
Abstract
Rett Syndrome (RTT) is a severe neurodevelopmental disorder, afflicting 1 in 10,000 female births. It is caused by mutations in the X-linked methyl-CpG-binding protein gene (MECP2), which encodes for the global transcriptional regulator methyl CpG binding protein 2 (MeCP2). As human brain samples of RTT patients are scarce and cannot be used for downstream studies, there is a pressing need for in vitro modeling of pathological neuronal changes. In this study, we use a direct reprogramming method for the generation of neuronal cells from MeCP2-deficient and wild-type human dermal fibroblasts using two episomal plasmids encoding the transcription factors SOX2 and PAX6. We demonstrated that the obtained neurons exhibit a typical neuronal morphology and express the appropriate marker proteins. RNA-sequencing confirmed neuronal identity of the obtained MeCP2-deficient and wild-type neurons. Furthermore, these MeCP2-deficient neurons reflect the pathophysiology of RTT in vitro, with diminished dendritic arborization and hyperacetylation of histone H3 and H4. Treatment with MeCP2, tethered to the cell penetrating peptide TAT, ameliorated hyperacetylation of H4K16 in MeCP2-deficient neurons, which strengthens the RTT relevance of this cell model. We generated a neuronal model based on direct reprogramming derived from patient fibroblasts, providing a powerful tool to study disease mechanisms and investigating novel treatment options for RTT.
Introduction
Rett Syndrome (RTT) is a postnatal neurodevelopmental disorder, caused by pathogenic variants in the X-linked human methyl-CpG-binding protein gene (MECP2) encoding the methyl CpG binding protein 2 (MeCP2), a global transcriptional regulator [1]. The cumulative incidence of RTT is 1 in 10,000 female births [2]. The disease is characterized by an apparently normal development in the first 6–18 months of life, followed by a period of regression with the loss of expressive language and purposeful hand use, as well as other debilitating symptoms [3,4].
The nature of RTT neuropathology was elucidated in human brain studies. Postmortem RTT brains weigh less and exhibit increased packing density of neurons, diminished dendritic arborization, and hence, a decreased number of synapses [5,6]. Moreover, in induced pluripotent stem cells (iPSCs)-derived neurons from Mecp2-deficient mice, as well as in human MECP2-null iPSC-derived neurons, alterations in electrophysiological activity were observed [7,8].
Currently, several mouse models carrying various pathogenic variants of the Mecp2 gene exist, which mimic the neurological phenotype of RTT [9]. Several studies have demonstrated a rescue of some RTT phenotypes through the restoration of biologically active MeCP2 levels in Mecp2-deficient mouse models [10 –13]. Nevertheless, these mouse models do not accurately recapitulate disease progression in humans.
One alternative and promising tool to study RTT is through the reprogramming and differentiation of patient-derived cells through iPSCs into neurons, as primary human neurons from patient and control subjects are not readily available. Usually, iPSCs are generated from human or murine fibroblasts, as well as blood cells, by virally delivering certain factors as previously described [14]. These iPSCs can then be differentiated into various cell types, including neurons, under specific growth conditions [14]. It has been demonstrated that neuronal cells derived from RTT-iPSCs exhibit a decrease in the number of synapses, spine density, and soma size, as well as altered electrophysiological properties, which are comparable to the phenotype observed in vivo [8,15]. The process of iPSC reprogramming, however, involves the introduction of epigenetic modifications, due to the fact that cells are reset to a pluripotent state [16]. Adult donor cells are hence rejuvenated, which is not optimal to study age-relevant diseases. Furthermore, reprogramming of fibroblasts into iPSCs is a prolonged process taking several months [17].
In contrast, direct reprogramming of fibroblasts into induced neuronal progenitors (iNPs) and further on into neurons through nonviral expression of reprogramming factors bypasses the pluripotent stage, thereby avoiding undesired epigenetic modifications, and reduces the risk of insertional mutagenesis. Furthermore, direct-to-iNP reprogramming prevents cell rejuvenation and maintains the aging memory of the donor human dermal fibroblasts (HDFs) [18 –21]. A method of somatic cell reprogramming using two transcription factors, SOX2 and PAX6, which play a major role in early neurogenesis and neuronal development [22 –25], to directly generate iNPs from adult human fibroblasts has been previously described [19,26]. This nonviral reprogramming method generates patient-derived iNPs, which can be further differentiated into neuronal cells using a two-step differentiation protocol.
This study presents a transient nonviral transfection-based strategy for the generation of patient-derived neuronal cells to investigate RTT. Direct conversion of fibroblasts [19,26] from a male individual with neonatal encephalopathy carrying a frameshift deletion in the MECP2 gene and a healthy control subject provides a novel opportunity to study phenotypic differences in neurons. Moreover, this in vitro model could be used to evaluate novel therapeutic options.
Materials and Methods
Mutation sequencing
For the reprogramming process, HDFs from a male individual with neonatal encephalopathy carrying a frameshift deletion (c.806delG) in the MECP2 gene (2 years old at sampling, “MeCP2-deficient,” “patient-derived”) resulting in a truncated MeCP2 protein [27] and an unaffected sex-matched HDF cell line (1 day old at sampling, AG21708, obtained from the NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research) were used. MeCP2-deficient fibroblasts were established for research purposes, approved by the Human Research Ethics Committee of the Children's Hospital at Westmead, Australia [27].
DNA was isolated from HDFs using the DNeasy Blood & Tissue Kit (No. 69504; Qiagen) according to the manual. Polymerase chain reaction (PCR) was performed using DreamTaq Green PCR Master Mix (No. K1081; Thermo Scientific), applying the forward primer 5′-GCAGCCAGGCAGTGTGACTCTC-3′ and the reverse primer 5′-GATGGGGAGTACGGTCTCCTGCAC-3′. PCR products were cleaned up with ExoSAP-IT™ PCR Product Cleanup Reagent (Applied Biosystems), and Sanger sequencing was performed using BigDye™ Terminator v3.1 Kit chemistry (Applied Biosystems). Sequencing products were run on an ABI 3500 Genetic Analyzer (Applied Biosystems), and the results were analyzed using SnapGene 6.2.1 software (
Reprogramming of HDFs
HDFs were grown in fibroblast proliferation medium consisting of DMEM (No. 41966-029; Thermo Scientific), 15% fetal bovine serum (FBS, No. F9665; Sigma-Aldrich), and 1% Penicillin/Streptomycin (No. 15140122; Thermo Scientific) and passaged 1:3 every 3–4 days when the cultures reached about 90% confluence. HDFs were regularly determined to be mycoplasma negative by quantitative real-time PCR (qPCR; ISO17025 certified, Eurofins Genomics) using 72 h conditioned media samples collected from cultures. Episomal DNA transduction was performed on cells below passage 17. The plasmid constructs EEV600-CAG-SOX2 and EEV600-CAG-PAX6 containing SOX2 or PAX6 complementary DNA (cDNA) were purchased from System Custom Constructs. Plasmid DNA was amplified in One Shot TOP10 chemically competent Escherichia coli cells (No. C404010; Thermo Scientific) and then isolated using the EndoFree Plasmid Maxi Kit (No. 12362; Qiagen).
