
Abstract
Allogeneic transplant organs are potentially highly immunogenic. The endothelial cells (ECs) located within the vascular system serve as the primary interface between the recipient's immune system and the donor organ, playing a key role in the alloimmune response. In this study, we investigated the potential use of recipient-derived ECs in a vein recellularization model. In this study, human iliac veins underwent complete decellularization using a Triton X-100 protocol. We demonstrated the feasibility of re-endothelializing acellular blood vessels using either human umbilical cord vein endothelial cell or human venous-derived ECs, with this re- endothelialization being sustainable for up to 28 days in vitro. The re-endothelialized veins exhibited the restoration of vascular barrier function, along with the restoration of innate immunoregulatory capabilities, evident through the facilitation of monocytic cell transmigration and their polarization toward a macrophage phenotype following transendothelial extravasation. Finally, we explored whether recellularization with EC of a different donor could prevent antibody-mediated rejection. We demonstrated that in chimeric vessels, allogeneic EC became a target of the humoral anti-donor response after activation of the classical immune complement pathway whereas autologous EC were spared, emphasizing their potential utility before transplantation. In conclusion, our study demonstrates that replacement of EC in transplants could reduce the immunological challenges associated with allogeneic grafts.
Introduction
Organ transplantation is currently the only curative therapy for end-stage organ failure [1,2]. However, the number of patients waiting for a suitable organ far exceeds the number of available organs leading to high wait list mortality. In Eurotransplant countries, wait list mortality can be as high as 14% [3]. Extended criteria donor (ECD, >60-year-old donors) organs are increasingly used to resolve the organ shortage. However, the use of these ECD in kidney transplant, which are typically higher risk organs, has affected (long term) transplant outcomes [4,5]. Furthermore, ischemia reperfusion injury (IRI) is inevitable in kidney transplantation and is widely regarded as an important mechanism for delayed graft function [6]. IRI leads to a proinflammatory environment, which can lead to immune cell infiltration. Subsequently, this inflammatory environment could lead to rejection of the graft through T cell-mediated rejection or antibody-mediated rejection (ABMR) [7 –10]. For these reasons, regenerative approaches have become promising experimental strategies to improve the quality of donor organs, thus increasing the donor pool.
Endothelial cells (ECs) are important for maintaining tissue homeostasis and form a barrier between blood and surrounding tissues under normal circumstances [11,12]. Additionally, they are the first donor cells to come into contact with the recipient's immune system, making them a primary target for rejection. Damaged endothelium, caused by IRI [13,14] exposes extracellular matrix (ECM) molecules, which aggravates inflammation, causes oxidative stress, and develops into thrombotic microangiopathy potentially leading to loss of graft function [15 –18]. Peritubular endothelium, for example, has limited regeneration capacity and permanent damage after IRI leads to progressive interstitial fibrosis [19]. Preservation and restoration of the endothelial barrier function may thus be beneficial for the optimal performance of the organ's vascular system after transplantation, thereby making the organ less immunogenic [20].
Advances in the field of ex situ liver perfusion could open up windows of opportunity for applying regenerative strategies [21,22]. Similar advances in the field of kidney perfusion could lead to the possibility to monitor the functionality of the organ, but could also allow for applying cell therapy aimed at preserving the endothelium [23,24]. The use of autologous ECs can reduce the alloreactive immune response and thereby improve transplant outcomes [25]. Creating “chimeric” organs could constitute an attractive solution to generate less immunogenic organs from allogeneic donors [26]. However, there are currently no adequate in vitro models available to study the effect of chimerism. Therefore, a robust in vitro model to study vasculature repair and chimerism is needed.
Creating such an in vitro model requires a bioactive and biomimetic scaffold, which can support ECs. Stiff and hard cell culture plastic might not adequately evoke similar cell–cell and cell–matrix interactions as the native ECM. Alternatively, the native blood vessel ECM can be used as a scaffold for tissue engineering purposes [27]. The native ECM can be obtained through decellularization. The aim of decellularization techniques is to remove all cells from a tissue without damaging the ECM, yielding bioactive and tissue-specific vascular scaffolds. These can subsequently be used to study vascular repair in vitro [28,29]. Previous studies have investigated the use of decellularized vessels to restore vasculature in rat, mouse, and human tissues [30 –36]. The majority of the studies utilize human umbilical cord vein endothelial cells (HUVECs) to repopulate both arteries and veins. However, due to their origin, HUVEC lack tissue-specific characteristics. Consequently, repopulating decellularized vein scaffolds with venous EC derived from the patient offers a more relevant in vitro model to study vascular recellularization.
To this end, we aimed to develop a robust in vitro model for veins by creating an effective decellularization method for human venous tissue. Subsequently, we repopulated it with patient-derived vein EC, allowing us to study the interaction between these EC and innate immunity. Our findings demonstrated that autologous ECs do not trigger the classical complement pathway activation, illustrating the feasibility of vascular repair and their potential utility before transplantation, and thereby reducing the immunogenicity of grafts.
Materials and Methods
Decellularization of human veins
Human common iliac vein samples were obtained from deceased liver donors. The veins (n = 36) were retrieved by specialized organ retrieval teams during liver procurement for the Erasmus MC. The iliac veins were retrieved as part of a back-up toolkit for surgical revascularization (vascular jump grafting) during liver transplantation. If jump grafting was not needed, veins were stored for 14 days at 4°C. After this 14-day period, the veins were discarded for clinical use, and upon consent of the recipient to use leftover materials, stored at −20°C (Erasmus MC Medical Ethics Committee MEC-2014-060).
The decellularization protocol was based on a previously published protocol for the decellularization of extrahepatic bile duct tissue [37]. The veins were first rinsed with saline solution. This was followed by a rinse with dH2O. Veins were then placed in 4% Triton X-100 (Brunschwig chemie, the Netherlands) +1% ammonia (Sigma-Aldrich, United Kingdom) (T × 100 solution) and incubated during constant shaking. The detergent solution was renewed every 30 min for up to 10 cycles. Subsequently, veins were rinsed with dH2O. The decellularized veins were incubated with DNase type I in 0.9% saline solution + 100 mM calcium chloride (CaCl2; Sigma-Aldrich)/100 mM magnesium chloride (MgCl2; Sigma-Aldrich) for 4 h at 37°C on an orbital shaker. Biopsies of the veins were taken before and after decellularization. These were either fixed in 4% paraformaldehyde (PFA; Fresenius-Kabi, The Netherlands) for histological analysis or snap frozen and stored at −80°C for DNA quantification and biochemical analysis. Decellularized veins were washed with 1 × phosphate-buffered saline (PBS) and Dulbecco's modified Eagle's medium (DMEM).
Subsequently, they were incubated overnight at 37°C and 5% CO2 with DMEM containing high concentrations of antibiotics (500 units/mL penicillin, 0.5 g/mL streptomycin, 1 g/mL primocin, 2.5 μg/mL antibiotic–antimycotic solution). Subsequently, the veins were washed in PBS and cut open along the sagittal plane. Circular ECM scaffold sections (8 or 10 mm in diameter) were cut using disposable epidermis biopsy punches. The circular ECM scaffolds were collected and washed in PBS three times. Decellularized veins were preserved at −20°C in PBS before use.
Histological evaluation of decellularized veins
Fixed vein samples were embedded in paraffin and sectioned. Tissue slides were stained with Hematoxylin and Eosin; or with the 4′,6-diamidino-2-phenylindole (DAPI) nuclear dye (VECTASHIELD).
