
Abstract
Although enhanced fibroblast growth factor (FGF) signaling has been demonstrated to be crucial in many cases of syndromic cleft palate caused by tongue malposition in humans, animal models that recapitulate this phenotype are limited, and the precise mechanisms remain elusive. Mutations in FGF9 with the effect of either loss- or gain-of-function effects have been identified to be associated with cleft palate in humans. Here, we generated a mouse model with a transgenic Fgf9 allele specifically activated in cranial neural crest cells, aiming to elucidate the gain-of-function effects of Fgf9 in palatogenesis. We observed cleft palate with 100% penetrance in mutant mice. Further analysis demonstrated that no inherent defects in the morphogenic competence of palatal shelves could be found, but a passively lifted tongue prevented the elevation of palatal shelves, leading to the cleft palate. This tongue malposition was induced by posterior spatial confinement that was exerted by temporomandibular joint (TMJ) dysplasia characterized by a reduction in Sox9+ progenitors within the condyle and a structural decrease in the posterior dimension of the lower jaw. Our findings highlight the critical role of excessive FGF signaling in disrupting spatial coordination during palate development and suggest a potential association between palatal shelf elevation and early TMJ development.
Introduction
Cleft palate is a prevalent congenital deformity in humans. Previous studies have demonstrated that both reduction and increase in the function of genes involved in the fibroblast growth factor (FGF) signaling pathway led to cleft palate in humans and mouse models.
Protein structure analyses and functional predictions have implicated nonsense mutations in FGF genes, such as FGFR1, FGFR2, FGFR3, and FGF8, in some cases of cleft palate in humans. 1 Similarly, various loss-of-function transgenic mouse models have underscored the intrinsic regulatory effects of FGF genes on palatogenesis. For instance, conditional deletion of Fgfr1 in the Wnt1-Cre lineage results in a cleft palate due to impaired proliferation and disturbed shelf fusion. 2 In Fgfr2b null mice, reduced proliferation in both the palatal mesenchyme and epithelium leads to a cleft palate without affecting the fusion potential. 3 In addition, impaired proliferation in Wnt1-Cre;Fgf10fl/fl mice and aberrant apoptosis in the shelf medial edge epithelium of Fgf10 null mice both contribute to cleft palate development. 3,4
Gain-of-function mutations in FGF genes usually result in cleft palate at a certain percentage in various human syndromes, such as FGFR2-related Apert syndrome, FGFR3-related Muenke syndrome, FGFR2- or FGFR3-related Crouzon syndrome, and FGFR1-related Hartsfield syndrome. 5 –8 Exploring the pathogenesis of these mutations is relatively complex because of the variations in ligand specificity and the activation by atypical ligands. For example, a gain-of-function mutation in Fgfr2 leads to decreased proliferation and reduced glycosaminoglycan levels in the palatal mesenchyme, 7 whereas another mutation in Fgfr2 causes abnormal proliferation, differentiation, and apoptosis. 5
Precise genetic diagnoses have identified partial FGF-related cleft palate cases in humans, and the role of additional FGF family members in palatogenesis is being explored using transgenic mouse models. Fgf9, a critical FGF ligand in craniofacial development, is expressed in the palatal epithelium and mesenchyme during palatogenesis in mice. 9 40% of Fgf9 knockout mice exhibited cleft palate. 10 Sox11 null mice that showed decreased Fgf9 expression in the palate and mandible during palatogenesis manifested an undersized mandible with a cleft palate, although the elevation and fusion potentials of the palatal shelf remained intact, 11 which resembles the cleft palate resulting from micrognathia and subsequent tongue malposition in the Pierre Robin sequence (PRS). A Ddx4-Cre-driven Fgf9 knockout mouse model revealed that both intrinsic mesenchymal proliferation defects and extrinsic micrognathia contribute to palate cleft, 11,12 implying that FGF9 signaling may play a comprehensive role in coordinating the development of maxillofacial structures.
