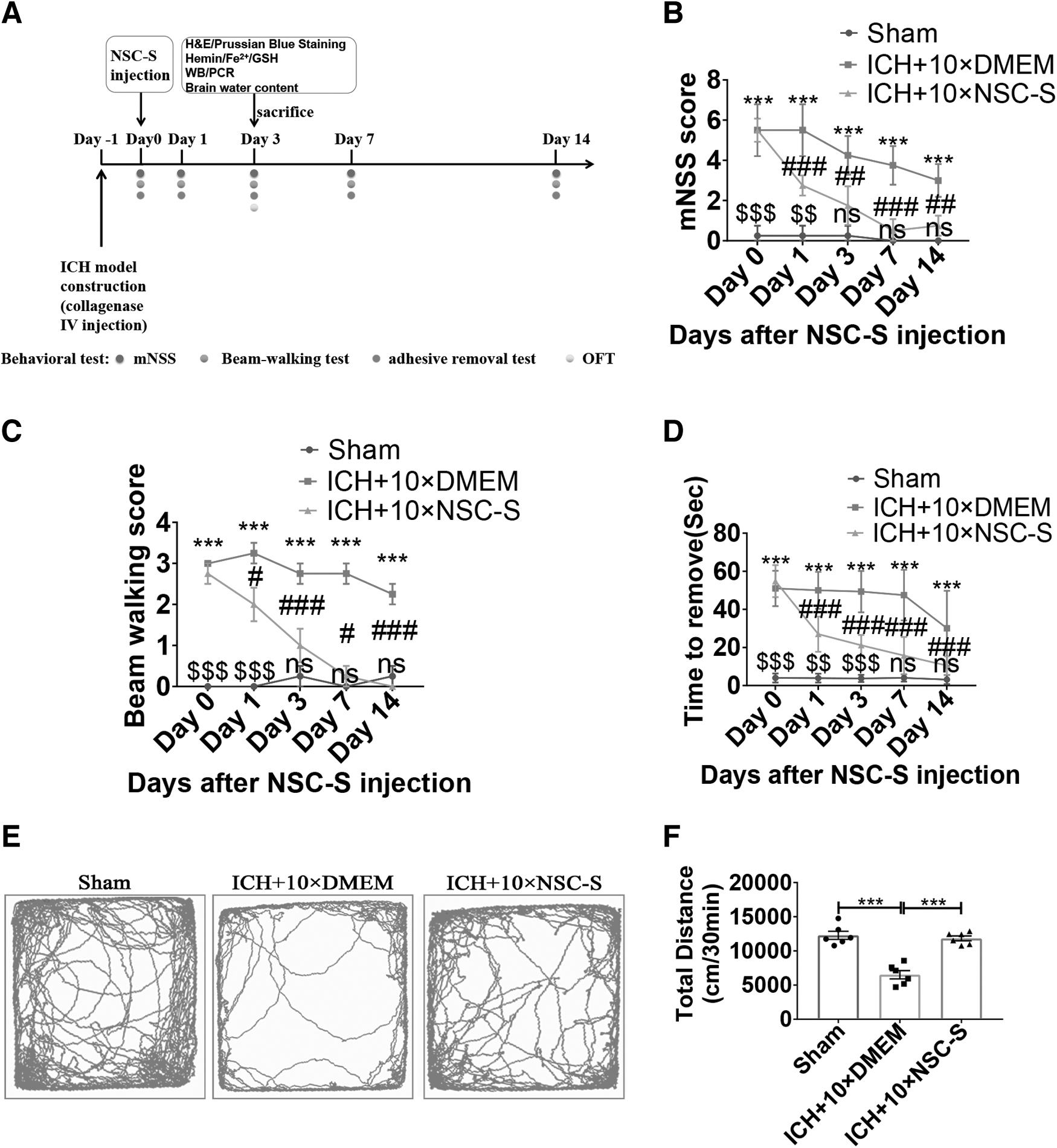
NSC-S improved neurological defects in ICH model mice.
NSC-S inhibited heme accumulation and attenuated brain injury
H&E staining revealed significant hematomas in the brains of ICH mice, but NSC-S treatment significantly reduced hematoma volume (Fig. 2A–C). Hemin content in the perihematomal brain tissue was measured to assess erythrolysis. Heme content in the perihematomal significantly increased after ICH and decreased after NSC-S treatment (Fig. 2D). Interestingly, however, HO-1, which is associated with heme metabolic clearance, increased gene and protein expression following NSC-S treatment (Fig. 2E–G). In addition, brain water content was significantly increased in the ICH group, and NSC-S treatment reversed the extent of ICH cerebral edema (Fig. 2H).