DNA quantification and biochemical analysis
DNA content before and after vein decellularization was assessed with the QIAamp DNA Mini Kit (Qiagen, Germany) according to the manufacturer's instructions. Briefly, wet weight of vein biopsies (n = 25 paired samples before and after decellularization) was determined before the samples were enzymatically digested in proteinase K at 56°C. Dissociated samples were then passed through a selective DNA-binding membrane. DNA content was analyzed spectrophotometrically (NanoDrop; Thermo Fisher Scientific) and normalized by the corresponding wet weight of the measured sample (nanograms DNA/milligram wet weight tissue).
The sulfated glycosaminoglycan (sGAG) content from native human veins and decellularized veins was determined through a Blyscan glycosaminoglycan assay (Biocolor, United Kingdom). Wet weight of the samples (n = 10) was determined and samples were digested by Papain solution (10 mg/mL; Sigma-Aldrich) at 65°C for 8 h. sGAG was extracted from the digested sample according to the manufacturer's instructions. Absorbance was measured at 680 nm using a Model 680 XR Microplate Reader (Bio-Rad).
Collagen content was measured using a Total Collagen Kit (QuickZyme Biosciences) according to the manufacturer's protocol (n = 11). Sample wet weight was determined before the samples were hydrolyzed in 6 M HCl at 95°C for 20 h. Absorbance was measured at 570 nm using an infinite M nano plate reader (TECAN, Switzerland).
Cell culture
HUVECs were purchased from PromoCell GmbH (Germany) and green fluorescent protein (GFP) HUVECs were purchased from Cellworks (United Kingdom). HUVECs were cultured in endothelial cell basal medium-2 (EBM-2; Lonza), and enriched with endothelial cell growth medium supplements-2 (EGM-2; Lonza), 5% heat-inactivated fetal bovine serum (HI-FBS; Corning, CA), 100 IU/mL penicillin, and 100 IU/mL streptomycin. HUVECs used in the experiments were between passage 3 and 10. During these passages, HUVECs maintained their phenotype and morphology. Cultures were maintained in a humidified atmosphere (5% CO2, 37°C).
The human monocytic cell line THP-1 (TIB-202; ATCC), originating from an acute monocytic leukemia patient, was cultured in RPMI 1640 medium (Thermo Fisher Scientific, The Netherlands) supplemented with 5% HI-FBS, 2 mM
Venous-derived EC isolation
Venous-derived ECs were isolated from kidney transplant-derived veins from three living donors with ages ranging from 21 to 62 years. The used vein segments were discarded during surgical procedure and could be used for research purposes, as approved by the Erasmus MC Medical Ethics Committee (MEC-2021-7918). Vein segments were washed with PBS and incubated in calcium-rich EBSS medium (Thermo Fisher Scientific) with 0.2% collagenase NB 4G from Clostridium histolyticum (Serva, Germany) for 7 min at 37°C. Subsequently, the cell suspension was centrifuged (850 g, 5 min). The cell pellet was suspended in enriched EBM-2 medium and plated. Potential EC colonies were enriched by mechanically removing colonies with non-EC morphology under the microscope. After expansion, EC identity was confirmed by flow cytometry (Supplementary Fig. S1) as described by Tejeda-Mora et al. [38], by using the following anti-human antibodies: CD45-PerCP (clone 2D1; BD Biosciences), CD31-PECy7 (clone WM59; BioLegend), CD34-APC (clone 8G12, BD Biosciences), CD146-AmCyan (clone P1H12; BD Biosciences), and CD105-FITC (clone 266; BD Biosciences).
FACS was performed using a FACS Symphony (BD Biosciences), and antibodies were titrated individually before use. The purified ECs were cultured as described for HUVEC.
Lentiviral transduction of venous-derived EC
Venous-derived ECs were transduced with recombinant lentiviral gene ontology (LeGo) vector LeGo-iG2 plasmid encoding GFP or recombinant LeGo-C plasmid encoding mCherry. Both LeGO-iG2 and LeGo-C were a gift from Boris Fehse (Addgene plasmid #27341 and #27339, respectively;
Recellularization of vein scaffolds
All recellularization was performed by adding 10 μL cell suspension containing 0.2 × 106, 0.4 × 106, or 1 × 106 cells/cm2 HUVEC or venous-derived ECs, to PBS-rinsed vein scaffolds. The vein scaffolds were incubated at 37°C for 30 min to allow cell attachment. Following this, enriched EBM-2 medium was added to the constructs and these were incubated in static conditions for at least 3 days before further use. The enriched EBM-2 medium was replaced every third day.
Immunocytochemical staining
To study the formation of recellularized vein scaffolds, the ECs were stained for nuclei (DAPI), the cytoskeleton (F-actin), and different molecules of interest (PECAM-1, Ki67, and ZO-1) at different time points after recellularization. The cells were washed with PBS, fixed in 4% PFA (25 min), permeabilized with 0.1% Triton X-100 in PBS (30 min), and blocked against unspecific binding with 5% bovine serum albumin (BSA) in PBS (60 min). Afterward, the cells were incubated with the corresponding primary antibodies overnight at 4°C. After washing with PBS, samples were incubated with secondary antibodies for labeling (AlexaFluor546, AlexaFluor647) in 1% BSA in PBS for 1 h at room temperature. Phalloidin-AlexaFluor488 and DAPI were used as a counterstain. Images were obtained with a fluorescence microscopy (Leica).
Quantification of Ki67 expression was done using Fiji Advanced distribution of ImageJ image analysis software (Version 1.53j; U.S. National Institutes of Health). Briefly, channels of interest were isolated, and image preprocessing was done (background subtraction, contrast adjustment, and denoising to enhance the image quality and make it easier for segmentation). Then thresholding was done to separate cells from the background, and noncells were discarded based on size/shape parameters. Finally, the overlap analysis was done with the “Image Calculator” function on the binary images of Ki67-positive cells and the DAPI channel. Cells were counted with the “Analyze Particles” function based on appropriate parameters for the overlap threshold.
Quantitative PCR gene expression analysis
Messenger RNA (mRNA) from recellularized vein samples (n = 3) was isolated with the High Pure RNA Isolation Kit (Roche Molecular Systems, South San Francisco, CA) according to the manufacturer's protocol. RNA content was measured using a NanoDrop and complementary DNA was synthesized from mRNA (500 ng) with random primers (Promega Benelux B.V., Leiden, The Netherlands). Quantitative gene expression was determined using TaqMan Gene Expression Assays-on-demand and SYBR select master mix for SFX (Applied Biosystems, Foster City, CA) on a StepOnePlus real-time PCR System (Applied Biosystems). Expression levels were normalized to glyceraldehyde-3-phosphate dehydrogenase. All the tested primer sets are listed in Supplementary Table S1.
Transendothelial electrical resistance assay
The transendothelial electrical resistance (TEER) assay was performed as previously described by Willemse et al. [37]. Briefly, larger vein scaffolds (L: 1 cm, W: 2 cm) were cut from the decellularized veins by hand using a scalpel. Vein scaffolds were recellularized with HUVEC or venous-derived ECs as previously described in the section “Recellularization.” The recellularized vein scaffolds (n = 4) were placed in an Ussing tissue slider (P2303A, area: 0.10 cm2; Physiologic Instruments) and placed in an Ussing chamber (Physiologic Instruments). The chambers were filled with Meyler's medium (128 mM NaCl, 4.7 mM KCl, 1.3 mM CaCl2, 1 mM MgCl2, 0.3 mM Na2HPO4, 0.4 mM NaH2PO4, 20 mM NaHCO3, 10 mM HEPES, and 10 mM glucose).