Interestingly, a human missense mutation in FGF9 induces cleft palate along with synostoses, in which both loss-of-function effects due to decreased FGFR3 binding and gain-of-function effects resulting from the reduced sequestration of functionally inactive dimers and consequently elevated FGF9 levels, is postulated as a pathogenic driver for cleft palate phenotype. 13 The pathogenic mechanisms of human FGF9 gain-of-function in palatogenesis warrant further exploration, especially considering the pivotal role of the ligand in the yet-to-be-fully understood ligand-dependent activation process. Herein, we generated a conditional mouse line using pMes-Fgf9 and Wnt1-Cre alleles to specifically enhance Fgf9 activity in cranial neural crest cells (CNCCs). Our findings demonstrated that Fgf9 overexpression consistently leads to cleft palate caused by tongue malposition rather than intrinsic impacts on the palate. The tongue malposition could potentially be driven by the posterior spatial confinement exerted by TMJ dysplasia.
Materials and Methods
Generation of Wnt1-Cre;pMes-Fgf9 mice
All animal care and experimental protocols were reviewed and approved by the Fujian Normal University Institutional Animal Care and Use Committee. Wnt1-Cre transgenic mice were sourced from Jackson Laboratory. 14 To generate the conditional Fgf9 transgenic mouse (pMes-Fgf9), we used pronuclear injection of the pMes-Fgf9-IRES-Egfp vector, constructed by inserting the full-length cDNA of mouse Fgf9 between the β-actin promoter and IRES-Egfp sequence, flanked by a transcription STOP cassette at the 3′ end (Supplementary Fig. 1A) 15 To specifically activate Fgf9 expression in the CNCC-derived mesenchyme, we crossed Wnt1-Cre mice with pMes-Fgf9 mice, resulting in Wnt1-Cre;pMes-Fgf9 embryos. Transgenic mice were identified by PCR genotyping and further validated by Green Fluorescent Protein (GFP) tracing, immunostaining, and quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR) detection. The primer for Fgf9 and β-actin is as follows: Fgf9-forward, ATGGCTCCCTTAGGTGAAGTT; Fgf9-reverse, TCATTTAGCAACACCGGACTG; β-actin-forward, GGCTGTATTCCCCTCCATCG; β-actin-reverse, CCAGTTGGTAACAATGCCATGT.
Skeletal preparation, histology, and immunohistochemistry
Littermates of Wnt1-Cre control and Wnt1-Cre;pMes-Fgf9 mutant embryos were collected at developmental stages ranging from E12.5 to 18.5. Heads of these samples were dissected for skeletal staining using Alcian blue/Alizarin red, following a protocol outlined previously 16 and prepared for either paraffin or frozen sectioning at 8 μm thickness. Slides were subjected to hematoxylin and eosin (H&E), Azan, or immunofluorescence staining. All the sections were prepared and stained according to the manufacturer’s guidelines. For immunofluorescence staining, a range of antibodies were used, including anti-FGF9 (Santa Cruz, sc-8413; 1:200), anti-MHC (DSHB, MF20; 1:200), anti-Ki67 (Spring, M3060; 1:200), anti-Sox9 (Abcam, ab3697; 1:200), anti-Runx2 (Santa Cruz, sc-390351; 1:200), anti-Col2 (Abcam, ab34712; 1:50), anti-Col10 (Abcam, ab260040; 1:100), and anti-caspase-3 (Abcam, ab44976; 1:200).
BrdU labeling
BrdU labeling was performed to detect cell proliferation rate using the BrdU Labeling and Detection Kit, as described previously. 17 Briefly, pregnant Wnt1-Cre mice mated with male pMes-Fgf9 mice and were given an intraperitoneal injection of BrdU labeling reagent (1 mL/100 g body weight) on E13.5. After 1 h of injection, mice were sacrificed, and embryos were collected. Embryonic heads were fixed in Carnoy’s fixative, dehydrated through an ethanol gradient, embedded in paraffin wax, and then sectioned at 10 µm. The BrdU immunodetection was conducted with a BrdU labeling and Detection kit (Roche Diagnostics Corporation) following the manufacturer’s protocol. The rate of cell proliferation was determined by counting BrdU-positive cells and total cells within defined arbitrary areas of the palatal epithelium and mesenchyme. Each of the three wild-type and mutant samples was assessed by counting three continuous sections. Student’s t-test was used to ascertain the significance of the difference.