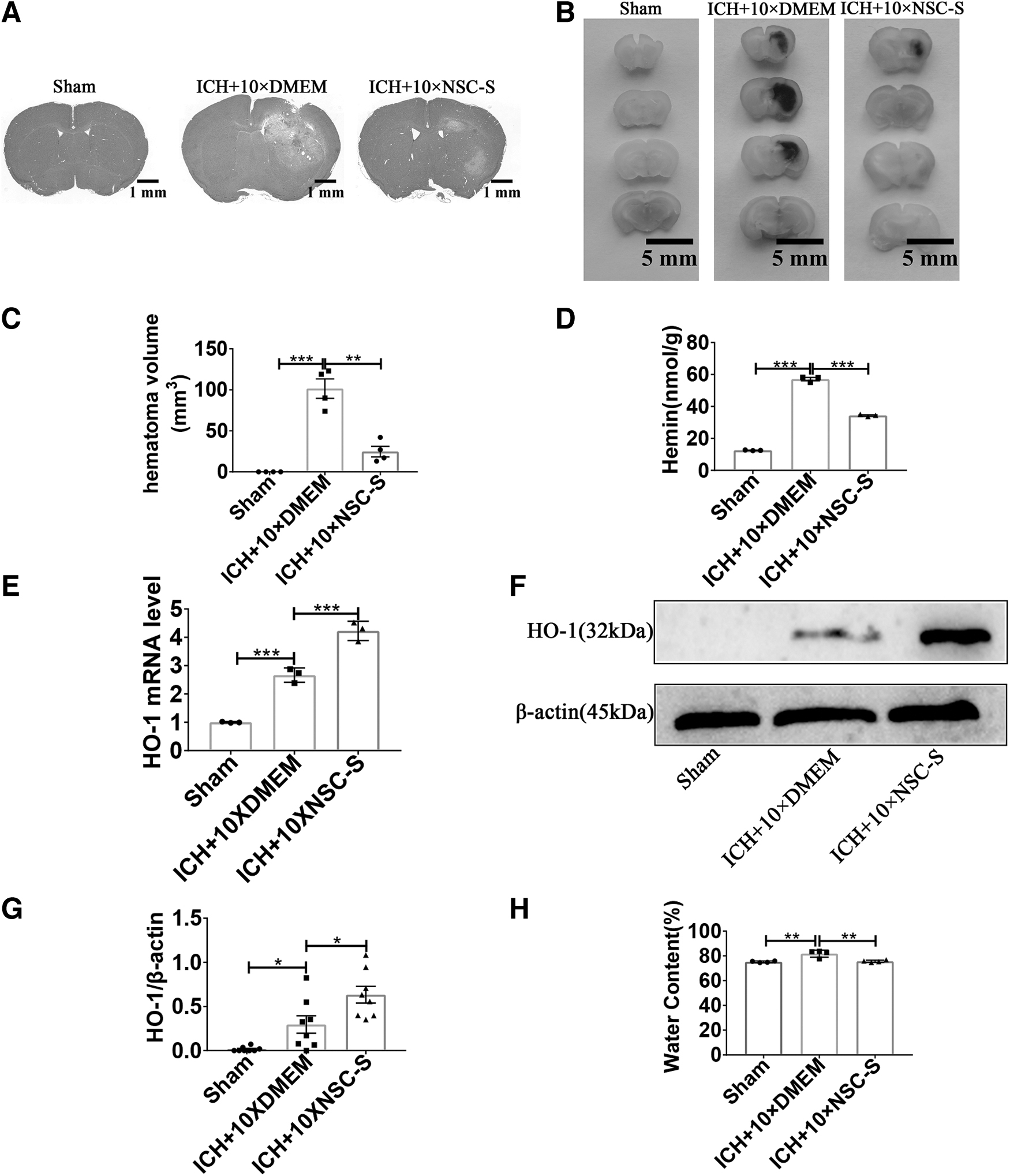
NSC-S inhibited heme accumulation and attenuated brain injury in ICH model mice.
NSC-S inhibited brain tissue ferroptosis
As shown in Fig. 3A, the brain GSH level of ICH mice was significantly decreased compared with that of the sham group, and the brain GSH level significantly recovered after NSC-S treatment. In contrast, ICH mice showed a significant increase in brain ferrous iron levels, and NSC-S treatment inhibited the upregulation of Fe2+ concentration (Fig. 3B), while Prussian blue staining showed a significant increase in iron deposition in perihematomal brain tissue in ICH, which was significantly reversed by NSC-S (Fig. 3C). Gene and protein levels of ferroptosis-associated GPX4 were significantly decreased in ICH mice compared with the sham group, whereas the gene levels of prostaglandin-endoperoxide synthase 2 (PTGS2) and the corresponding protein levels of COX2 were increased, which could be reversed by NSC-S treatment (Fig. 3D–H).