HDFs were cultured until the desired number of cells had been reached and split 1 day before transfection. For electroporation, cells were trypsinized, collected, counted, and washed with Dulbecco's phosphate-buffered solution (DPBS, No. D8537; Sigma-Aldrich). Pelleted cells were resuspended in NEON resuspension buffer at 1 × 107 cells/mL. Per transfection assay, 106 cells and 15 μg of each plasmid DNA were used. Electroporation was carried out using 100 μL pipette tips of the Neon Transfection System (No. MPK10096; Invitrogen) according to the manufacturer's instructions. Electroporation conditions used were one pulse with 1,000 V for 60 ms. Electroporated cells were seeded in polystyrene six-well plates (No. 140675; Thermo Scientific) containing 2 mL fibroblast proliferation medium and allowed to recover for 3 days.
The medium was changed 3 days post-transfection to neural reprogramming medium comprising Neurobasal-A medium (NBA, No. 10888-022; Thermo Scientific), 0.3%
Neuronal differentiation of iNP cells
For neuronal differentiation, the cells were mechanically dissociated and seeded onto poly-
After 10 days, culture conditions were changed to neural differentiation medium stage 2 comprising 3%
Treatment with TAT-MeCP2 fusion protein
TAT-MeCP2, purified as previously described [28], was added to the neuronal differentiation medium stage 2 on days 23, 25, and 28 of neuronal differentiation at a concentration of 50 nM. Control cells were incubated with MeCP2 storage buffer [DPBS, 200 mM NaCl, 10% (v/v) Glycerol, 0.05% (v/v) CHAPS, pH = 7.2].
RNA extraction and quantitative reverse transcription polymerase chain reaction
Total RNA was extracted from reprogrammed iNPs and nontransfected control cells using the RNeasy Plus Mini Kit as per the manufacturer's instructions (No. 74134; Qiagen). Total RNA from neurons was isolated on day 30 of neuronal differentiation using the RNeasy Plus Micro Kit (No. 74034; Qiagen). iScript™ genomic DNA (gDNA) Clear cDNA Synthesis Kit (No. 1725035; Bio-Rad) was used to synthesize cDNA from total RNA. For qPCR the iTaq Universal Probes Supermix (No. 1725131; Bio-Rad) was used. qPCR analyses were carried out using comparative Ct (ΔΔCt) method. PPIA was used as the internal standard. The used TaqMan® Assays are listed in Supplementary Table S2.
Immunofluorescence staining
HDFs and iNPs were fixed with ice-cold 4% (vol/vol) formaldehyde (No. 28906; Thermo Scientific) in DPBS for 20 min at 4°C and permeabilized in DPBS with 0.2% Triton X-100 (No. T8787; Sigma-Aldrich) twice for 5 min. After blocking for 30 min with 3% bovine serum albumin (BSA, No. A9647; Sigma-Aldrich) in DPBS, primary antibodies with 3% goat serum (No. G9023; Sigma) were applied overnight at 4°C. Before and after incubation with secondary antibodies (Goat anti-Mouse IgG Alexa Fluor™ 488 No. 11029, Goat anti-Rabbit IgG Alexa Fluor 568™ No. 11011; Invitrogen, 1:250 in DPBS with 3% goat serum) for 1 h at room temperature, cells were washed with DPBS thrice for 5 min. Coverslips were mounted using ProLong™ Diamond Antifade Mountant with DAPI (No. P36966; Invitrogen).
For immunofluorescence staining of neurons, a slightly different protocol was used. Cells were fixed for 10 min at 4°C and permeabilized thrice for 2 min. Blocking was carried out overnight at 4°C with 30% goat serum in DPBS. The used primary antibodies are listed in Supplementary Table S3.
All fluorescent images were acquired using a confocal microscope (DMI6000; Leica Microsystems) and LAS × 3.5 software. The following objectives were used: HC PL APO CS 20 × /0.70 UV (No. 506513; Leica Microsystems) and HCX PL APO CS 40 × /1.25 OIL PH3 UV (No. 506181; Leica Microsystems). Images were deconvolved with Huygens Essential version 23.04 (Scientific Volume Imaging;
Electrophysiological recordings
For whole-cell patch clamp recordings, an Axopatch 200B amplifier, a Digidata 1320A digitizer, and the Clampex 10.7 software (Molecular Devices) were used. The external solution contained 120 mM NaCl, 3 mM KCl, 2 mM MgCl26H2O, 20 mM glucose, 10 mM HEPES, and 2 mM CaCl2H2O (pH = 7.3, 290–310 mOsmol). Patch electrodes were filled with 130 mM CsCl, 20 mM TEACl, 0.24 mM CaCl2H2O, 5 mM EGTA, 10 mM glucose, and 10 mM HEPES (pH = 7.3). The recordings were performed at room temperature. The resistance of the patch pipettes was between 2 and 5 MOhm. For blocking voltage-gated sodium channels, 1 μM tetrodotoxin (TTX) was added through a drug application system. The holding potential was set to −80 mV, and the current was recorded by a depolarizing voltage step to −60 mV.
Neuronal morphology analysis
For quantification of the number of neurites, branches, and the total neurite outgrowth per neuron, 30-day differentiated cells were stained with TUJ1 as described. For image acquisition, 10 z-stacks with 1 μm z-stack resolution were acquired. Images were analyzed using the machine learning software AIVIA 12.0.0 (Leica Microsystems). First, a three-dimensional (3D) pixel classifier training set was created, which recognized soma and neurites. Before using the “3D Neuron Analysis” recipe to quantify the number of neurites, branches, and total neurite outgrowth per neuron, an average filter was applied. From three independent experiments at least 100 neurons per group were analyzed.
The number of vesicular glutamate transporter 1 (VGLUT1) puncta per μm dendrite was quantified using the Fiji plugin “Analyze Particles” [29], as described previously [30]. Before analysis, images were subjected to background subtraction with a rolling ball radius of 10. Subsequently, thresholding was applied consistently to all images of each experiment. To separate puncta in close proximity to each other, the watershed algorithm was applied. Anti-microtubule-associated protein 2 (MAP2) images served as dendritic mask. Regions of interest (ROIs) were chosen in the dendritic mask at a width of 20 μm. The length of the dendrite in the ROI was measured, and the puncta were quantified. The puncta were subsequently calculated as puncta per μm of dendrite.