In vitro roller culture and organ culture
Mouse embryos E13.5 were collected for in vitro roller culture and palate fusion assay. The lower part of the head, including the tongue and mandible, was removed, whereas the remaining upper portion containing the palatal shelf was used for in vitro roller culture, as detailed in a previous report. 18 Both mutant and control embryos were positioned in a glass bottle containing the culture medium and roller cultured for 24 h. Subsequently, the samples were fixed and stained with H&E. For the palate fusion assay, the palatal shelves were carefully separated from the maxilla, as previously described. 19 Paired palatal shelves from both mutant and control embryos were placed on filter paper and cultured for 48 h before being processed for fixation and H&E staining. Both in vitro roller culture and palate fusion assay were performed using three different embryos from different pregnant mice.
Measurement
Littermates of Wnt1-Cre control and Wnt1-Cre;pMes-Fgf9 mutant embryos were compared in all statistical evaluations. Experiments were performed with at least three biological replicate pairs. The coronal observations were aligned to the reference section crossing the 4 first molars, and the sagittal observations were made with the reference to the central sagittal plane of the body. The obtained images were calibrated and imported into ImageJ software for precise digital calculations. Statistical evaluation of the measurements in this study was performed using either the paired Student’s t-test or unpaired two-sample t-test. A statistical significance threshold of P < 0.05 was used to determine the significance of the observed results. The paired Student’s t-test results were displayed with connected control-and-mutant plots to show the details of different pairs.
Results
Cleft palate is not caused by intrinsic defects in the palatal shelves
To confirm the overexpression of Fgf9, the presence of GFP that was expressed by an Ires-Egfp cassette flanked downstream of the Fgf9 transgene in cells of CNCC-derived tissues such as the craniofacial mesenchyme in the maxillary and mandibular processes, periocular mesenchyme, and midbrain at E13.5 was identified (Supplementary Fig. S1C–F). qRT-PCR assay further confirmed ectopic Fgf9 expression in the palate mesenchyme (Supplementary Fig. S1B). All Wnt1-Cre;pMes-Fgf9 embryos suffered postnatal mortality. Mutant embryos from E15.5 to E18.5 consistently displayed a cleft of the secondary palate accompanied by an open mouth with 100% penetrance (n = 55, Fig. 1A’, B’).

Augmented FGF9 signaling induces cleft palate.
To explore whether overexpression of Fgf9 alters the intrinsic morphogenic competence of the palate, we examined cell proliferation, apoptosis, shelf elevation, and fusion potential, which are crucial for palate formation. BrdU staining showed comparable levels of cell proliferation and inconspicuous apoptosis in the epithelium and mesenchyme of control and mutant palatal shelves at E13.5 (Fig. 1C–D’, Supplementary Fig. S2). Results from in vitro roller culture indicated that, as controls, mutant palatal shelves could normally elevate from vertical to horizontal after 1 day of culture (Fig. 1E–E’). The palate fusion assay demonstrated that paired shelves from both groups initiated fusion within 2 days of culture with comparable efficiency (Fig. 1F–F’). These findings suggest that the cleft palate defect in Wnt1-Cre;pMes-Fgf9 mice is not the outcome of intrinsic developmental defects in the palatal shelves.
