
Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive lung disease with no cure except transplantation. Abnormal alveolar epithelial regeneration is a key driver of IPF development. The function of Yes1 Associated Transcriptional Regulator (YAP) in alveolar regeneration and IPF pathogenesis remains elusive. Here, we first revealed the activation of YAP in alveolar epithelium 2 cells (AEC2s) from human IPF lungs and fibrotic mouse lungs. Notably, conditional deletion of YAP in mouse AEC2s exacerbated bleomycin-induced pulmonary fibrosis. Intriguingly, we showed in both conditional knockout mice and alveolar organoids that YAP deficiency impaired AEC2 proliferation and differentiation into alveolar epithelium 1 cells (AEC1s). Mechanistically, YAP regulated expression levels of genes associated with cell cycle progression and AEC1 differentiation. Furthermore, overexpression of YAP in vitro promoted cell proliferation. These results indicate the critical role of YAP in alveolar regeneration and IPF pathogenesis. Our findings provide new insights into the regulation of alveolar regeneration and IPF pathogenesis, paving the road for developing novel treatment strategies.
Introduction
Idiopathic pulmonary fibrosis (IPF) represents a chronic, progressive, and irreversible interstitial lung disorder characterized by the relentless accumulation of fibrotic tissue within the pulmonary interstitium, culminating in architectural distortion and compromised gas exchange. 1 The clinical trajectory of IPF is underscored by substantial morbidity and mortality, as patients undergo a gradual deterioration in pulmonary function and quality of life, typically accompanied by a median survival duration ranging from 3 to 5 years. 2 Despite recent advancements in the understanding of IPF pathogenesis, therapeutic options for IPF remain limited, and the disease continues to confer a grave prognosis. 3 Therefore, there is an urgent need for the development of novel therapeutic strategies targeting the underlying pathophysiological mechanisms driving fibrosis progression.
Defective injury repair in alveolar epithelium initiates IPF. 4 Alveolar epithelium 2 cells (AEC2s) can self-proliferate and differentiate into alveolar epithelium 1 cells (AEC1s) upon injury. 5 –8 However, mechanisms governing AEC2 regeneration have yet to be elucidated.
The Hippo pathway, originally identified in Drosophila melanogaster, has emerged as a crucial signaling cascade governing diverse cellular processes across species, including organ development, tissue homeostasis, and regeneration. 9 Yes1 Associated Transcriptional Regulator (YAP) and its paralog Transcriptional Coactivator With PDZ-Binding Motif (TAZ) serve as the effectors of the Hippo pathway, acting as primary regulators of organ size while also being involved in regeneration. 10,11 Previous studies indicate removal of both Yap and Taz in mice greatly impedes alveolar regeneration upon bacterial infection. 12,13 However, conflicting findings have been reported regarding the specific roles of YAP and TAZ in pulmonary fibrosis. Sun et al. reported that only TAZ contributes to the alleviation of fibrosis by promoting alveolar regeneration, while YAP does not play a significant role in regulating alveolar regeneration. 14 These observations echo previous reports highlighting the differential functions of YAP and TAZ, despite sharing numerous functional similarities. 15,16 Therefore, whether singular YAP regulates alveolar regeneration and thus IPF pathogenesis remains unclear. Additionally, the molecular mechanisms underlying the promotion of alveolar regeneration by YAP and TAZ have yet to be fully elucidated.
In this investigation, we delineate the pivotal role of YAP in AEC2 in alveolar regeneration. We revealed the activation of YAP in IPF patients. Subsequently, deletion of YAP in AEC2 in mice exacerbated pulmonary fibrosis. Notably, alveolar regeneration was impeded in the mutant mice and alveolar organoids with YAP deletion. Mechanistically, YAP regulated AEC2 stemness by promoting cell proliferation and AEC1 differentiation by directly inducing regulators of cell cycle progression and AEC1 differentiation. Our study provides the first evidence that YAP facilitates alveolar regeneration by enhancing AEC2 self-renewal and promoting differentiation into AEC1, thereby mitigating pulmonary fibrosis.