NSC-S inhibited brain tissue ferroptosis in ICH model mice.
NSC-S inhibited the accumulation of hemin and the uptake of hemin
Next, to investigate the specific mechanism of the protective effect of NSC-S on neurons in ICH, a coculture model of BV2 microglia and N2a cells was established, and the culture mode is shown in Supplementary Fig. S3A. The hemin concentration with cell viability reduced to 50% was chosen by the MTT method for subsequent experiments, and the concentration of hemin was 150 μM. Cell viability was positively correlated with the concentration of NSC-S, and N2a cells were treated with NSC-S at a concentration of 100% in the subsequent experiments (Supplementary Fig. S3B, C). Then, according to the experimental time line shown in Fig. 4A, the heme content in supernatants was measured 24 h after NSC-S treatment of heme-damaged N2a cells, and NSC-S was found to decrease hemin content in supernatants (Fig. 4B).
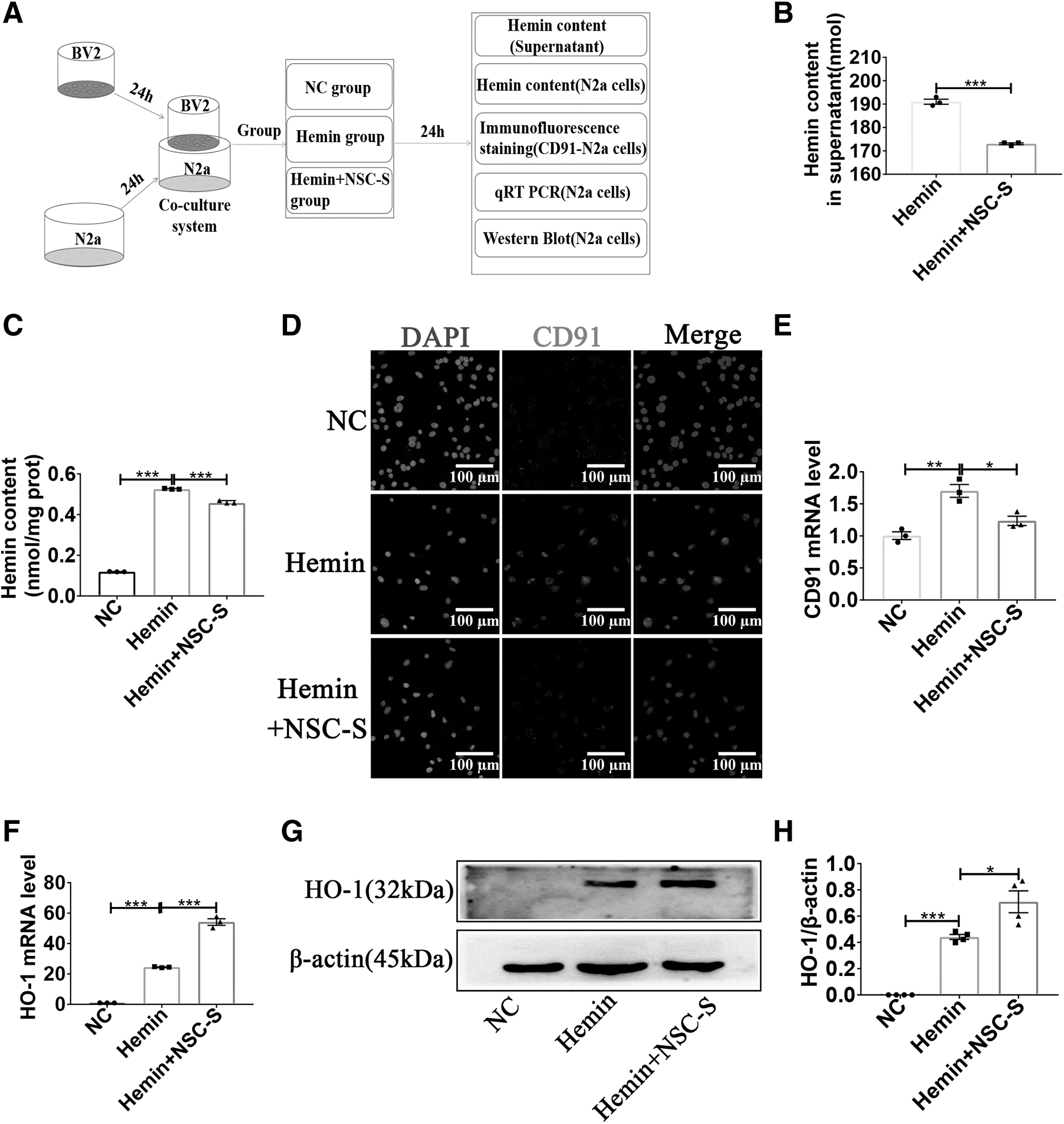
NSC-S inhibited the accumulation of hemin and the uptake of hemin by N2a cells in vitro.