Tongue malposition prevents palatal elevation
During palatogenesis, further histological analysis found that both control and mutant palatal shelves were formed laterally alongside the tongue in comparable positions at E13.5 (Supplementary Fig. S3). Meanwhile, a slightly deformed tongue could be seen in the mutant at E13.5 (Supplementary Fig. S3). By E14.5, when both sides of lateral palatal shelves in controls were successfully elevated to a horizontal position and fused with each other, leaving an epithelial seam in between (Fig. 2A, B, C), the mutant shelves remained vertically oriented and separated from each other with the tongue in between (Fig. 2A’, B’, C’), indicating that cleft palate was hampered by the tongue during palatal shelf elevation in Wnt1-Cre;pMes-Fgf9 embryos. Moreover, compared with the control, mutant tongues were significantly higher than control ones from anterior to posterior by E14.5 (Fig. 2A’, B’, C’, D), and an unexpectedly shorter tongue was observed at E18.5 (Fig. 2E’), both supporting the occurrence of tongue malposition in the mutants. Obviously, this tongue malpositioning prevented palate elevation.
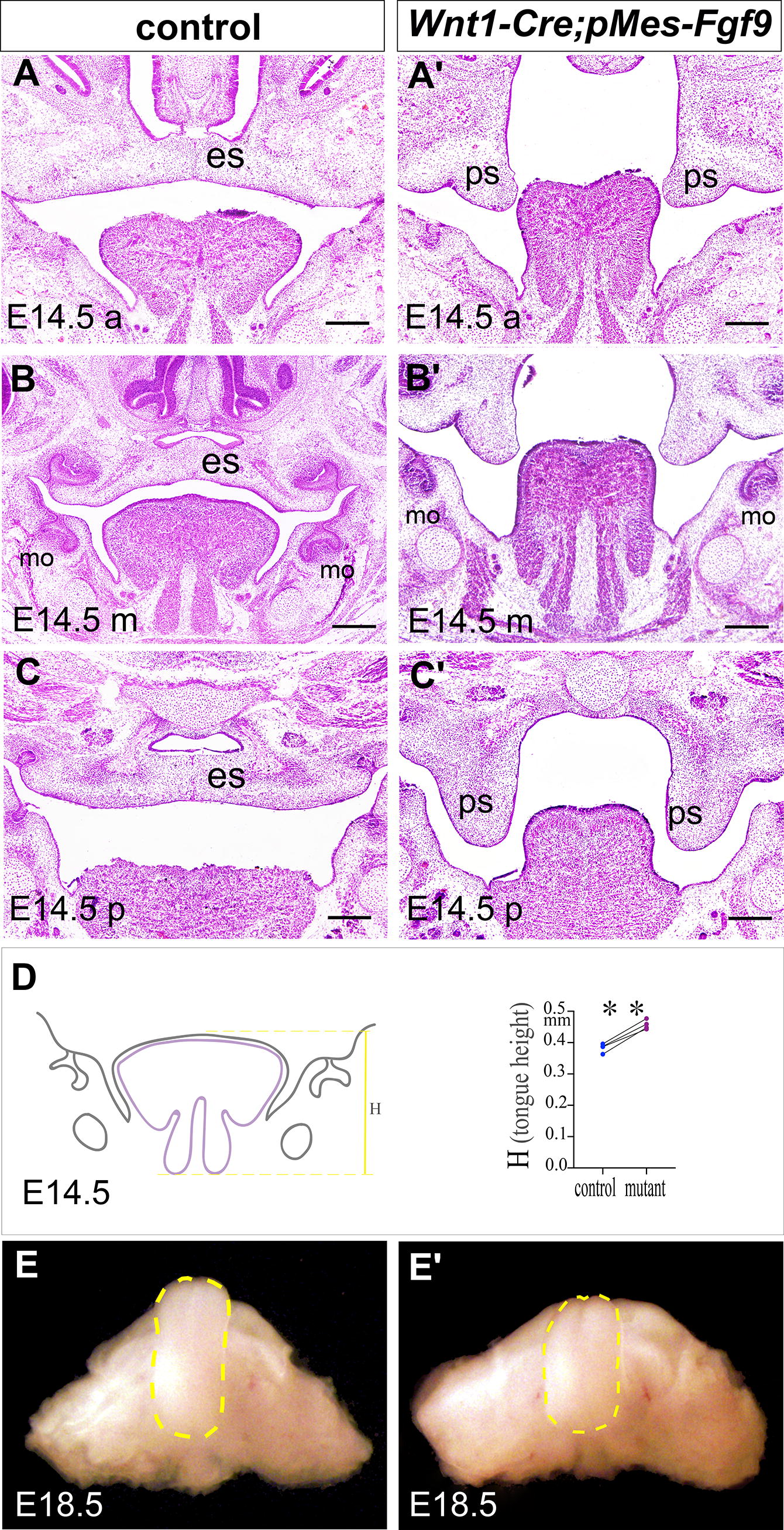
The malpositioning tongue is responsible for the cleft palate. (
Passive malpositioning of the tongue
We further explored the main reasons contributing to tongue malpositioning. It is well known that the tongue is mainly composed of muscle tissues and is derived from muscle progenitor cells instead of CNCCs, meaning that its differentiation behavior would probably not be disturbed by augmented FGF9 signaling in CNCCs. To test this notion, we examined the arrangement and attachment of tongue muscles using the muscle-specific marker MF20 during tongue formation. No abnormalities in myogenic arrangement and muscle attachment in mutant tongues were observed at E14.5 (Fig. 3A–B’), suggesting no myogenic disorders occurred during tongue positioning. Meanwhile, despite the height enlarged in the mutant tongue, the tongue area had no significant difference between controls and mutants (Fig. 3C), implying the normal differentiation behavior and the passive malpositioning of the tongue in the mutant. This phenotype is supported by the observation of the comparable rate of cell proliferation evaluated by the density of Ki67+ cells in the mutant tongue at E13.5 compared with the control one (Fig. 3D–D’), further supporting the idea of passive malpositioning of the tongue in the mutant.

The normal differentiation of the tongue.