In-Cell Western Assay
In-Cell Western Assay was used to quantify the protein acetylation level of H3K9 (No. H9286, 1:200; Sigma) and H4K16 (No. 07-329, 1:200; Sigma). iNPs were seeded in 96-well plates (No. 655090; Greiner Bio-One). After 30 days, differentiated neurons were fixed with Histofix (No. P087.5; Roth) for 20 min at room temperature and permeabilized thrice in Tris-buffered saline (TBS) with 0.1% Triton X-100 at 30 rpm agitation. After blocking for 90 min using the Intercept® Blocking Buffer (No. 927-60001; LI-COR), primary antibodies were diluted in Intercept Antibody Diluent (No. 927-65001; LI-COR) and incubated overnight at 4°C.
On the following day, cells were incubated with secondary antibody (IRDye® 800CW Goat anti-Rabbit IgG Secondary Antibody, No. 926-32211, 1:1,000; LI-COR) and CellTag™ 700 (Stain for In-Cell Western™ Assays, No. 926-41090, 1:500; LI-COR), diluted in Intercept Antibody Diluent, for 1 h at room temperature. Before and after the 1-h incubation, cells were washed five times in TBS with 0.1% Tween-20 (No. P9416; Sigma). Once the plate was completely dry, the plate was imaged using the Odyssey CLx Imager (LI-COR). Blank wells were used for background subtraction. Fluorescence intensity was normalized to CellTag 700.
RNA-sequencing and data analysis
Total RNA was extracted from each of three independent samples of MeCP2-deficient and wild-type HDF cells, iNPs, and neurons as described above. Sequencing libraries were prepared at the Core Facility Genomics, Medical University of Vienna, using the SMARTer Stranded Total RNA Seq Kit—Pico v2 according to the manufacturer's protocols (Takara Bio). Libraries were QC-checked on a Bioanalyzer 2100 (Agilent) using a High Sensitivity DNA Kit for correct insert size and quantified using Qubit dsDNA HS Assay (Invitrogen). Pooled libraries were sequenced on a NextSeq 500 instrument (Illumina) in 1 × 75 bp single-end sequencing mode. Approximately 22 million reads were generated per sample. Reads in fastq format were aligned to the human reference genome version GRCh38 [31] with Gencode 29 annotations [32] using STAR aligner [33] version 2.6.1a in 2-pass mode. Reads per gene were counted by STAR, and differential gene expression was calculated using DESeq2 [34] version 1.20.0.
Differentially expressed genes (DEGs) were defined as those with log2-fold change above 1 or below −1 and an adjusted P value threshold of 0.05. Pathway analysis of DEGs was performed using the web-based tool NetworkAnalyst [35,36] or g:Profiler [37]. The principal component analysis (PCA) plot and clustered heatmaps are based on regularized log-transformed count data generated in DESeq2 [34]. Heatmaps were generated using the coolmap function of the R package limma [38]. VolcaNoseR web app was used for creation of the volcano plot [39]. Estimation of the cell identity was performed through