Experiments were performed at 37°C and 95% O2 5% CO2 gas mixture was added to the chambers. The potential difference was clamped at 0 mV using a VCC MC8 voltage clamp module (Physiologic Instruments). The short circuit current (Isc) was recorded using Acquire and Analyze 2.3 (Physiologic Instruments) TEER was measured by applying 5 V spikes (n = 7) and the resistance was calculated using Ohm's law, Equation (1):
The resistance created by the cells was subsequently determined by subtracting the measured resistance of the decellularized vein ECM from the resistance of the recellularized vein ECM as shown in Equation (2).
Subsequently, Equation (3) shows that TEER was determined by correcting for the surface area of the tissue slider (A = 0.10 cm2).
Dextran permeability assay
To assess barrier leakage through recellularized vein scaffolds, a clamp system for vein tissue was designed and 3D-printed. The clamp consists of a 3D printed plate and ring that holds the vein scaffolds. The design was made using FreeCAD software (FreeCAD) and printed using an Original Prusa i3 MK3S printer (Prusa, Czech Republic) with 0.05 mm print settings and polylactic acid filament. The clamp system was compatible with 24-well plates.
All vein scaffolds were clamped (n = 3 for each condition) and inspected for leaks by PBS addition. Recellularized vein scaffolds were inspected after 10 days of culture after recellularization. FITC-labeled dextran (4 and 70 kDa; Sigma-Aldrich) was used to determine permeability of recellularized vein scaffolds. Dextran was dissolved in PBS to a concentration of 0.2 mg/mL and diluted 1:10,000 before being used. Both products contained a FITC:glucose ratio of 1:250.
Clamped vein scaffolds were placed in a 24-well plate containing 500 μL PBS per well. One hundred microliters of either 4 or 70 kDa dextran was added to the apical side of the wells. At set times (30, 60 and 120 min) samples from the basolateral side were collected (100 μL) and their fluorescence intensity was measured using a fluorescent plate reader. Afterward, the samples were returned to basolateral compartments of the corresponding wells.
Nitric oxide production assay
To determine the nitric oxide (NO) production of reseeded EC in vein scaffolds (n = 3), a NO assay (Thermo Fisher, Austria) was used according to the manufacturer's instructions. In brief, supernatant was taken from EC cultures in vein scaffolds 3 days after recellularization. Both nitrate and nitrite concentrations were measured colorimetrically at 450 nm using a plate reader. The interaction of NO in the cells was calculated by determining both nitrate and nitrite concentrations and normalized to the DNA concentration of each vein scaffold [39].
THP-1 monocyte adhesion assay
Vein scaffolds recellularized with HUVECs were stimulated with tumor necrosis factor-α (TNF-α) (10 ng/mL; Thermo Fisher Scientific) for 24 h. Unstimulated scaffolds were used as a control. Then the repopulated scaffolds were washed (3 × ) with PBS and fluorescent PKH26 (Sigma-Aldrich)-labeled THP-1 cells were added on top of the EC monolayer and incubated for 30 min at 37°C (2 × 105 THP-1 cells/scaffold were added). Unbound monocytes were removed by washing (3 × ) with PBS. Adhered monocytes were visualized with confocal microscopy (Leica TCS SP5 system). From each sample three randomly selected areas were imaged. The number of monocytes in each area was estimated by counting PKH26-positive cells using ImageJ (v. 1.53j) (FIJI, U.S. National Institutes of Health).
THP-1 monocyte polarization assay
THP-1 monocytes were added to decellularized vein scaffolds and recellularized vein scaffolds with venous-derived EC (1:5 EC:THP-1 monocyte cell ratio). Recellularized scaffolds stimulated with TNF-α (10 ng/mL, 24 h; Thermo Fisher Scientific) were washed with PBS. The co-cultures were kept in culture for 4 h, 3 and 7 days at 37°C. At the specified times, cells were dissociated from the scaffolds by 0.05% trypsin-EDTA (Life Technologies, The Netherlands). The monocyte immunophenotype was assessed by flow cytometry. Cells were stained with mouse-anti-human monoclonal antibodies (BioLegend ) against CD206-BV421 (clone STA), CD16-BV510 (clone 3G8), CD80-BV650 (clone 2D10), CD14-BV785 (clone M5E2), CD163-PeCy7 (clone GHI/61), and CD45-APCR700 (clone H130). Separately, EC phenotype was assessed by antibodies against ICAM-1-PE (CD54, clone HA58), VCAM-1-BV421 (CD106, clone STA), PECAM-1-PECy7 (CD31, clone WM59), and CD45-APCR700 (clone H130). Antibodies were titrated individually before use.
Flow cytometry was performed on a FACS Symphony (BD Biosciences). At the specified time, scaffolds were fixed in 4% PFA, processed, embedded in 1% agarose, embedded in paraffin, and sectioned. Tissue sections were stained for histological and immunohistochemistry evaluation.
THP-1 cell transmigration system
Recellularized vein scaffolds with venous-derived EC or HUVECs were stimulated with TNF-α (10 ng/mL; Thermo Fisher Scientific) for 24 h. Vein scaffolds were then rinsed with PBS. Subsequently, vein scaffolds were secured in the vein clamp as previously described. Prepared PKH26-labeled THP-1 cells (150 μL, 2 × 105 cells) were added to the upper chamber of the system. Human recombinant monocyte chemoattractant protein-1 (MCP-1, 50 ng/mL; Invitrogen Molecular Probes) in enriched RPMI-1640 or RPMI = 1640 medium (500 μL) was added to the basolateral compartment of the well creating a chemotaxis chamber. The system was subsequently incubated at 37°C for 8 h. At the end of the incubation period, the ECM scaffolds were retrieved, washed with PBS, and fixed with 4% PFA for 15 min at room temperature. Migrating monocytes were visualized by a Leica TCS SP5 confocal microscopy (Leica Microsystems B.V., The Netherlands). The number of transmigrated monocytes was estimated by counting PKH26-positive cells in two randomly selected fields of view (only monocytes were quantified) using ImageJ (v. 1.53j).
Complement activation in recellularized scaffolds
To examine complement-mediated HLA-specific lysis of chimeric HLA-A2 (+)/HLA-A2 recellularized vein ECM, a complement activation assay in the presence of HLA-A2 antibody was performed. Scaffolds were seeded with a 1:1 ratio of venous-derived EC, which were HLA-A2 (+) and HLA-A2 (−). After 7 days of culture, recellularized scaffolds were incubated with an HLA-A2 antibody (igG2b, 0.2 μg for 106 cells; Abcam, United Kingdom) for 30 min. Samples were washed and a mix of EBM-2 medium and rabbit complement (4:1 ratio) (Cedarlane, Canada), was added to the samples and incubated at 37°C for 20 min. Samples were then washed and fixed in 4% PFA. Complement binding was investigated using immunohistochemistry for C1q (Santa Cruz Biotech). Fixed scaffolds were incubated with primary antibody overnight at 4°C. Binding of the primary antibody was visualized with the appropriate secondary antibody.