Subsequently, the hemin content in N2a cells was detected, and it was found that the hemin content in N2a cells was lower than that in hemin group after NSC-S treatment (Fig. 4C). Immunofluorescence staining results showed that hemin administration increased the expression of the heme clearance receptor CD91 on the surface of N2a cells, whereas NSC-S treatment significantly decreased CD91 expression (Fig. 4D). qRT-PCR results showed that CD91 mRNA expression was decreased in NSC-S-treated N2a cells compared with hemin groups (Fig. 4E). Consistent with the in vivo results, the mRNA and protein expression of HO-1, which is associated with heme metabolic clearance, increased after NSC-S treatment (Fig. 4F–H).
NSC-S reduced oxidative stress and ferroptosis induced by hemin
According to the experimental time line shown in Fig. 5A, N2a cells were incubated with DCFH-DA, a cell-permeable oxidative fluorescent dye, and ROS production in N2a was assessed using fluorescence microscopy, and NSC-S treatment significantly reduced hemin-induced ROS levels (Fig. 5B, C). It could also be seen from the microscope picture that treatment with NSC-S reversed the hemin-induced decrease in the number of N2a cells, cell body shrinkage, and axonal shortening. NSC-S treatment inhibited LDH release compared with the hemin-injured group. MAP2 immunofluorescence results also showed that hemin significantly reduced axonal length in N2a cells and NSC-S improved the axon shortening (Supplementary Fig. S3D–F). Meanwhile, hemin-induced GSH depletion and Fe2 + accumulation were significantly increased in N2a cells, whereas NSC-S reversed this phenomenon (Fig. 5D, E).
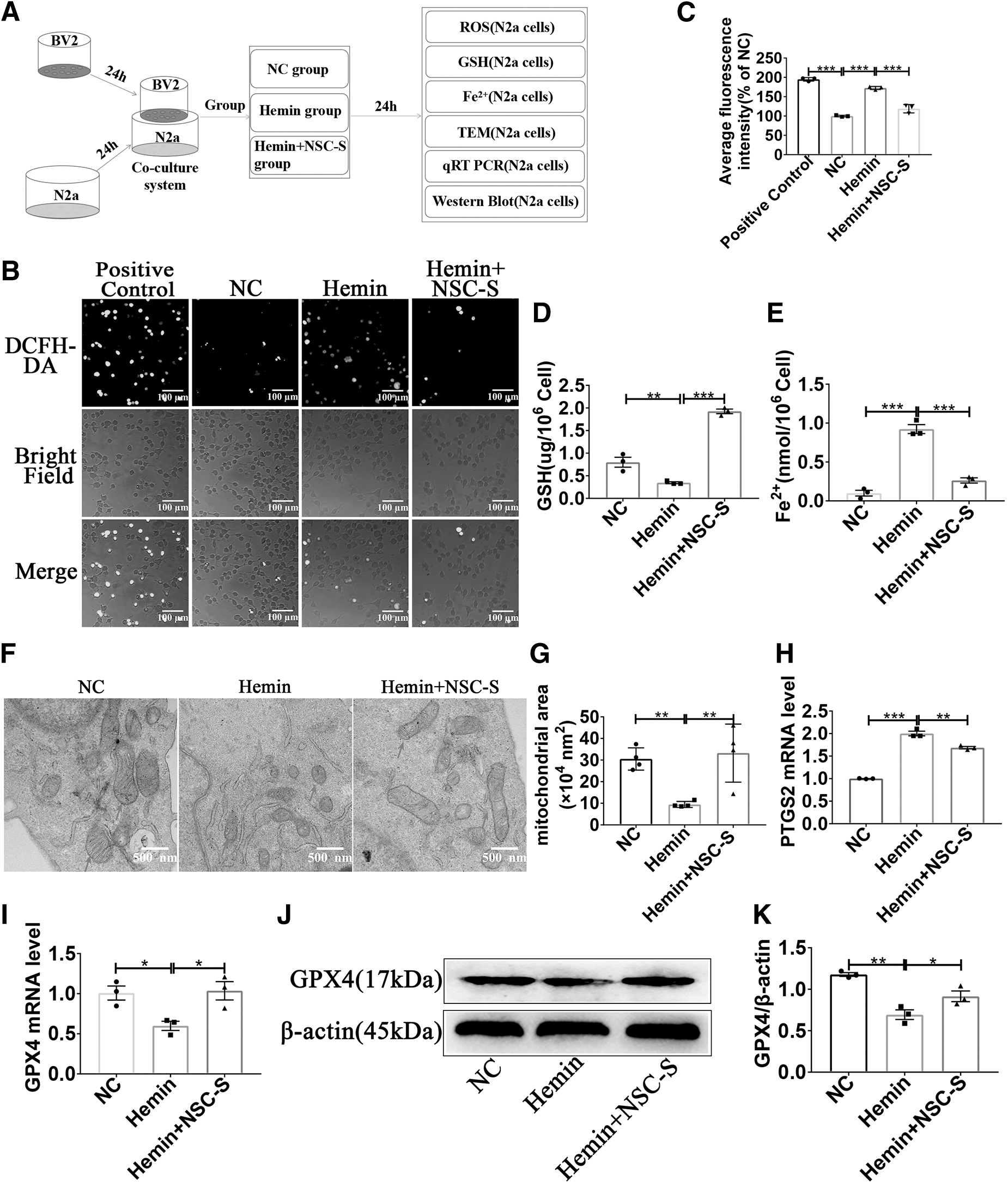
NSC-S reduced oxidative stress and ferroptosis level of N2a cells induced by hemin in vitro.