Although it was encouraging to observe an expanded bony mandibular body that was Runx2-labeled and may provide the spatial squeezing effect to induce the passive tongue malpositioning in the mutant (Fig. 3A, A’), we found that the bilateral mandibular bodies in mutants provided a larger area with statistical significance when measuring the accommodating area in coronal sections crossing the 1st molar buds (Supplementary Fig. S4). These data ruled out the coronal extrusion provided by mutant mandibular bodies, suggesting that no coronal spatial confinement exists to act as a peripheral factor in disturbing tongue positioning during palate shelf elevation in the mutant.
TMJ dysplasia results in posterior spatial confinement
Sagittal spatial confinement exerted by the mandible is regularly investigated as a common cause of secondary cleft palate. Therefore, we further examined jaw structure in the sagittal plane to see if it is associated with the palatal elevation failure in Wnt1-Cre;pMes-Fgf9 mutants. Skeletal staining in the sagittal plane revealed defects in the cranial vault and craniofacial bones in the mutant at E18.5 (Fig. 4A–B’). The control condyle rested in the joint fossa, which works as the rotation center for the whole mandible in the wild type. However, the rested condyle exhibited a shortened posterior height in the mutant with a passive mouth opening (Fig. 4B’). It is known that the TMJ-determined posterior development of the lower jaw is crucial in the coordination of craniofacial development. 20 Then, we defined the posterior and anterior interjaw heights as H1 and H2, respectively, based on the distance from the posterior or anterior point of the mandibular body to the fossa reference plane (Fig. 4B, B’, C). The mutant posterior height (H1) was severely reduced compared with the control one, resulting in posterior vertical confinement against tongue accommodation (H2, Fig. 4B, B’, C). Detailed examination of the lower jaw samples further revealed an undersized and deformed condylar process of the TMJ with thinner condyle cartilage in the mutant at E18.5 (Fig. 4D–D’). Although the anterior length of the mandibular body (horizontal, labeled as L3) was almost unaffected in mutants (Fig. 4D–D’, E), the posterior length of the lower jaw was significantly decreased, including both the ramus length (oblique, labeled as L2) and condyle-cartilage length (oblique, labeled as L1, Fig. 4E). These reduced posterior oblique lengths signify their shortened projections onto the vertical and horizontal directions. The above length analyses from the sagittal plane suggest that mandibular body length does not contribute to spatial confinement in the mutants. Instead, TMJ dysplasia appears to be the primary factor leading to decreased posterior dimension against tongue accommodation, resulting in abnormal tongue shaping and positioning as well as, ultimately, cleft palate.