Additional samples were kept in culture for 1, 3, and 7 days. Samples were collected at every time point and fixed in 4% PFA. Cell proliferation after complement activation was followed by confocal microscopy.
Bioinformatic analysis
Bioinformatics analyses were performed in R (version 4.0.4). Flow cytometry data were preprocessed using the flowCore package [40]. Thresholds were set on FSC and SSC to remove nonsingle cells from the data. Data were compensated using a spill over matrix generated with single labeled cells. Data quality was checked for anomalies regarding flow rate, signal acquisition, and dynamic range using flowAI [41]. Fluorescence signal data were transformed to logicle scale. Unbiased and unsupervised data clustering was performed using a nonlinear generalization of principal component analysis using FlowSOM [42]. Data were then displayed using Uniform Manifold Approximation and Projection.
Statistical methods
For all the experiments included in this study, two or more biological replicates were performed or stated otherwise; the exact sample size (n) for each experimental group/condition is given. Statistical analysis was performed using GraphPad Prism (version 8.4.2; GraphPad Software, CA) or R (version 4.0.4). Graphs were performed using GraphPad Prism. For a comparison of coupled experimental groups, two-sided Student's t-tests (for the parametric datasets) and the Mann–Whitney test (for the nonparametric datasets) were used. The data are presented as mean ± standard error of the mean. A P value <0.05 was considered statistically significant.
Results
Decellularized human veins preserve microanatomy and ECM components
Human veins were decellularized using a 4% Triton X-100 solution (Fig. 1a). During the decellularization procedure, the color of the veins changed from red/pink to pale yellow. The decellularization process did not affect the macroscopic outlook of the luminal side of the veins. The effectiveness of the decellularization process was subsequently evaluated through histological analysis and DNA quantification. Both DAPI and Hematoxylin staining revealed no visible DNA after decellularization (Fig. 1b, c). The dsDNA content of decellularized samples was decreased from 61.17 ± 7.80 ng DNA per milligram wet ECM weight to 7.15 ± 1.43 ng DNA per milligram wet ECM weight (Fig. 1e).

Triton X-100 effectively decellularizes human veins, while retaining the integrity of the ECM.
The total collagen content of decellularized samples showed no significant change indicating preservation of collagens (Fig. 1f; P-value: 0.34). This was also shown by a collagen type I and collagen type IV staining on both samples (Fig. 1d). The decellularization procedure did reduce sGAG from the tissue (Fig. 1g; 3.9-fold reduction, P < 0.05). This reduction could be due to the removal of the endothelial glycocalyx, which contains sGAG as well. These results demonstrated the successful decellularization of whole human veins while keeping original collagen content and anatomical ECM structure.
HUVEC fully repopulate the luminal surface of decellularized vein scaffolds
After decellularization, we tested whether the vein ECM could support repopulation with EC. At day 1, confocal analysis showed that HUVEC adhered to the decellularized vascular construct. After 7 days, the two highest cell seeding densities (0.4 × 106 and 1.0 × 106 cells/cm2) completely covered the surface of the vein ECM (Fig. 2a). Restoration of the EC barrier function was tested for the two highest cell concentrations. We observed that after 10 days of culture, the 0.4 × 106 and 1.0 × 106 cells/cm2 concentrations had TEER measurements above background of 4.94 ± 1.69 and 15.12 ± 7.04 Ω/cm2, respectively. Although not significantly different (P value: 0.2), this indicates a trend where higher seeding densities created a higher resistance within the 10-day culture period (Fig. 2b). In addition to the TEER measurements, we also evaluated the tight junction integrity of recellularized scaffolds at day 7 by staining for tight junction protein 1 [zonula occludens-1 (ZO-1)] (Fig. 2c).
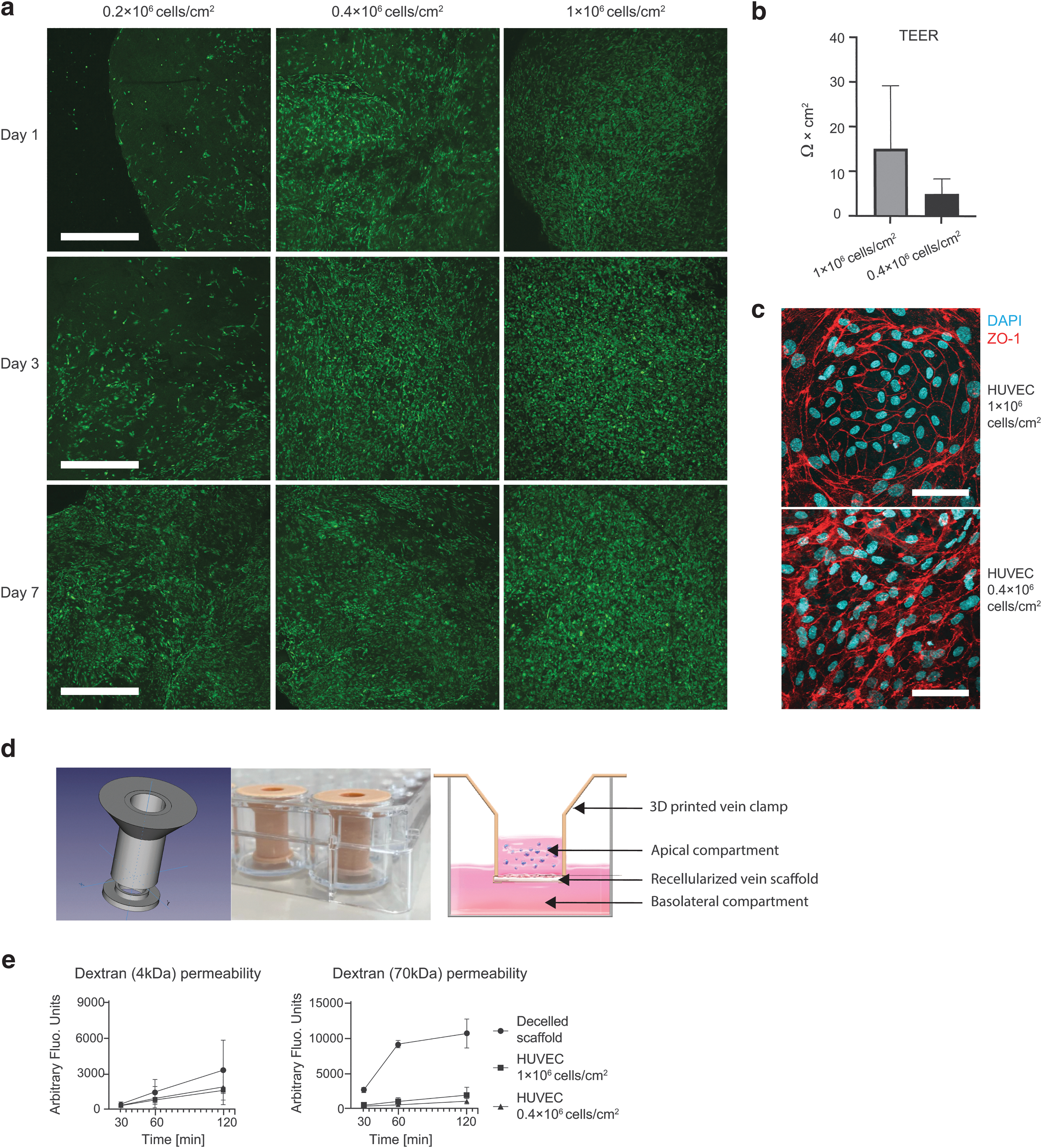
HUVEC effectively repopulate decellularized vein ECM.