Statistical analysis
All experiments were performed at least independent times from three independent differentiations. Analysis of the data was carried out with GraphPad Prism 9. Data are given as mean ± standard deviation. Unpaired t-tests were used to test for significance, with *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
HDFs for reprogramming and differentiation
We used patient-derived HDFs of a male individual with a deletion of a guanine residue at position 806 of the MECP2 gene for reprogramming and neuronal differentiation, henceforth referred to as “MeCP2-deficient” or “patient-derived” (Fig. 1A). This in turn leads to a frame-shift mutation at amino acid residue 269 at the transcriptional repressor domain (TRD) portion of MeCP2, resulting in a truncated protein (Fig. 1B). To confirm this finding, we investigated the cellular localization of the mutated protein and stained sex-matched MeCP2-deficient and wild-type fibroblasts with an N-terminal and a C-terminal MeCP2 antibody as a control. In wild-type fibroblasts, MeCP2 could be detected in the cell nucleus with both the N- and C-terminal MeCP2 antibodies. In MeCP2-deficient cells, MeCP2 was detected using only its N-terminal antibody, while no staining was observed with the C-terminal antibody, pointing to the truncated MeCP2 presence in MeCP2-deficient fibroblasts (Fig. 1C).
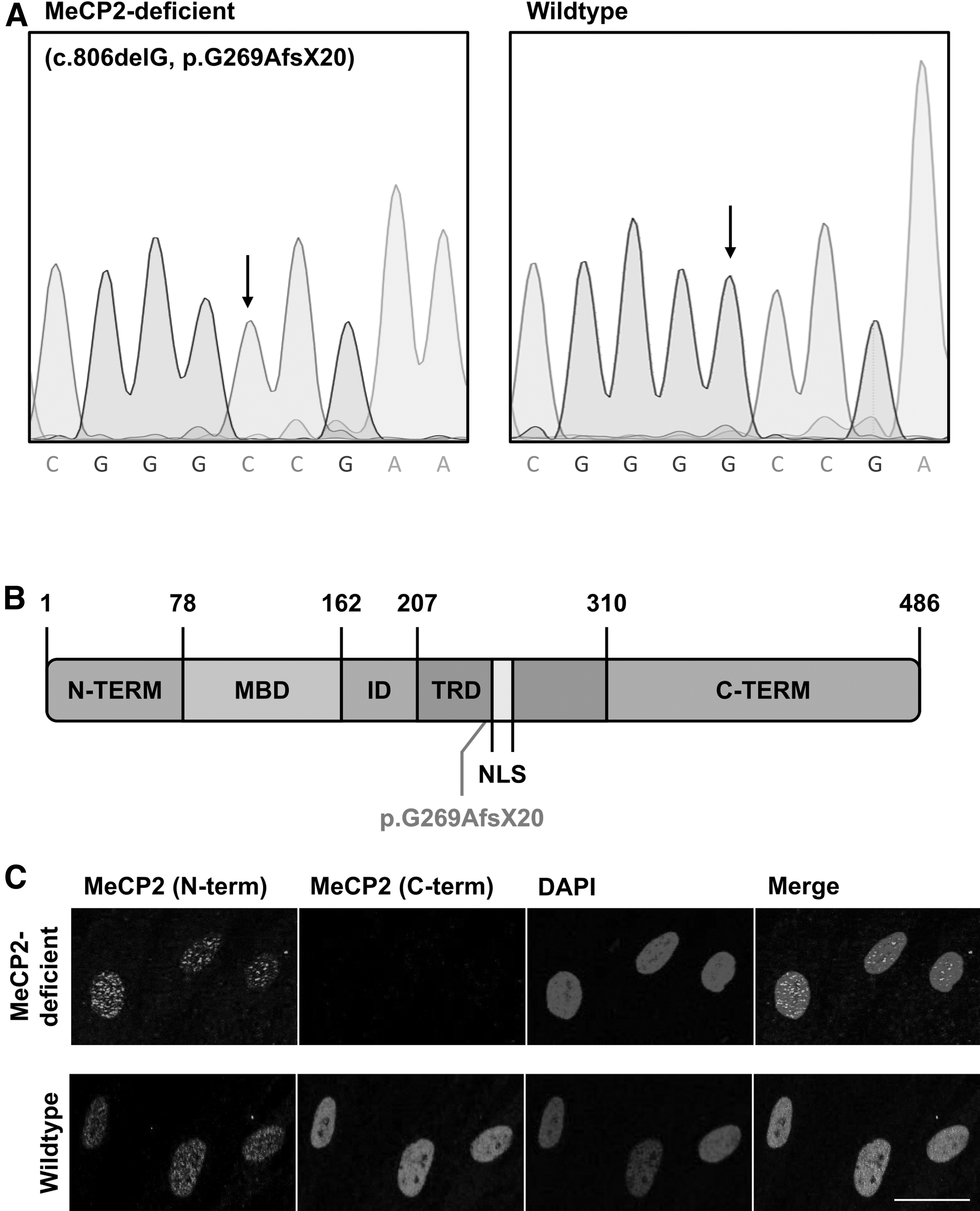
Mutational analysis of MeCP2-deficient HDFs. (
Direct reprogramming of HDFs into iNPs through episomal expression of PAX6 and SOX2
MeCP2-deficient and wild-type fibroblasts were transfected through electroporation with two episomal plasmids encoding the transcription factors PAX6 and SOX2. Reprogramming of patient-derived and wild-type fibroblasts resulted in the formation of iNPs, which were then further differentiated into neuronal cells. An overview of the reprogramming and differentiation process is shown in Fig. 2A.
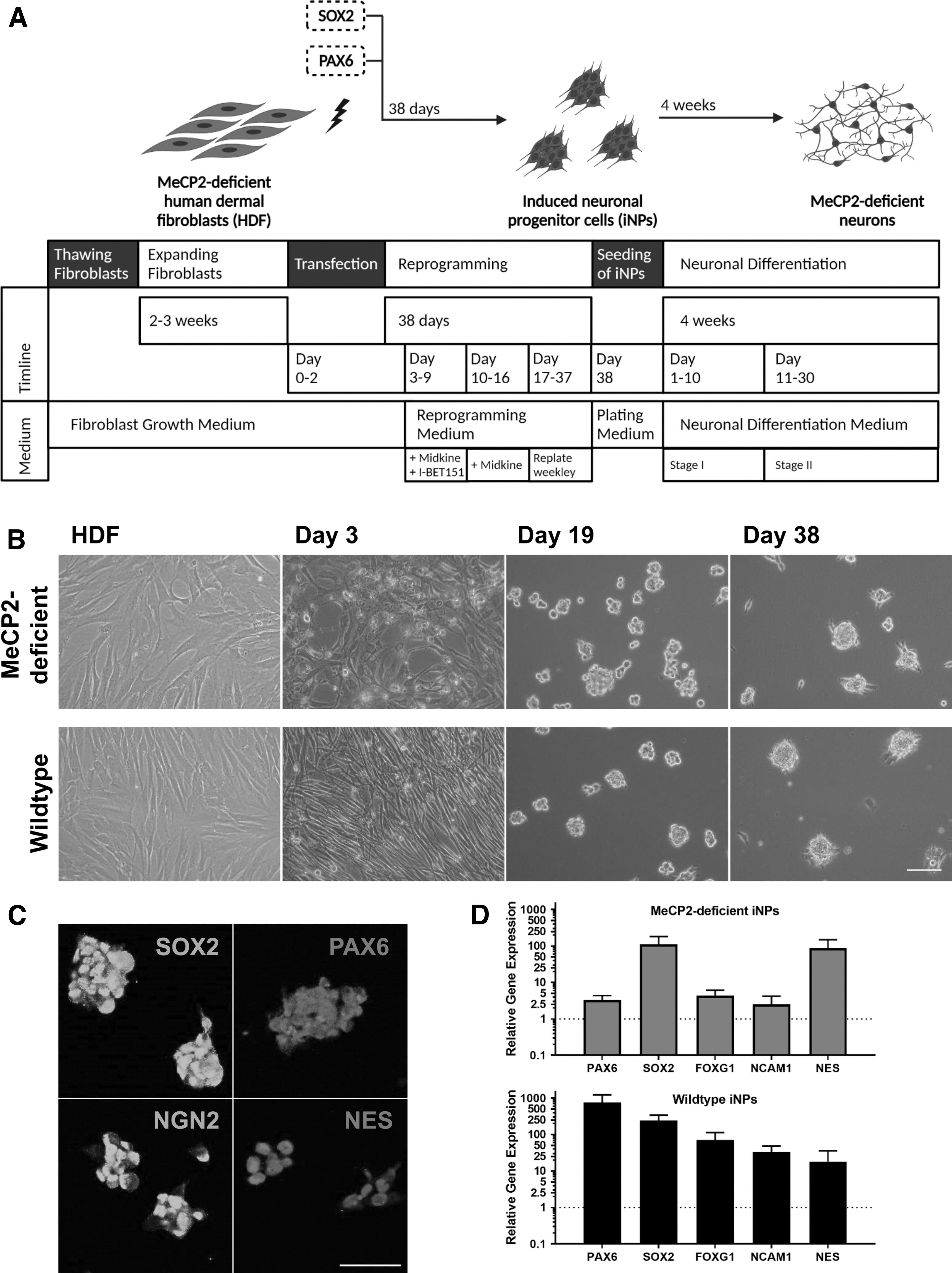
Direct conversion of HDFs into iNPs and neurons. (
Three days post-transfection, the cells were stained for PAX6 and SOX2 to assess the transfection efficiency (Supplementary Fig. S1). Positive PAX6 and SOX2 staining was observed, pointing to the fact that the transfection was indeed successful. The medium was changed from fibroblast growth medium to neuronal reprogramming medium. Weekly passaging from day 17 on led to the generation of iNPs. Bright-field microscopy images showed the morphological changes of the reprogramming process from elongated and flat fibroblasts to neurosphere-like colonies of iNPs over time (Fig. 2B).