Reduced posterior dimension in TMJ.
To verify that the early TMJ development could serve as a pathogenic cause of palate elevation, we conducted additional analyses. At E13.5, the mutant lower jaw exhibited a similar passive rotation (Supplementary Fig. S1D), an earlier deficiency in the posterior height. At E14.5, when FGF9 expression was detectable in the condyle and peripheral muscles of controls and was obviously elevated in mutants (Fig. 5A–A’), TMJ dysplasia with an obscured condylar anlage was seen (Fig. 5B, B’). A remarkably thinner and shorter condyle process was found at E15.5 (Fig. 5C, C’) in mutants. Moreover, by setting the maxillofacial midline as the reference vertical line, the posterior height from the apex of the condyle to the bottom point of the mandibular body was obviously reduced in mutants at E15.5 (Fig. 5C, C’) and E18.5 (Fig. 5D, D’). These data suggest that posterior confinement caused by TMJ dysplasia occurs during palate shelf elevation.

TMJ dysplasia.
Impairments of chondrogenesis in condyles
Since the reduced cartilage formation was observed in the mutant condyle (Fig. 4D–D’), we further investigated the impact of augmented FGF9 on condylar morphogenesis. Our findings revealed that the number of Sox9+ progenitors of condylar cartilage was largely reduced in the mutant condyle during the critical period of palatogenesis from E13.5 to E15.5 (Fig. 6A–C’). In addition, we observed that the differentiation of chondrocytes, indicated by the Col2 antibody, was relatively advanced at E14.5 in the mutant condyle (Fig. 6D–D’). This accelerated differentiation can deplete the smaller pool of Sox9+ progenitors before their proliferation. Later in TMJ development, chondrocyte hypertrophy, marked by the Col10 antibody, was significantly delayed in the mutant condyles (Fig. 6E–E’). These data suggest that although chondrogenesis initiation occurs simultaneously at E13.5, continuous impairments accumulate during chondrogenesis of the condyle in mutants prior to E15.5, the final timing for spatial adjustment for palate shelf elevation. Furthermore, examination of condylar apoptosis and proliferation revealed minimal apoptosis in control and mutant condyles at E13.5 and E14.5 (Fig. 6F–G’). However, a notable reduction in the proportion of proliferating cells labeled with the Ki67 antibody was observed in the mutant condyle at E14.5, a crucial time point for condylar proliferation and palate shelf elevation (Fig. 6I, I’, J). Collectively, our evidence indicates that during the critical period of palatogenesis, both impaired chondrogenesis and decreased proliferation in the mutant condyle contribute to TMJ dysplasia.

Impaired chondrogenesis and proliferation in the TMJ condyle.
Discussion
Mutations in FGF9 have been linked to cleft palate in humans. 13 FGF9 insufficiency secondary to Sox11 deficiency may contribute to influencing the mandible dimension rather than causing the loss of elevation and fusion potential of palatal shelves, ultimately resulting in cleft palate. 21 This phenotype resembles cleft palate in humans with PRS, which is caused by micrognathia and subsequent tongue malposition. Fgf9 deletion has been shown to exert an intrinsic influence on palatogenesis along with its extrinsic impacts. 11 Our study demonstrated that augmented FGF9 activity in CNCC-derived tissues induced cleft palate caused by tongue malposition rather than intrinsic impacts. Moreover, our data also suggest that the elevated FGF9 level can induce TMJ dysplasia, which may act as an extrinsic cause of cleft palate by undermining the spatial coordination between tongue descent and palatal shelf elevation.