Next, dextran permeability assays were performed using dextran with two different molecular weights (4 and 70 kDa). We used 3D printed vein clamps to allow dextran leakage measurements (Fig. 2d). Dextran (both molecular weights) can permeate through decellularized vein scaffolds as increasing concentrations were measured over a 2-h period at the basolateral side of the scaffold (Fig. 2e). The 4 kDa dextran transport concentration reported to be lower after 120 min for the reseeded scaffolds compared with decellularized vein ECM (44% for 0.4 × 106 cells/cm2 and 53% for 1.0 × 106 cells/cm2). The 70 kDa Dextran permeates slower through the repopulated vein ECM, as the intensity of the fluorescent units was 84% for 0.4 × 106 cells/cm2 and 93% for 1.0 × 106 cells/cm2 lower after 120 min when compared with the decellularized vein ECM.
The increased TEER and lower dextran permeability indicates that repopulation of the vein ECM with HUVEC restores endothelial barrier function. Moreover, higher seeding densities led to faster coverage of the vein ECM and increased barrier function. Consequently, the highest seeding density (1.0 × 106 cells/cm2) was used for subsequent experiments.
Cell proliferation was further examined with histology. The HUVECs were capable of maintaining complete coverage over the vein ECM for up to 28 days. The proliferation marker Ki-67 was detectable up until the end of experiment at day 28, showing that the proliferative ability of HUVEC is maintained on vein ECM (Fig. 3a and Supplementary Fig. S2a). The EC marker PECAM-1 was also present at day 28 of culture (Fig. 3b). The previous observations were confirmed with specific gene expression analysis, where the highest cell seeding density used for recellularization showed a higher expression for several vascular endothelial growth markers (PECAM-1, VIM, ENG, VEGFA), and endothelial activity (ACOX2, CDH5) genes across time points (Fig. 3c and Supplementary Fig. S2b). Fluctuations in gene expression after recellularization could be explained by the changes in cell density, cell–cell interactions and nutrient availability [43,44]. In the case of Ki-67, the late increase in expression may stem from the proliferation of EC that start to replace dead cells as a natural turnover mechanism.
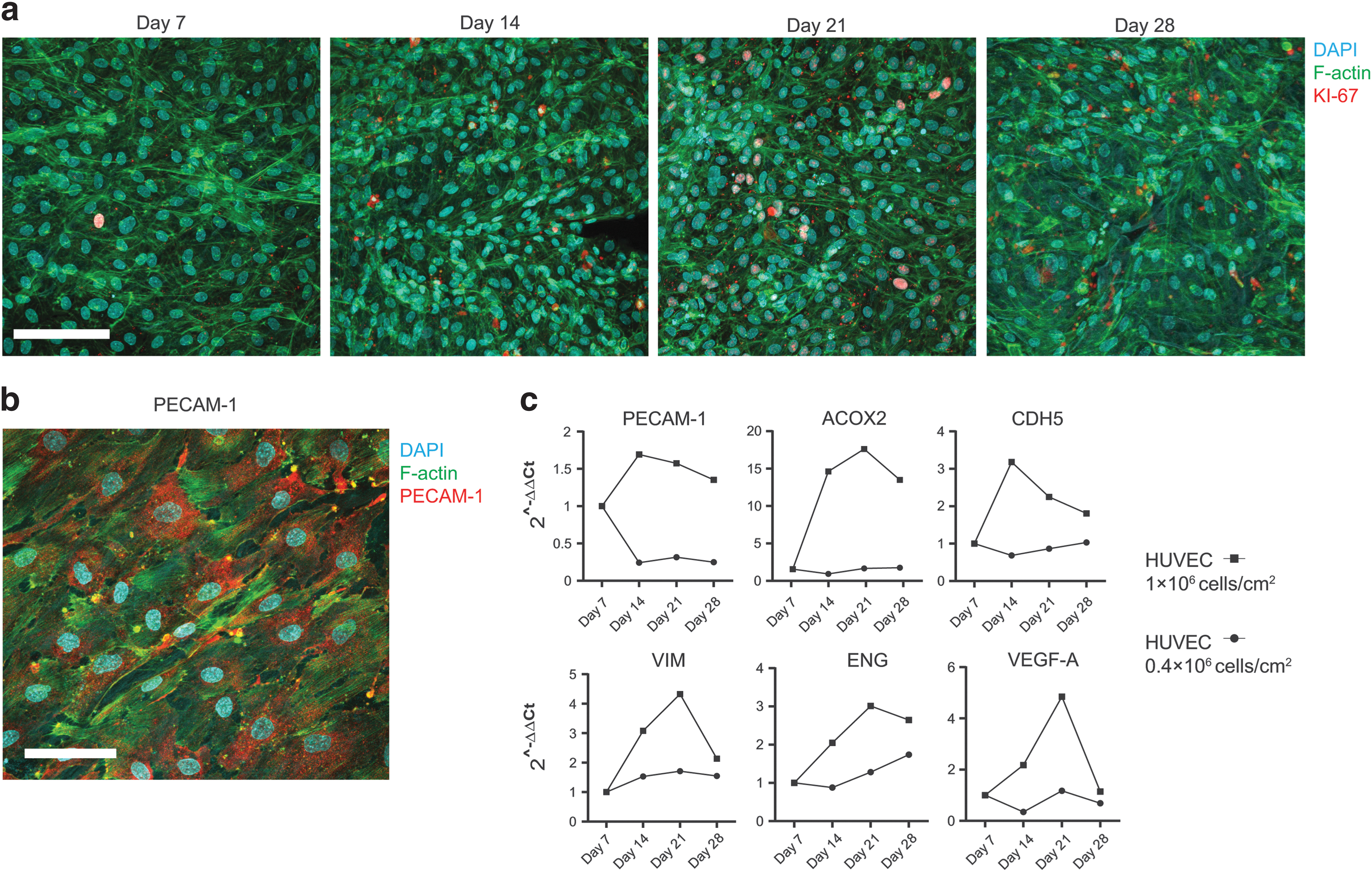
Proliferation of HUVEC on vein scaffolds.
Venous-derived ECs show similar repopulation capacity as HUVEC
HUVECs are among one of the most popular models used for ECs in vitro, nevertheless due to their origin, they differ in properties from vascular EC and do not entirely reflect the specific functions of specialized EC from organs and tissues [45]. Therefore, we examined vascular recellularization using adult human vein EC.
Human venous-derived ECs were capable of completely covering the luminal surface area of the decellularized vein at day 7 (Fig. 4a–c). Lower cell seeding densities than 1.0 × 106 cells/cm2 did not yield similar results (data not shown), therefore, all experiments with EC were performed with a 1.0 × 106 cells/cm2 seeding density. We observed a similar expression of vascular endothelial growth genes (PECAM-1, VIM, ENG) and endothelial activity genes (CDH5, ACOX2) in the different venous-derived EC seeded on the same decellularized vein scaffold after 10 days in culture (Fig. 4d). Despite the trend for increase in relative gene expression for most of the genes related to EC activity when compared with EC grown on plastic, differences were not significant (P values >0.1 for all genes), demonstrating that there is no change in EC phenotype after recellularization.