For validation of the neuronal progenitor identity, iNP protein levels were examined on day 38 of the reprogramming process. Using immunocytochemistry, sustained expression of the transcription factors PAX6 and SOX2, which were originally used to initiate the reprogramming process, was confirmed. In addition, the neuronal progenitor markers Neurogenin-2 and Nestin were expressed in MeCP2-deficient and wild-type iNPs (Fig. 2C). At the same timepoint, RNA was isolated and subsequent qPCR showed increased gene expression levels of neuronal progenitor associated genes FOXG1, NCAM1, and NES in iNPs, compared to HDFs. In addition, the gene expression of PAX6 and SOX2 was strongly upregulated in iNPs (Fig. 2D).
Generation of patient-derived neurons from iNPs
On day 38 of reprogramming, iNPs were seeded for neuronal differentiation according to a two-step differentiation protocol. The morphology of the cells changed to a neuron-like phenotype with a soma and branched extensions (Fig. 3A). MeCP2-deficient, as well as wild-type, neurons expressed neuron-specific proteins such as MAP2 and neuronal nuclei (NeuN) in addition to glutamatergic (VLGUT1) and GABAergic [glutamate decarboxylase (GAD65/67)] markers (Fig. 3B). In addition, the presence of voltage-gated sodium channels in iNP-derived neurons was demonstrated using the patch clamp technique in the whole cell configuration. To isolate inward currents, potassium channels were blocked with an intracellular solution containing cesium chloride. To confirm the presence of voltage-gated sodium channels, currents in response to depolarizing voltage steps were blocked with 1 μM TTX, followed by a washout period of TTX with recovery of the channel activity (Fig. 3C).
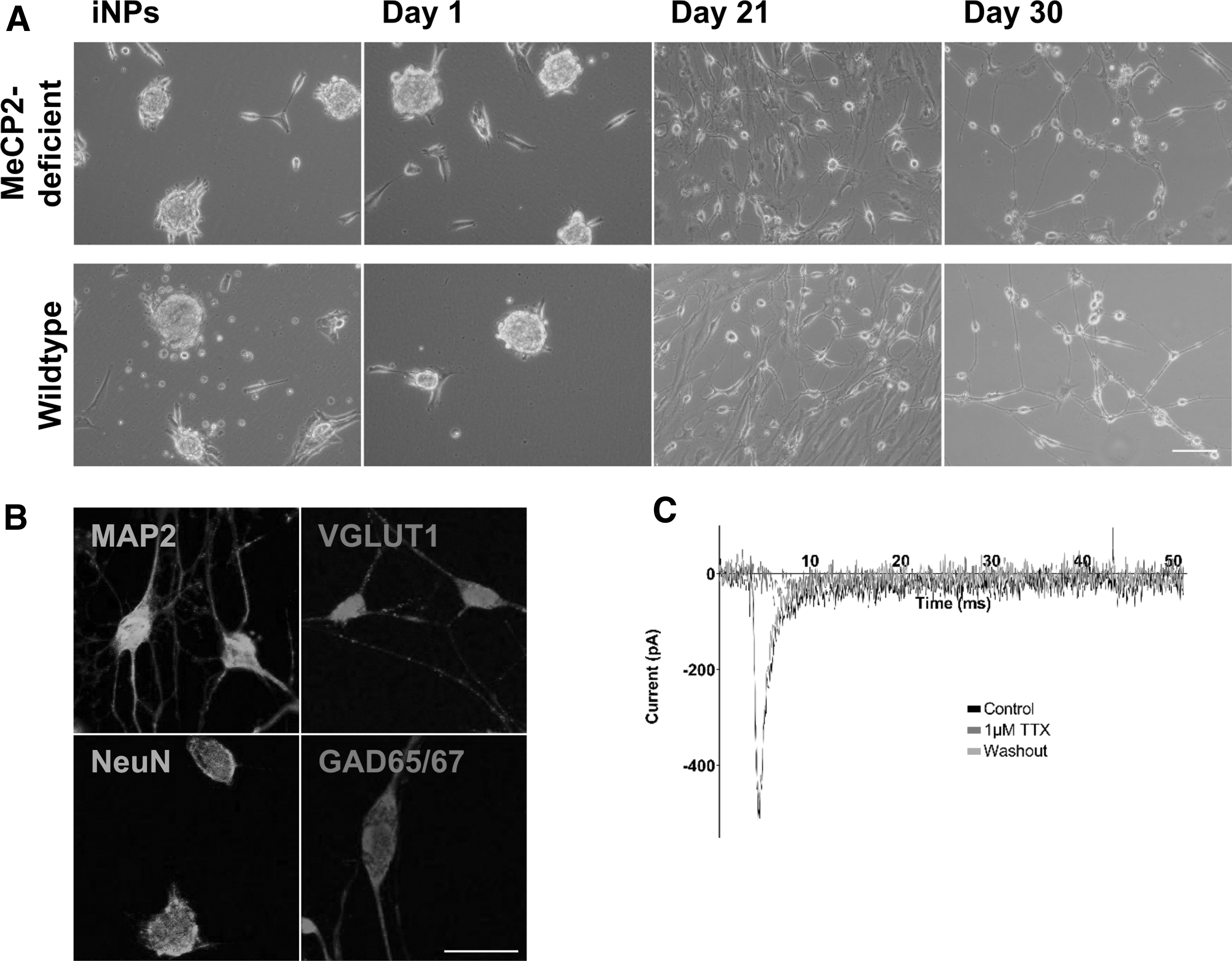
Differentiation of iNPs into neuronal cells. (
RNA-Seq confirms neuronal identity
RNA-Seq analysis was performed in triplicates in MeCP2-deficient and wild-type HDFs, iNPs, and neurons to demonstrate successful differentiation and to compare patient-derived and wild-type cells. Initially, PCA based on regularized log-transformed count data was used to get an insight of similarities and differences between each sample group. The first principal component (PC1), which represents the largest possible variance, clearly distinguished fibroblasts from iNPs and neurons. The second principal component (PC2) defined iNPs and neurons as two distinct clusters (Supplementary Fig. S2).
To demonstrate gene expression changes during differentiation, pathway analysis was carried out on DEGs between fibroblasts and neurons of MeCP2-deficient, as well as wild-type, cells (Fig. 4A and Supplementary Table S4). Using Protein ANalysis THrough Evolutionary Relationships (PANTHER) gene ontology database, cellular component analysis displayed a significant upregulation of neuron-specific cellular components such as “axon,” “neuronal cell body,” and “dendrite” in both patient-derived and wild-type neuronal cells, whereas expression of genes related to “cytosol,” “cytoplasm,” “cytoskeleton,” and “plasma membrane” was diminished (Fig. 4B). These similar expression patterns showed that patient-derived and wild-type neurons were comparable, and differentiation resulted in enrichment of neuron-specific cellular components.