Mitochondrial morphology with shrinking and crista reduction was found in hemin-induced N2a cells using TEM, whereas NSC-S treatment significantly attenuated mitochondrial morphological damage (Fig. 5F, G). In addition, the expression level of PTGS2 gene was significantly increased in hemin-injured N2a cells, while the expression level of GPX4 gene and protein was inhibited, which was greatly reversed by NSC-S treatment (Fig. 5H–K).
NSC-S ameliorated hemin-induced ferroptosis by activating Nrf-2 signaling pathway
A protein–protein interaction analysis revealed that Nrf-2 was involved in the regulation of ferroptosis (Fig. 6A). According to the experimental time line shown in Fig. 6B, immunofluorescence staining results showed that hemin induced nuclear transfer of Nrf-2 in N2a cells compared with the NC group, whereas NSC-S treatment significantly increased nuclear transfer of Nrf-2 compared with the hemin group (Fig. 6C, D). In NSC-S-treated N2a cells, Nrf-2-specific antagonist ML385 was used to inhibit the activation of Nrf-2 signaling pathway, and the concentration of ML385 was 25 μM. Western blot results showed that NSC-S significantly promoted hemin-induced nuclear translocation of Nrf-2 in N2a cells, and increased the expression of KEAP1 protein compared with the hemin group.
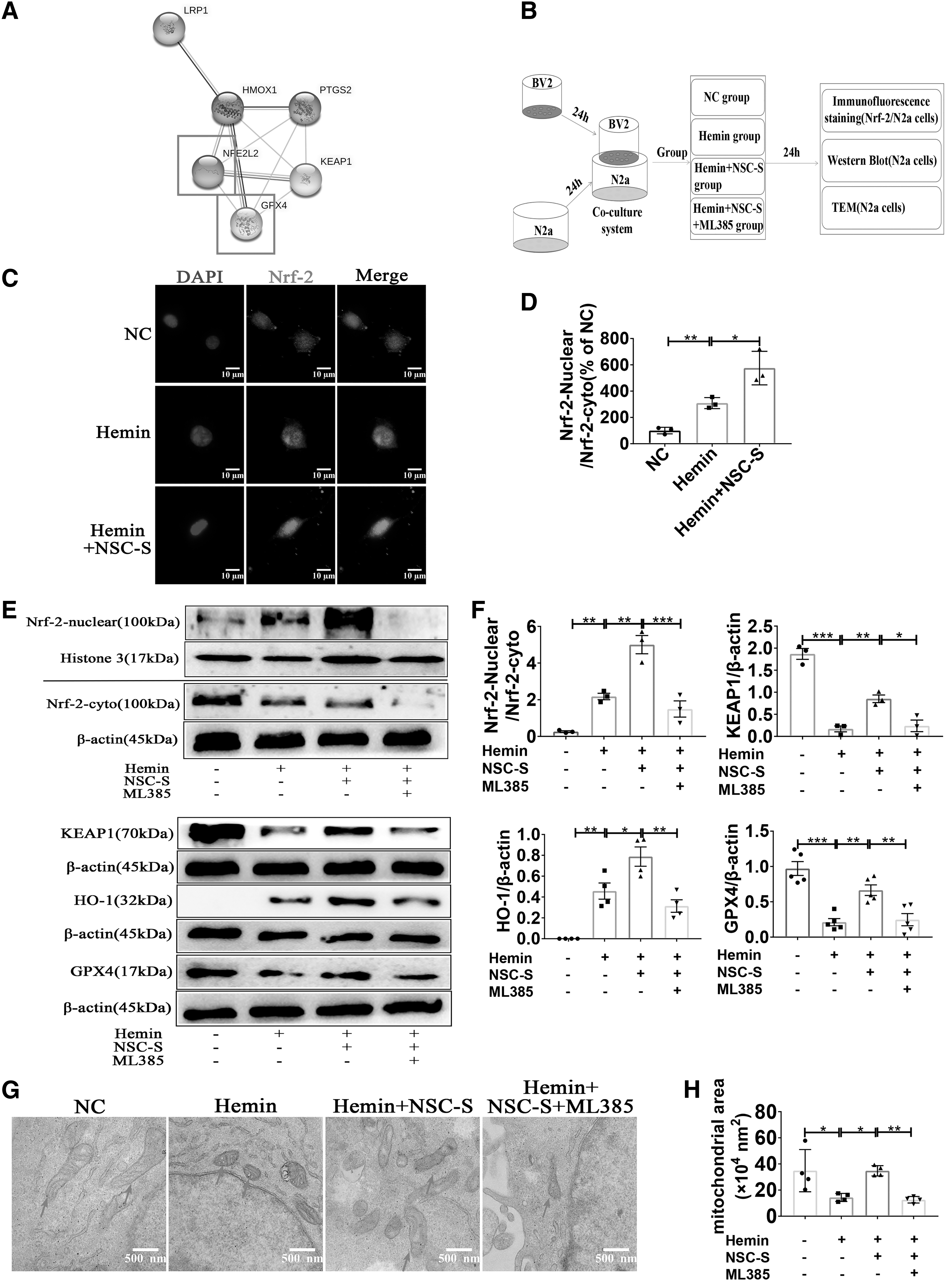
NSC-S ameliorated hemin-induced ferroptosis in N2a cells by activating Nrf-2 signaling pathway in vitro.
However, ML385 pretreatment blocked the promoting effect of NSC-S on nuclear translocation of Nrf-2 in N2a cells, and partially eliminated the repair effect of NSC-S on hemin-induced KEAP1, HO-1, and GPX4 ferroptosis-related proteins in N2a cells (Fig. 6E, F). TEM results showed that ML385 weakened the protective effect of NSC-S on mitochondrial morphology (Fig. 6G, H). In addition, other ferroptosis-related endpoints also showed that ML385 weakened the protective effect of NSC-S on N2a cells (Supplementary Fig. S4A, B).