Materials and Methods
Human samples
The pulmonary fibrosis lung section was from a postmortem sample obtained from the collection of the Shenzhen Third People’s Hospital (one COVID-19 patient). The control lung tissues were from normal tissues far from the tumor of lung adenocarcinoma patients, obtained from the First People’s Hospital of Guangzhou City.
Organoid culture
We isolated primary AEC2 using fluorescence-activated cell sorting. Briefly, lung tissue was minced and digested at 37°C with a specific enzyme cocktail (15 U/mL dispase, 225 U/mL collagenase, and 50 U/mL DNase) for 45 min. Subsequently, red blood cells were lysed using a red blood cell lysis buffer following filtration through a 70-µm filter. The resulting single-cell suspension was then further filtered through a 40-µm filter for flow sorting to obtain GFP-labeled AEC2. Subsequently, 5,000 AEC2 were co-cultured with 30,000 MEF, centrifuged, and resuspended in 45 µL of small airway growth medium (SAGM, LONZA, CC-3118). The cell mixture was then combined with Matrigel (Corning, 356234) and inoculated in Transwell inserts. After allowing the Matrigel to solidify in the Transwell by placing it in a cell incubator for 20 min, SAGM supplemented with a Rho kinase inhibitor (Abmole, M1817) was added along the Transwell’s side wall. The medium with the Rho kinase inhibitor was replaced with inhibitor-free SAGM medium 24–48 h later, and subsequently refreshed every 48–72 h. After 14 days of incubation, spheroids were collected for downstream experiments.
Animals
All mice were housed and treated in accordance with the animal protocol approved by the Laboratory Animal Research Center of Sun Yat-sen University. SpcCreER, 17 Rosa26mTmG [Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J; RRID: IMSR_JAX:007576], Yap flox/flox18 mice were previously described. All the mice used in the experiment were male, 12–16 weeks of age, with a C57BL/6 background. Intraperitoneal injection of tamoxifen activating Cre recombinase for 3 consecutive days was used to specifically knock out the Yap gene in AEC2 of Spc-CreER;Yap-cKO; Rosa26mTmG mice and perform AEC2 lineage tracing in Spc-CreER; Rosa26mTmG and Spc-CreER;Yap-cKO; Rosa26mTmG mice. Tamoxifen was dissolved in corn oil with a storage concentration of 50 mg/mL and an injection dose of 200 mg/kg in mice.
Results
YAP accumulated in the nucleus in response to lung injury
To explore the role of YAP in IPF, the expression of YAP in normal and pathological tissues of both humans and mice was analyzed. Analysis of previously published single-cell RNA-Seq (scRNA-Seq) datasets curated in the database IPF cell atlas (https://p2med.shinyapps.io/IPFCellAtlas) showed that the expression of YAP is increased in both AEC2 cells and fibroblasts in human IPF samples, with a particularly prominent increase observed in AEC2 cells (Fig. 1A). 19

YAP is activated (nuclear localized) in response to lung injury. (
YAP enters the cell nucleus upon activation. 10,11 Immunohistochemistry analysis revealed enhanced nuclear localization of YAP in the lungs of human IPF patients compared to normal lungs (Fig. 1B). Similarly, nuclear YAP was increased in the fibrotic region of bleomycin-induced mouse lungs compared to control (Fig. 1C and D). These results demonstrated YAP activation in fibrotic lungs in both patients and mice. Sun et al. has demonstrated the ability of TAZ to facilitate AEC2 regeneration, and presented data suggesting activation of TAZ, but inactivation of YAP, in alveolar organoid. We thus also probed their status in alveolar organoids and pulmonary fibrosis. YAP and TAZ were both activated in AEC2 in these two conditions (Fig. 1D–G), suggesting that YAP may have a similar role with TAZ in alveolar regeneration.