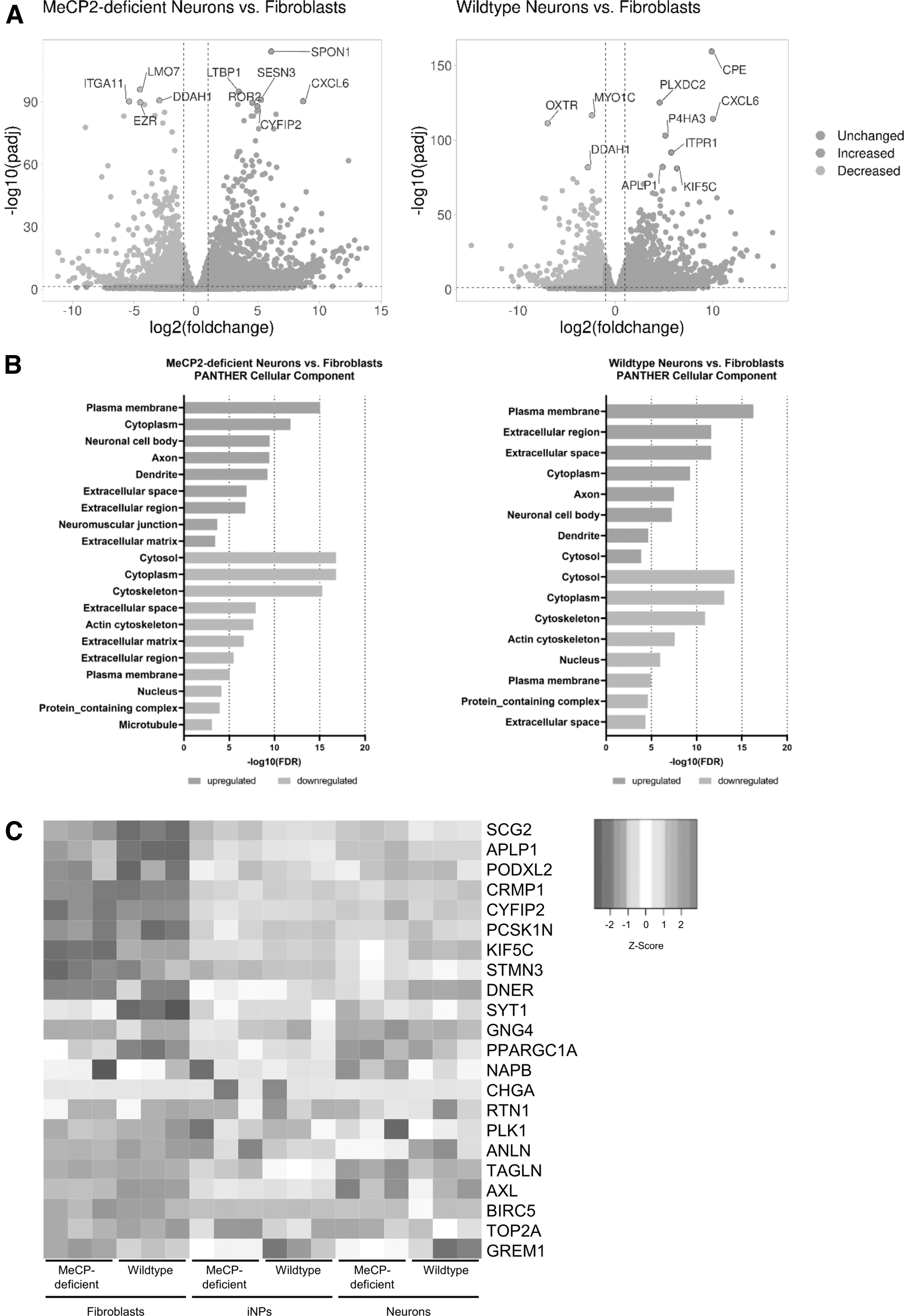
Gene expression profile of the differentiation process from fibroblasts to neurons. (
To further confirm neuronal identity, we compared our RNA-Seq expression dataset with a meta-analysis of 10 high throughput expression datasets of induced neurons from human and mouse fibroblasts demonstrating a common gene expression pattern of differentiation [42]. Comparing our dataset with the 22 common DEGs described in this meta-analysis, we found similar gene expression changes. Gene expression of iNPs, which already underwent a transformation to a neuronal lineage, was more comparable to neurons than fibroblasts (Fig. 4C).
To estimate the cell identity after the reprogramming and differentiation process, we compared RNA-Seq data from wild-type and MeCP2-deficient neurons with published data from neurons and glia cells [41]. MeCP2-deficient and wild-type cells revealed 74.3% and 72.4% neuronal cell identity, respectively (Supplementary Fig. S3). Hence, our data indicate that there is no significant difference in the differentiation potential of these two cell lines.
Morphological and gene expression analysis of neurons
The c.806delG pathogenic variant was stable throughout the reprogramming and differentiation processes, and MeCP2 could not be detected through immunofluorescence staining using the C-terminal MeCP2 antibody after 4 weeks of neuronal differentiation. For quantification of the morphological structure of MeCP2-deficient and wild-type neurons, cells were stained with TUJ1, a marker for mature neurons (Fig. 5A). Using the machine learning based software AIVIA, the number of neurites, branches, and the total neurite outgrowth were determined (Fig. 5B). Statistical analysis of the morphological structure confirmed a reduction in the number of neurites, branches, and total neurite outgrowth per neuron in MeCP2-deficient cells, compared to their wild-type counterparts (Fig. 5C). These results resemble the observations in postmortem human brain samples of RTT patients [43], which also showed a diminished dendritic arborization.