Liquid chromatography tandem mass spectrometry analysis on NSC-S
NSC-S components detected by liquid chromatography tandem mass spectrometry (LC-MS/MS) were ranked by relative content. It was shown that most of the proteins differentially expressed from control medium were cytoskeleton-associated proteins or enzymes involved in glucose metabolism, while HSPE1 ranked 24th (Fig. 7A). Gene Ontology enrichment analysis of overlapping differentially expressed protein fractions from multiple assays showed that HSPE1 was present in extracellular exosomes, and HSPE1 was also involved in biological processes such as cell death and apoptosis (Fig. 7B, C). HSPE1-interacting proteins were predicted by STRING on the website and HSPE1 was found to be closely associated with Nrf-2 and ferroptosis-related proteins (Fig. 7D).

Bioinformatic analysis of LC-MS/MS detection result on NSC-S.
HSPE1 recombinant protein ameliorated hemin-induced ferroptosis through Nrf-2
According to the experimental time line shown in Fig. 8A, the viability of hemin-damaged N2a cells treated with HSPE1 human recombinant protein (rhHSPE1) at different concentration gradients was significantly increased, and HSPE1 at concentrations of 50 and 100 ng/mL acted close to NSC-S, and N2a cells were treated with HSPE1 at a concentration of 50 ng/mL in subsequent experiments (Fig. 8B). Meanwhile, hemin-induced morphological damage in N2a cells was also ameliorated (Fig. 8C). rhHSPE1 was also observed to inhibit hemin-induced ROS (Fig. 8D, E). In addition, western blot results showed that rhHSPE1-treated N2a cells had increased nuclear Nrf-2 protein levels and significantly increased protein expression levels of Keap1, HO-1, and GPX4 compared with hemin-induced N2a cells (Fig. 8F–H).