Although TMJ morphogenesis received limited attention in previous cleft palate studies, our study highlights its potential role in spatial coordination during palatogenesis. It is known that the TMJ condyle is detectable as early as the 7th week of gestation in humans 22,23 and E12.5 in mice, 20,24 indicating the overlap of TMJ morphogenesis and palatogenesis prior to the elevation of the palatal shelf. The TMJ-determined posterior development of the lower jaw is crucial in coordinating craniofacial development. TMJ dysplasia can compress the capacity of the oral cavity to accommodate the descending tongue during the time of shelf elevation, as demonstrated in our model. Locally, the posterior region of the secondary palate is particularly vulnerable to cleft owing to molecular heterogeneity and varied elevation mechanisms along the palatal anterior-posterior axis. 25 –27 In our model, TMJ dysplasia confines the posterior dimension and exerts additional stress on this area, leading to complete penetrance of the cleft palate in Fgf9 mutants.
The co-occurrence of TMJ dysplasia and cleft palate has been reported in several human FGF-related syndromes, such as achondroplasia and Muenke syndrome. 28 –30 However, no specific FGF ligand has been associated with the embryonic chondrogenesis of the TMJ condyle, a secondary cartilage with distinct molecular regulation. 20,31 FGF9 has been demonstrated to be an essential regulator of chondrogenesis and is involved in mesenchymal condensation, chondrocyte proliferation, and initiation of hypertrophy in the stylopod (primary cartilage). 32 In this study, we identified FGF9 expression in the condyle and peripheral muscle tissue during early TMJ morphogenesis. Given that FGF9 can activate all identified FGF receptors in the condyle, 33,34 the biological significance of this ligand in condyle morphogenesis warrants further exploration. Our findings reveal the pathological effects of enhanced FGF9 signaling on condyle chondrogenesis, including reduced Sox9+ progenitors, accelerated chondrogenic differentiation, decreased proliferation, and impaired chondrogenic maturation. These observations are consistent with the impairments observed in gain-of-function mutations in Fgfr3, an affinity-optimized receptor for FGF9. 28 –30 These similarities suggest a potential binding of FGF9 to FGFR3 underlying TMJ dysplasia. Considering FGF9 is expressed in the normal condyle as well, we propose that the FGF9-FGFR3 signaling may play important roles in spatially coordinating maxillofacial development via regulating condyle chondrogenesis and disruption of this signaling would result in TMJ dysplasia and associated cleft palate.
Furthermore, even in some human syndromes that are classified as non-isolated PRS, such as auriculo-condylar syndrome and Treacher–Collins syndrome, cleft palate occurs in a certain proportion in the context of TMJ malformation. 35 –38 Notably, cephalometric data have shown significant posterior height loss along with variable decreases in mandibular length in infants with isolated PRS and children with multiple non-isolated PRS. 39,40 This suggests that posterior dimension loss may be another interpretation in other human syndromes besides the generally focused mandible length reduction in micrognathia. Our animal model suggests that TMJ dysplasia alone could trigger palato-oropharyngeal disharmony and cleft palate without a reduction in mandibular length. In addition, cephalometric data from humans further emphasize the strong correlation between posterior facial height, mandible length, and the clinical severity of palato-oropharyngeal defects, 41 highlighting the importance of precise structural analysis for personalized treatment and prognosis in individuals with PRS.
Footnotes
Ethics Statement
The animal study was reviewed and approved by the Fujian Normal University Institutional Animal Care and Use Committee.
Data Availability Statement
The original contributions presented in this study are included in this article. Further inquiries can be directed to the corresponding author.
Author Disclosure Statement
The authors declare that the research was conducted without any commercial or financial relationships that could be construed as potential conflicts of interest.
Funding Information
This study was supported by the National Natural Science Foundation of China (82170917 and 82001002), the Health Education Joint Research Project of Fujian Province (2019-WJ-14), and the Fujian Provincial Engineering Research Center of Oral Biomaterial, School and Hospital of Stomatology, Fujian Medical University (2023GC-B01 and 2022GXA01).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4