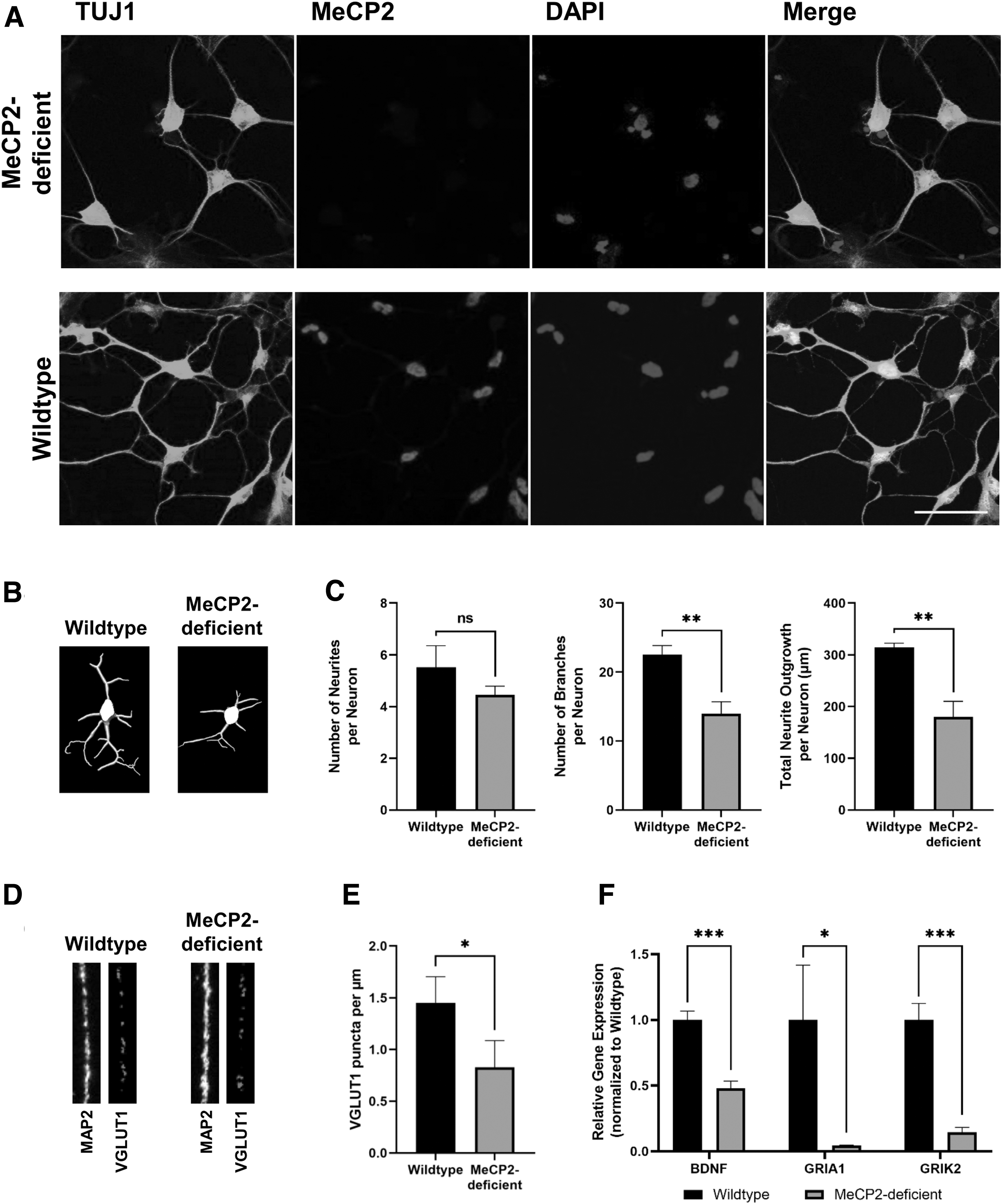
Morphological and gene expression analysis of MeCP2-deficient and wild-type neurons. (
Another characteristic of RTT is the decreased protein expression of VGLUT1, a vesicular glutamate transporter [15,44]. MeCP2-deficient neurons showed a significant reduction of VGLUT1 puncta per μm MAP2-positive dendrite, compared to their wild-type counterparts (Fig. 5D, E).
Impaired MeCP2 function is also known to affect BDNF levels. BDNF is a prominent target gene of MeCP2 [45 –47]. Gene expression analysis using qPCR revealed a significant downregulation of BDNF in MeCP2-deficient neurons, compared to their wild-type counterparts (Fig. 5D), in line with previous work [48]. RTT is also associated with dysfunctions in the glutamatergic system [15,49,50]. GRIA1 and GRIK2, which encode ionotropic glutamate receptors, were significantly downregulated in MeCP2-deficient neurons (Fig. 5F).
KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis of DEGs between MeCP2-deficient neurons revealed enrichment in “calcium signaling pathway,” “ECM-receptor interaction,” or “axon guidance” (Supplementary Fig. S4). These pathways were also found to be enriched in motor cortex and cerebellum samples of RTT patient autopsies [51].
TAT-MeCP2 treatment reduces histone hyperacetylation in MeCP2-deficient neurons
A hallmark of RTT pathophysiology on the cellular level is the hyperacetylation of histone H3 and H4 [52 –54]. For investigation of acetylation levels in patient-derived neurons, an In-Cell Western Assay was performed. Increased acetylation levels of H3K9 and H4K16 were detected in MeCP2-deficient neurons (Fig. 6). As treatment with TAT-MeCP2 was shown to be sufficient to ameliorate histone hyperacetylation in RTT-relevant cells [53], we investigated whether our cell model is a suitable tool to quantify the therapeutic potential of TAT-MeCP2 fusion protein by treating MeCP2-deficient neurons with TAT-MeCP2 fusion protein on days 23, 25, and 28 of neuronal differentiation. Following treatment, H4K16 amounts have reverted to wild-type levels (Fig. 6B). This reduction of hyperacetylation indicates that the observed differences between wild-type and MeCP2-deficient occur due to MeCP2 deficiency.

(
However, the morphological abnormalities of MeCP2-deficient neurons were not ameliorated within 7 days of TAT-MeCP2 treatment at the used dose (Supplementary Fig. S5).
Discussion
Disease recapitulating cell culture models are a vital tool for the investigation of RTT in vitro, as postmortem human brain samples are scarce and cannot be used for preclinical testing of novel treatment approaches. Furthermore, mouse models cannot accurately reflect human diseases due to species specific differences [55,56]. In this study, we present for the first time the application of a direct reprogramming method [19,26] to generate neurons from fibroblasts of an individual carrying a pathogenic variant in the X-linked MECP2 gene. To initiate fibroblast reprogramming, the transcription factors PAX6 and SOX2 were exogenously introduced to initiate the reprogramming process, as described previously [19,26].
To date, most of the human neuronal cell culture models for RTT were based on iPSCs or embryonic stem cells (hESCs) [15,57,58]. Using a direct reprogramming method, in contrast to iPSCs, the pluripotent cell state is bypassed and epigenetic aging signatures are maintained, leading to a more accurate reflection of the genetic and epigenetic background of the patient [21,59]. In the case of MeCP2, it is particularly important as this protein is a critical epigenetic regulator [60,61], and as such, a direct programming method—without the generation of a pluripotent intermediate—is preferential.