HSPE1 recombinant protein ameliorated hemin-induced ferroptosis in N2a cells through Nrf-2 in vitro.
Discussion
Intracerebral hemorrhage is a fatal type of stroke, and there is no effective treatment in clinical practice. Stem cell therapy has attracted increasing attention in recent years. As seed cells of the CNS, NSC transplantation appears to be a promising therapeutic approach. However, there are still some challenges limiting its application to be a routine option for clinical treatment. Many studies have shown that NSC paracrine substances are abundant in composition and these substances have been shown to play an important role in alleviating disease [34,35]. At the same time, NSC-S is easier to obtain and has the advantages of high biological activity and low immunogenicity. Thus, the NSC-S is considered a promising alternative tool for cell therapy.
In this study, we examined the neuroprotective effects of NSC-S by injecting collagenase IV into the mouse striatum, which is a classic drug for establishing mouse models of ICH [36]. NSC-S has been found to significantly improve neurological deficits in ICH mice. Specifically, NSC-S treatment significantly reduced the total distance traveled in the open field, balance beam score, and tape removal time in ICH mice. Interestingly, hematoma volume, heme accumulation, and brain edema were also improved in the ICH group by NSC-S treatment. These results tentatively confirmed the therapeutic effect of NSC-S on ICH.
Cerebral edema is a fatal pathological condition in secondary ICH injury, especially within the first 72 h [37,38]. At this time, a large amount of heme accumulates at the site of hematoma injury and can enter neurons faster to cause direct damage compared with astrocytes and microglia. Excess heme can also be broken down into ferrous ions by HO, which may produce iron-dependent oxidative damage to cell populations with limited capacity for iron chelation, such as neurons [39]. Studies have shown that iron overload in ICH injury is associated with various processes leading to neuronal injury following ICH, including ROS production, lipid peroxidation, and others [40], and ultimately leads to ferroptosis and aggravates brain edema. Ferroptosis was first described in 2012. Morphologically, ferroptosis has typical features, such as dysmorphic small mitochondria with reduced cristae and membrane condensation.
Biochemical features include GSH antioxidant dysfunction, GPX4 depletion, and lipid peroxide accumulation. Increasing evidence suggests that ferroptosis may be a major factor inducing secondary injury in ICH [41,42]. Studies have found that the use of ferroptosis inhibitors, such as deferoxamine, can reduce heme accumulation in the brain after ICH in pigs and can reduce ICH-induced brain injury, including edema formation, neuronal death, and neurological defects [43,44]. Collectively, these findings highlight the potential therapeutic value of targeting heme metabolism and ferroptosis in the treatment of ICH injury.
Therefore, we hypothesize that the therapeutic effect of NSC-S on mouse models of ICH may be achieved by inhibiting heme accumulation in the brain and neuron ferroptosis. Heme was used to mimic the in vitro model of coculture of ICH neurons and BV2 microglia. We adopted the indirect coculture model, in which BV2 cells simulated microglia in vivo, located in the upper inserts, and N2a cells simulated neurons, located in the lower well. The model induced oxidative damage and ferroptosis in neurons, while ROS were decreased after NSC-S treatment. Importantly, heme levels in the coculture system were significantly decreased after NSC-S treatment, heme levels in N2a cells were also relatively decreased, and mitochondrial morphology in N2a cells was also close to that in normal controls. However, strangely, NSC-S treatment decreased the expression of CD91 (a receptor involved in heme uptake) in N2a cells, but the level of heme in the supernatant decreased.
As we all know, microglia are the main cells for removing heme in the human body, and their tolerance to heme is also stronger. Therefore, we guess that the decrease of heme level in supernatant may be related to microglia in the coculture system. Our research group also found that after NSC-S treatment, the content of heme in BV2 microglia in the coculture system increased, and the level of Fe2+ also increased, which preliminarily confirmed our conjecture (the results are not shown). At the same time, microglia, as immune cells, can secrete a large number of inflammatory factors when stimulated, thus causing inflammatory damage to the surrounding cells. In this coculture system, is the effect of NSC-S on N2a cells related to BV2-mediated inflammatory reaction?