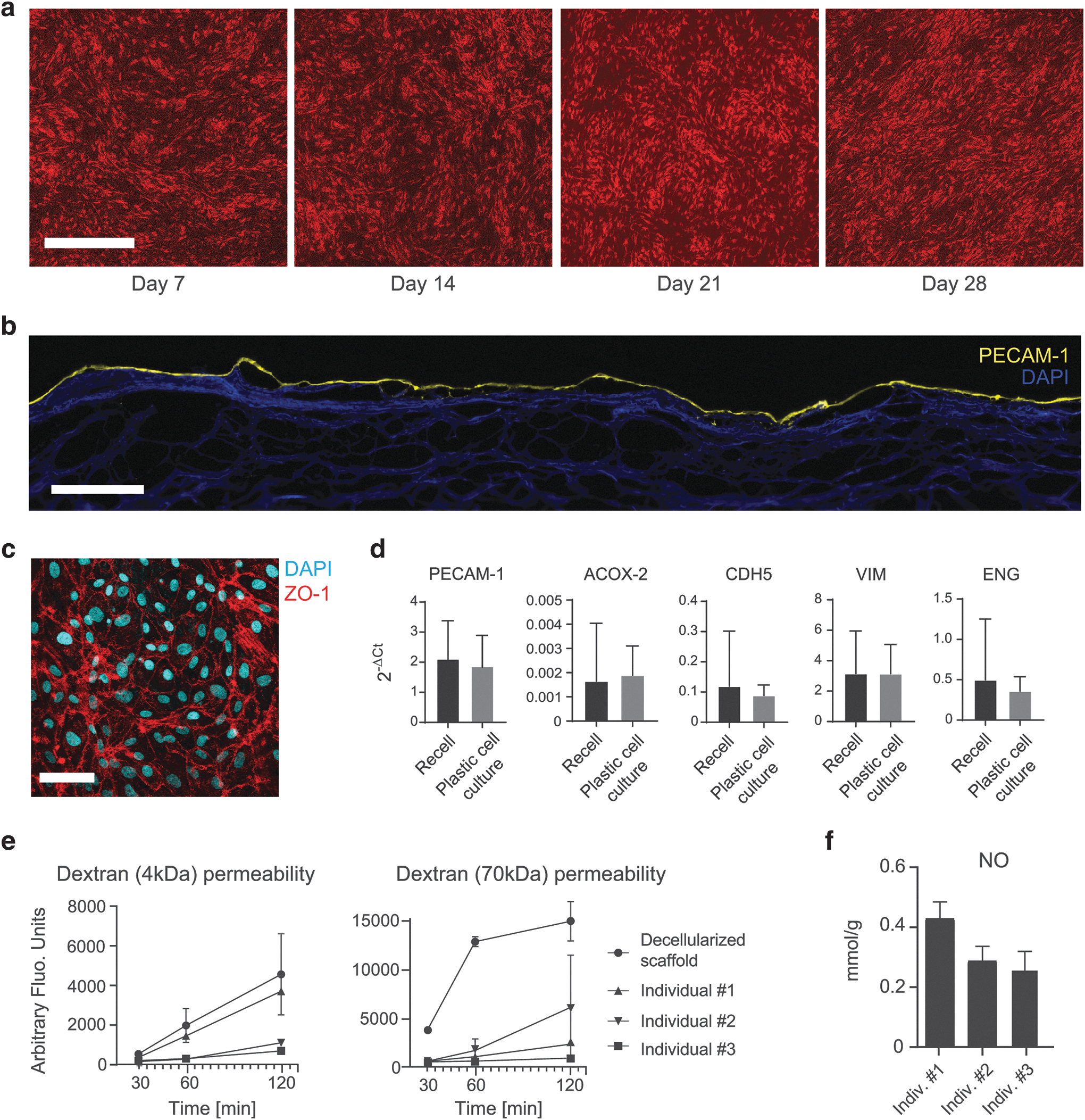
Recellularization of human iliac vein scaffolds with venous EC.
After 10 days in culture, monolayer integrity was examined by TEER and dextran permeability assays. All recellularized vein scaffolds recorded a higher resistance than decellularized scaffolds, and we did not detect significant differences among the various EC donors (Supplementary Fig. S3; P > 0.05). Specifically, EC from individual #3 displayed a nine-fold increase in resistance (21.35 Ω/cm2) compared with the other ECs. Similarly, dextran (4 and 70 kDa) permeability was significantly reduced after recellularization (Fig. 4e; P value: 0.03 for 4 kDa, P value: 0.001 for 70 kDa at minute 120). Individual #3 EC achieved the lowest leakage over time for both dextran molecular weights. Next, we measured EC activity by NO production in recellularized scaffolds after 10 days in culture (Fig. 4f). Notably, EC from individual #3 recorded the highest NO activity (0.42 mmol/g), and no significant difference in NO production was recorded among the various ECs.
These findings suggest that venous-derived ECs can effectively restore a functional endothelium in vein tissue. Furthermore, the confluency measurements revealed the presence of heterogeneity among venous-derived donor ECs
Recellularized vein scaffolds enable innate immune response
Once we established that venous-derived EC could repopulate decellularized vein tissue and restore the endothelial barrier function, we examined whether these constructs allowed innate immune cell interaction. We focused on monocytic cells due to their high sensitivity for foreign molecules and residual tissue damage. To examine this interaction, we added PKH26-labeled THP-1 monocytic cells to vein scaffolds that were repopulated with venous-derived EC. Monocyte phenotype changes due to EC culture media (EBM-2) usage were not observed (Supplementary Fig. S4). The inflammatory cytokine TNF-α was used to activate the venous-derived ECs and upregulate the surface expression of cell adhesion molecules crucial for monocyte adhesion.
The recellularized scaffolds were subsequently co-cultured for 1, 30, or 60 min with PKH26-labeled monocytes. No monocyte adhesion was observed after 1 min, discarding the possibility of monocytes' adherence to the vein's loose connective tissue. However, significant monocyte adhesion was observed on the recellularized vein scaffolds under both noninflammatory and inflammatory conditions (Fig. 5a, b).

Monocytes adhere and transmigrate across recellularized scaffolds with venous-derived EC.
The TNF-α stimulation significantly increased the number of adherent monocytes compared with nonstimulated EC and the decellularized vein scaffolds at 30 min (123 ± 15.2 and 70.5 ± 12.0 monocytes/mm2, respectively) and 60 min time points (177.5 ± 33.9 and 93.88 ± 20.6 monocytes/mm2, respectively) (Fig. 5b). No significant differences were observed across conditions between the 30- and 60-min time points (all P values >0.05). These findings gave the first hint that recellularization restores monocytic interactions with vein scaffolds.
We then investigated whether recellularized vein scaffolds may alter monocyte transendothelial migration and monocyte polarization. We used the vein clamps described in Fig. 2d. We first corroborated the null effect of the clamp system over the THP-1 monocyte immunophenotype and EC (Supplementary Fig. S5). Then, we tested monocyte transmigration under noninflammatory and inflammatory conditions (TNF-α stimulated recellularized vein scaffolds).