Yap deficiency in AEC2 exacerbated pulmonary fibrosis
To decipher the role of YAP in AEC2 in pulmonary fibrosis, Yap was conditionally deleted in AEC2 by crossing SftpcCreER;Rosa26mTmG mice to convert Yap floxed allele, generating SftpcCreER;Yap flox/flox;Rosa26mTmG (hereafter named Spc-CreER;Yap-cKO;mTmG) mice. Spc-CreER;Yap-cKO;mTmG and the control Spc-CreER;mTmG mice were injected with tamoxifen and 14 days later, were subjected to oropharyngeal injection of bleomycin to induce pulmonary fibrosis. At 14 days postinjury (dpi), the lungs were harvested for analysis (Fig. 2A). Immunohistochemistry indicated decreased YAP level in Spc-CreER;Yap-cKO;mTmG (Fig. 2B). YAP was further demonstrated to be successfully deleted in AEC2 by immunofluorescence (Fig. 2C). Compared to the control, Spc-CreER;Yap-cKO;mTmG exhibited increased alveolar damage, heightened collagen deposition, and augmented immune cell infiltration (Fig. 2D), demonstrating worsened fibrotic conditions in Spc-CreER;Yap-cKO;mTmG lungs.
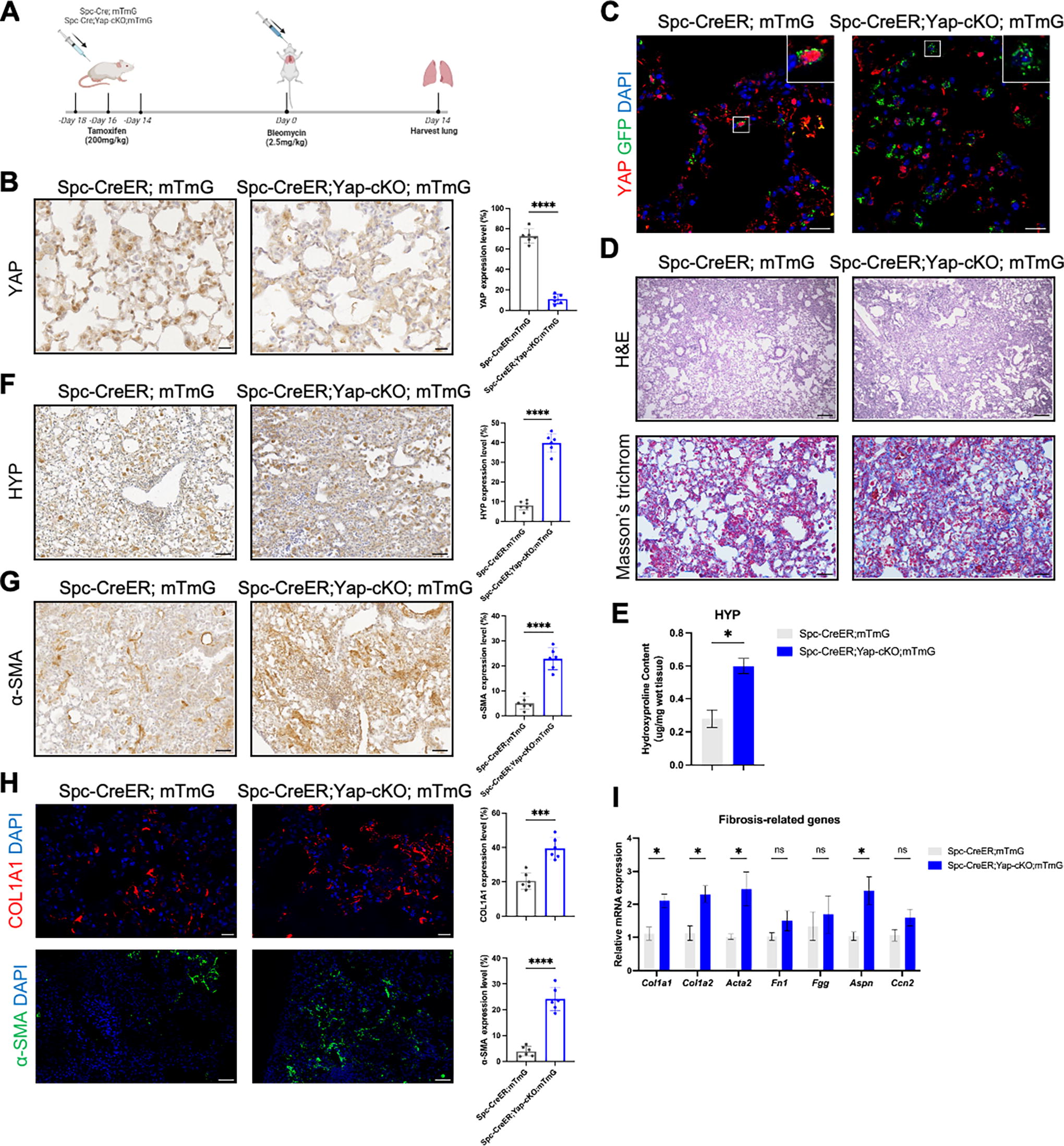
Yap deficiency in AEC2 exacerbated pulmonary fibrosis. (
Measuring hydroxyproline (HYP) by means of immunohistochemistry has been shown to be a more accurate method of assessing the degree of fibrosis than traditional biochemical methods. 20 We thus employed both immunohistochemistry and traditional biochemical methods to examine HYP. In line with the histological staining, results from both methods indicated elevated HYP content in Spc-CreER;Yap-cKO;mTmG lungs (Fig. 2E and F), demonstrating enhanced collagen deposition. The marker of myofibroblasts, α-SMA, was increased in the lungs of Spc-CreER;Yap-cKO;mTmG, suggesting elevated myofibroblast differentiation (Fig. 2G). Consistently, protein levels of collagen and α-SMA were increased in the mutant lungs (Fig. 2H). These results corroborate aggravated pulmonary fibrosis upon Yap deletion in AEC2. Consistent with histological results, fibrosis-related molecules including Col1a1, Col1a2, Acta2, and Aspn, were also upregulated upon Yap deficiency in AEC2 (Fig. 2I). In summary, these results indicated that Yap deficiency in AEC2 exacerbates pulmonary fibrosis in mice.