We used HDFs from a male individual with neonatal encephalopathy carrying a frameshift deletion in the MECP2 gene (c.806delG), although RTT is primarily diagnosed in females. As the MECP2 gene is located on the X chromosome, the use of male cells with only one X chromosome is beneficial to study pathophysiological changes caused by the complete loss of functional MeCP2. A case report about a male individual with an insertional MECP2 mutation presents with a clinically similar phenotype to female RTT patients [62].
In this study, we could demonstrate the successful reprogramming and differentiation of MeCP2-deficient and wild-type fibroblasts. iNPs showed a typical neurosphere-like morphology with the expression of neuronal progenitor markers Neurogenin-2 and Nestin. Furthermore, when comparing these iNPs with untransfected HDFs, an increase in gene expression of the neuronal precursors FOXG1, NCAM1, and NES could be shown (Fig. 2), consistent with similar reprogramming methods [19,20].
The successful differentiation of iNPs to neuronal cells was confirmed using immunocytochemistry depicting the expression of neuronal markers such as TUJ1 or MAP2. Further evidence for differentiation of iNPs toward a neuronal lineage was the PANTHER cellular component analysis, which showed an enrichment of neuron-specific cell structures, indicating the presence of a neuronal cell population, compared to untransfected fibroblasts. Moreover, we could demonstrate that wild-type, as well as MeCP2-deficient, neurons exhibit a typical gene expression pattern, which was also found in other direct reprogramming approaches for the generation of induced neurons from mouse and human fibroblasts [42].
So far, the c.806delG pathogenic variant was only studied in human iPSC-derived neural cells by Andoh-Noda et al. [63]. Human iPSCs were derived from RTT-monozygotic female twins. Differentiation of these two pairs of isogenic mutant and wild-type iPSCs resulted in an enhanced astrocytic differentiation of MeCP2-deficient cells. In contrast, using the direct reprogramming approach here described did not result in a significant difference in the astrocyte population between MeCP2-deficient and wild-type cells. However, according to the cell identity population estimation of our cell model, the percentage of astrocytes is very low.
Analysis of neuronal morphological structure of MeCP2-deficient neurons showed a decrease in the number of neurites, branches, and total neurite outgrowth per neuron in comparison to their wild-type counterparts. Similar observations were made in postmortem human brain samples from RTT patients [43], mouse models [64,65], and iPSC-derived neurons [15,58].
In addition, gene expression levels of BDNF, a gene important for neuronal development and survival [66], were downregulated in MeCP2-deficient neurons, in line with a meta-analysis of RTT transcriptomic data from different species, models, and MECP2 pathogenic variants [48]. In MeCP2-deficient neurons, a significant downregulation of GRIA1 and GRIK2 gene expression was also observed. Analysis of postmortem human brain samples of RTT patients also showed a decrease of GRIA1 gene expression [67]. A similar decrease was found in patients with schizophrenia [68]. Pathogenic variants in the GRIK2 gene, which led to loss of function of the encoded protein, are linked to autosomal recessive intellectual disability [69]. Moreover, puncta density of the vesicular glutamate receptor VGLUT1 was diminished in MeCP2-deficient neurons, which is in line with previous findings in other RTT disease models [15,44] and points to a general dysregulation of the glutamatergic system.
Another characteristic of RTT is the hyperacetylation of H3K9 and H4K16 [52 –54], which was also present in the used MeCP2-deficient neurons. Furthermore, the treatment of MeCP2-deficient neurons with recombinant TAT-MeCP2 fusion protein resulted in a decrease of H4K16 acetylation levels, underlining the applicability of this neuronal cell model for the study of novel treatment options such as protein replacement therapy [28,53,70,71], reactivation of the inactive X-chromosome [72], or gene therapy [73 –75]. In addition, the use of a direct reprogramming strategy would be beneficial for the study of DNA methylation patterns in RTT, as the epigenetic state of the cells is maintained [21,76,77]. Considering the benefits of direct reprogramming, this method serves as a valuable tool to study neurological diseases as it was shown for Huntington's disease [78], fragile X syndrome [30], or Parkinson's disease [18].
Although TAT-MeCP2 treatment ameliorated acetylation levels in MeCP2-deficient neurons, no significant improvement of the dendritic arborization was observed. For the rescue of morphological abnormalities, we hypothesize that a more extended treatment period may be required. This would be in line with findings of treatment with VPA for 20 days [57] or Nefiracetam for 14 days [79], which showed beneficial effects on the morphology of iPSC-derived RTT neurons.
One limitation of the study is the lack of further electrophysiological characterization of MeCP2-deficient and wild-type neurons. Although the presence of voltage-dependent sodium channels was confirmed, no action potentials could be recorded. To quantify action potentials, a longer differentiation time could be required. However, in RTT-iPSC-derived neurons, a significant impairment of action potential properties was observed [7,15]. Therefore, a more rigorous electrophysiological analysis of directly reprogrammed MeCP2-deficient and wild-type neurons will be the subject of further studies.
Conclusion
In conclusion, we have demonstrated that MeCP2-deficient HDFs can be reprogrammed and differentiated into neuronal cells through transient expression of the transcription factors SOX2 and PAX6. The generated MeCP2-deficient neurons express typical neuronal markers and the appropriate morphology. The MeCP2-deficient neurons exhibit properties typical for RTT, including diminished dendritic arborization and histone hyperacetylation, compared to their wild-type counterparts. Therefore, this cell model can act as a reliable proxy for the study of RTT and serve as an important platform for investigating prospective RTT treatment approaches.
Footnotes
Acknowledgments
The authors acknowledge the Core Facilities of the Medical University of Vienna, a member of VLSI, for the contribution of RNA-Seq experiment. The authors also thank Britta Kluge, Mario Mikula, Reinhard Lehner, and Klaus Schicker for their technical and consultative assistance. The research conducted at the Murdoch Children's Research Institute (MCRI) was supported by the Victorian Government's Operational Infrastructure Support Program. The Chair in Genomic Medicine awarded to J.C. is generously supported by The Royal Children's Hospital Foundation. ![]() was created with BioRender.com
was created with BioRender.com
Author Disclosure Statement
F.L. is inventor of the patent WO2007115578 A8, “Synthetic mecp2 sequence for protein substitution therapy”; holder of the patent: Georg August University, Universitätsmedizin Göttingen. All other authors declare no conflicts of interest.
Funding Information
This research was mainly funded by the Italian Rett Syndrome Association (AIRETT ETS) grant (H.S.). The initial phase of the study was supported by the German “Elternhilfe für Kinder mit Rett Syndrom.” I.G. was funded by Austrian Science Fund (FWF) grant I 3778, which is part of the ERANET programme iPS&Brain.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4