Monocyte transmigration across venous-derived EC recellularized scaffolds was recorded after 8 h in response to MCP-1. MCP-1 significantly increased the number of transmigrated monocytes found on the basolateral surface of the vein, marking a 3.5-fold increase in TNF-α-stimulated venous-derived EC (Fig. 5c; TNF-α-stimulated ECs with MCP-1: 92.2 ± 29.1 migrated monocytes/mm2; without MCP-1: 26.5 ± 14.2 migrated monocytes/mm2, Supplementary Fig. S6). This significant change was not observed for nonstimulated venous-derived ECs (nonstimulated EC with MCP-1: 32.5 ± 19.9 migrated monocytes/mm2, without MCP-1: 22.0 ± 18.1 migrated monocytes/mm2). Moreover, monocyte transmigration across decellularized scaffolds was exclusively observed in the presence of MCP-1, and the transmigration levels were significantly lower compared with the EC recellularized conditions (16.25 ± 3.26 migrated monocytes/mm2).
To examine the polarization of monocyte, THP-1 cells were co-cultured with venous-derived EC recellularized scaffolds for 4 h, 3 and 7 days. Co-culture was done under both noninflammatory and inflammatory conditions (TNF-α-stimulated recellularized vein scaffolds). At each time point, we assessed the cell populations for the expression of monocyte markers (CD14, CD16), M1 macrophage markers (CD80), and M2 macrophage markers (CD163, CD206) through flow cytometry. We observed changes in the monocyte/macrophage markers in a time-dependent manner (Fig. 5d). By day 3, monocytic cells were predominantly, with some cells beginning to exhibit macrophage marker expression (CD16, CD80, CD163, CD206; Fig. 5d; cluster 1). By day 7, all monocytic cells had polarized toward proinflammatory (M1) and anti-inflammatory (M2) macrophages. This included a subset of cells exhibiting a mixed M1/M2 phenotype, characterized by the absence of CD14 and coexpression of CD80 and CD163 markers (cluster 6 in Fig. 5d).
Similar results were observed by immunohistochemistry, demonstrating the presence of CD80+ and CD163+ macrophages in both noninflammatory and inflammatory conditions after 7 days (Fig. 5e). Moreover, Fig. 5d shows distinct cell populations within the EC cluster. This change in EC phenotype over time was due to reduced expression of adhesion molecules (PECAM-1, ICAM-1, and VCAM-1; Supplementary Fig. S7).
Modeling vascular repair alloantibody-dependent complement-mediated cellular cytotoxicity
The successful restoration of EC functions through recellularization in vein tissue indicates that this technique may be suitable for repairing injured endothelium in donor organs before transplantation. The use of autologous EC holds the potential to reduce alloimmune responses against donor organ EC. Recipient donor-specific antibodies play an important role in allograft rejection by binding to donor HLA molecules present on EC, subsequently triggering the classical complement cascade, leading to cell injury and graft damage.
To examine whether autologous EC could avoid potential donor-specific humoral immunity, we created chimeric recellularized veins composed of venous-derived EC with a HLA-A2 mismatch (1:1 ratio). The chimeric recellularized vein was then exposed to anti-HLA-A2 antibodies and then treated with rabbit complement. We detected strong C1q binding on HLA-A2 (+) EC but not on HLA-A2 (−) EC (Fig. 6a). Following C1q binding, we observed the detachment of HLA-A2 (+) EC (labeled with PKH26 dye; Fig. 6b). Both on day 3 and 7, only remnants of the PHK-26 dye were visible, likely due to cell membrane remnants.
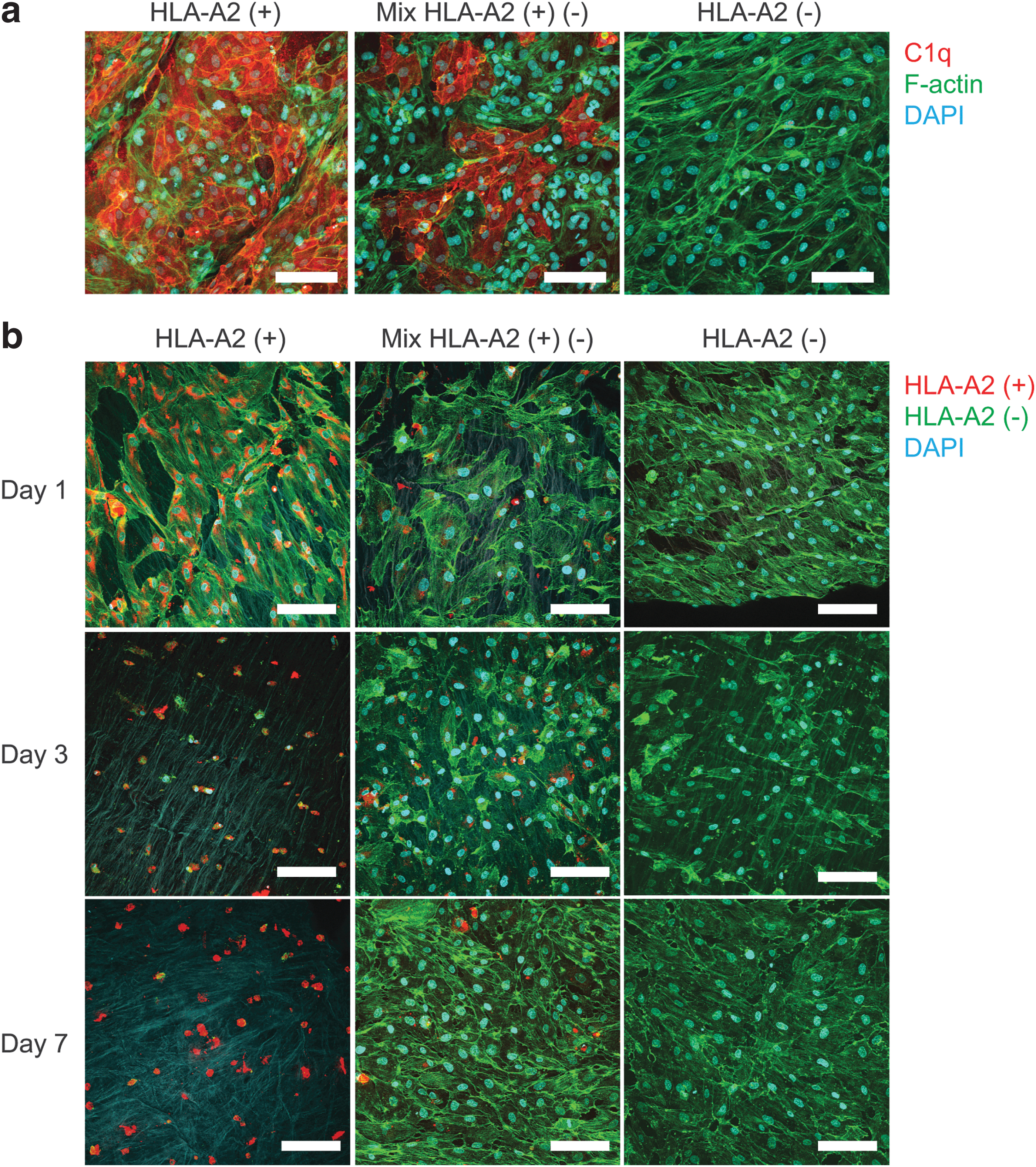
Venous-derived EC can repair vein ECM after HLA-specific complement activation.