In the previous research of our group, it was also confirmed that NSC-S can inhibit the expression of inflammatory factors in BV2 cells stimulated by LPS, thus playing a protective role in neurons [20], but the specific role of inflammatory reaction in BV2 cells needs further study.
In a complex network of heme metabolism, oxidative stress, and ferroptosis, Nrf-2 is right in the center. Nrf-2 is a transcription factor that normally binds cytoskeletal protein Keap1 to the N-terminal Nrf-2-ECH homology 2 domain of Nrf-2. Upon exposure to oxidative stress, Nrf-2 dissociates from Keap1 and translocates into the nucleus. Nrf-2 subsequently binds to antioxidant response elements and activates antioxidant proteins, including HO-1, superoxide dismutase (SOD), and others, thereby exerting cytoprotective effects [45]. Numerous studies have demonstrated that treatment with Nrf-2 activators such as methylbazedoxolone and nicotinamide improves behavioral impairment and brain injury in ICH mouse models [46]. In contrast, knockdown of Nrf-2 resulted in oxidative DNA damage, cytochrome C release, and increased lesion volume [47]. Thus, Nrf-2 may be a potential target for ICH therapy.
Emerging studies have shown that Nrf-2 activation effectively inhibits ferroptosis in neurons and astrocytes [48]. We observed that NSC-S promoted nuclear transfer of Nrf-2, and inhibition of Nrf-2 using ML385 resulted in diminished protection of NSC-S from impaired mitochondrial morphology. Thus, Nrf-2 may be a downstream pathway through which NSC-S exerts protection.
To explore substances that specifically protect damaged neurons in ICH models in NSC-S, we used nano liquid chromatography tandem mass spectrometry (nanoLC-MS/MS) to identify protein fractions in NSC-S and performed bioinformatic analysis. We ranked the protein fractions of NSC-S based on relative quantification of differential proteins. In the top 30, most of the proteins were cytoskeletal and structural, or basic enzymes involved in sugar metabolism. The HSPE1 protein at the 24th position caught our attention. HSP10, normally present in cellular exosomes, is a stress-induced mitochondrial matrix protein that forms chaperone protein complexes with HSP60 and is required to correct misfolded or denatured proteins [49]. HSP10 is closely associated with CNS diseases, and reduced mitochondrial HSP10 levels lead to mitochondrial dysfunction and ultimately neuronal damage in Parkinson's disease [50].
Studies have shown that HSPE1 plays an active role in a variety of pathological situations, including improving mitochondrial dysfunction, regulating inflammation, upregulating antiapoptotic proteins (eg, b-cell lymphoma-2), and affecting caspase processing, thereby preventing cell death [51,52]. HSPE1 can also upregulate Nrf-2, which in turn enhances gene expression of SOD and other antioxidant enzymes, thereby reducing ROS production [53,54].
Notably, we found a connection between HSPE1 and Nrf-2 in the protein interaction network and observed improved viability and decreased ROS levels in hemin-injured N2a cells treated with exogenous recombinant HSPE1. In addition, nuclear Nrf-2 protein levels increased in rhHSPE1-treated N2a cells, as did protein expression levels associated with ferroptosis. However, the composition of NSC-CM is complex, including metabolites. HSPE1 may just be a link in its cellular protection network. In addition, the absolute content of HSPE1 in NSC-S was not determined, so the effect concentration of HSPE1 could not be determined. In addition, it is not clear whether HSPE1 has the same protective effect on BV2 microglia in the coculture system. Therefore, in the following study, we will discuss it in more depth and detail.
Conclusion
In conclusion, NSC-S could directly inhibit the accumulation of heme and reduce oxidative stress levels in neurons, thereby protecting the damaged neurons and improving the neurological defects in ICH models. NSC-S might promote nuclear transfer of Nrf-2 to inhibit ferroptosis. Also, the protective effect of NSC-S might be related to HSPE1 in the NSC-S. In addition, it also indirectly exerts protection through the inflammatory reaction and heme metabolism regulation of microglia in the coculture system. NSC-S has an ideal cytoprotective effect, which may open up a new way for the treatment of ICH (Supplementary Fig. S5).
Footnotes
Data Availability Statement
The data sets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
The authors thank Professor Yongmin Yan, Professor Weining Zhang, and Associate Professor Jia Wang of Jiangsu University for their guidance on this study.
Author Disclosure Statement
The authors declare that they have no competing interests.
Funding Information
This study was supported by the Science and Technology Cooperation Foundation of Health bioMed (Grant No. 20200605) and a project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.