Similar observations can be seen for the “chimeric” condition. As expected, HLA-A2 (−) ECs remained unaffected by the complement cascade and maintained the cell monolayer over 7 days in culture. Interestingly, 7 days after complement activation, the untargeted HLA-A2 (−) ECs were capable of reoccupying the exposed ECM area left by the complement attacked HLA-A2 (+) EC (Supplementary Fig. S8).
Discussion
The availability of suitable donor grafts is a key problem in the field of organ transplantation. In effort to decrease waitlist mortality rates, more ECD organs are transplanted [3,46]. This, however, is associated with an increase in early allograft dysfunction and rejection, as these ECD organs are more prone to damage. IRI affects the entire organ, including the endothelium. At the level of the vasculature, injury can lead to increased vascular permeability, increased adherence of leukocytes and immune cell infiltration. Ultimately, this can lead to T cell-mediated or ABMR-mediated rejection of the graft. Ex vivo cell therapy aiming at repair of the endothelium could reduce the risk of delayed graft function or rejection by improved preservation of the endothelium.
In this study, we explored the potential of HUVEC- or patient-derived EC to repair vascular damage in vitro. We showed that EC can fully repopulate bare vein ECM and can restore endothelium barrier function. Moreover, in a chimeric model, we showed that autologous ECs have advantages over donor HLA mismatched EC. Our study opens up a window of opportunity for repair of damaged endothelium ex situ before implantation.
Decellularization of blood vessel tissue is a promising strategy to obtain tissue-specific ECM, which can be used as a scaffold for in vitro experiments to study cell–cell and cell–matrix interactions, such as the repair of damaged endothelium. Triton X-100 has been successfully applied for decellularization of whole livers [36,47] and extrahepatic bile duct [37]. For whole livers, Triton X-100 appeared to retain more ECM components than a combination of Triton X-100 and sodium dodecyl sulfate, however, the decellularization procedure did remove sGAG from the tissue. A similar decrease in sGAG content was seen in the decellularization of vein tissue. We hypothesize that the removed sGAG are part of the glycocalyx [48]. This is supported by the 1.6-fold increase (from 0.49 ± 0.03 to 0.82 ± 0.02 μg sGAG per milligram ECM) in sGAG after recellularization compared with decellularized vein ECM (Supplementary Fig. S9). The decellularization protocol was also tested on iliac arteries and we were able to decellularize these arteries with slight adaptation of the protocol (Supplementary Fig. S10).
The repopulation of the decellularized arteries was not further pursued as the cells used in all experiments were derived from veins.
Creating functional vasculature inside decellularized tissue or organs is often seen as one of the biggest challenges in the field of tissue engineering [49]. Recellularization techniques have been applied to different ECM organ tissues [29,32 –35]. In this study, we aimed to create a small-scale in vitro model for repopulation of decellularized vein ECM. Knowledge gained from this relatively simple repopulation model can be used to build more complex three-dimensional repopulation models, which ultimately can be used for complete repopulation of the vasculature in decellularized tissue and/or damaged ECD organs. To do so, several important considerations have to be taken into account. First of all, the decellularization of the vasculature has to be scaled up and performed under good manufacturing practices. At the same time, the decellularization process should minimize damage to the vein ECM. Simple decellularization models, like the one described in this study, can aid in optimizing decellularization methods by further investigating the effect of the procedure on the integrity of the ECM.
However, perhaps more importantly, the ECM has to be fully repopulated with EC for the construct to be functional [50].
Repopulating human-scale decellularized organs does require a large number of cells. However, the exact required number of cells depends on various factors, such as attachment rate, degree of differentiation, and the proliferation speed of the cells. Moreover, for the vasculature, the required number also depends on the surface area to be repopulated. Two-dimensional models are too simplistic to fully resemble the complexity of repopulation inside organs, but they can still be used to further investigate these factors. For example, they can be used to optimize the culture medium to improve repopulation efficiency. Subsequently, this knowledge can be incorporated into more complex and biomimetic three-dimensional models, including flow and pressure. Data obtained from these models could, in the future, be used to build computational models capable of simulating repopulation and making estimations on the number of cells required for a certain organ or surface area using patient-derived cells.
Among different cell sources [35,51,52], are patient-derived cells [53 –55]; these autologous cells are of interest from an immunological point of view. Although vein-derived EC might not be the most trivial cell source for recellularization purposes, we opted to use them, to match vein ECM and favor cell adhesion and proliferation [56,57]. This choice opened the opportunity to test EC isolated from individuals, which would favor less immunogenicity and are the ideal choice to repair the graft's vasculature.
Moreover, we have shown that EC can be isolated from individuals with minimally invasive techniques [38], and that these can be easily expanded. Nevertheless, working with an individual's EC highlighted the heterogeneity in results used to benchmark the EC functionality. Our measurements, performed after 7 or 10 days of seeding in vein scaffolds, reflected a snapshot and could well be that different EC reach ideal results at different time points. This disparity could be explained due to the age of the individual from which the ECs were isolated. We observed better proliferation in EC isolated from younger individuals. Therefore, this is a critical variable to take into consideration for developing repopulation models and future clinical applications, translating into a higher cell yield during cell isolation or longer culture expansion periods.
We showed that our decellularized blood vessels are not immunogenic and that the presence of an EC monolayer initiates monocyte patrolling and trafficking across the recellularized blood vessels. Contrary to our expectations, the recellularized blood vessels polarized monocytes mainly in a M2 and mixed M1/M2 macrophage phenotype. We can only speculate the reason for this, since we missed the exact time point when M1 and M2 changes occur. Previous work has shown that polarization toward M2 phenotype is involved in the fast resolution of the inflammatory state [58,59], which would be beneficial for any recellularized tissue. Further investigation is needed to assess the reason behind triggering a mixed M1/M2 phenotype in vein recellularization.
Reperfusion damage caused to the graft's endothelium spawn the loss of regulators of the complement from the cell surface, making it prone to be targeted by the complement system [60]. Complement activity post-transplantation has a role in tissue damage, particularly during acute and chronic ABMR. Therefore, avoidance of complement activation in recellularized material is desirable. In this study, we showed that even after complement activation in a chimeric recellularized vein setup, the untargeted and remaining ECs are capable to recover the EC monolayer after 7 days in culture. This gives the first hint for the possibility to repair ECD organs before transplantation with autologous EC. These EC could aid in later graft repair by signaling and recruitment of circulating EC [38,61] and help to reduce the immunogenicity of the graft.
Conclusion
Given the key role that EC plays in graft rejection, the generation of autologous endothelium could render donor organs more immunologically acceptable. In this study, we set up a methodology to decellularize human veins and a recellularization approach with HUVECs or patient-derived vein ECs. These cells were able to restore the barrier function and retained EC phenotype over 28 days. Moreover, in a chimeric model we showed that untargeted cells were able to repopulate the ECM after elimination of allogeneic EC by complement activation. The results from the two-dimensional model can, in the future, be used to build more complex and biomimetic models capable of mimicking the processes during and after transplant.
Footnotes
Acknowledgments
Figure 1a and the third panel from ![]() were drawn by Bernart Visuals.
were drawn by Bernart Visuals.
Data Availability
All data used in this article are available upon request to the corresponding author.
Author Disclosure Statement
No competing financial interests exist.
Funding Information
No funding was received for this article.
Supplementary Data
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Figure S6
Supplementary Figure S7
Supplementary Figure S8
Supplementary Figure S9
Supplementary Figure S10
Supplementary Table S1