Knockout Yap in AEC2 inhibits its regeneration and differentiation
Next, we investigated the mechanism by which Yap deficiency in AEC2 exacerbates pulmonary fibrosis. Removal of both YAP and TAZ in AEC2 enhances the inflammation and impairs alveolar regeneration upon bacterial infection, 12 and TAZ deletion in AEC2 worsens bleomycin-induced fibrosis through suppressing alveolar regeneration. 14 We thus surmised that Yap deficiency exacerbated fibrosis through modulating AEC2 stemness and alveolar regeneration. Immunostaining of the proliferation marker EdU showed that proliferation was decreased in the AEC2 of the lungs of Spc-CreER;Yap-cKO;mTmG (Fig. 3A). To trace the differentiation of AEC2 into AEC1 upon injury, we utilized Spc-CreER;mTmG mice for lineage tracing. AEC1 differentiation was examined by immunofluorescence at 14 dpi and 28 dpi. Importantly, compared to control Spc-CreER;mTmG, much fewer GFP-traced AEC1s were observed in Spc-CreER;Yap-cKO;mTmG following bleomycin injection, indicating that differentiation from AEC2 to AEC1 was impeded in the mutant (Fig. 3B and Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/scd). Keratin 8 (KRT8) in alveoli marked a population of cells transitioning from AEC2 to AEC1, 21 and it is reported that KRT8 expression was associated by greater regenerative capacity of AEC2. 22 Upon Yap deficiency in AEC2, AEC2-traced KRT8+ cells were significantly reduced (Fig. 3C). Altogether, these findings demonstrated that the proliferation of AEC2 and AEC2 to AEC1 differentiation was impaired upon Yap deletion in AEC2. Therefore, YAP was essential for the stemness of AEC2 and alveolar regeneration.
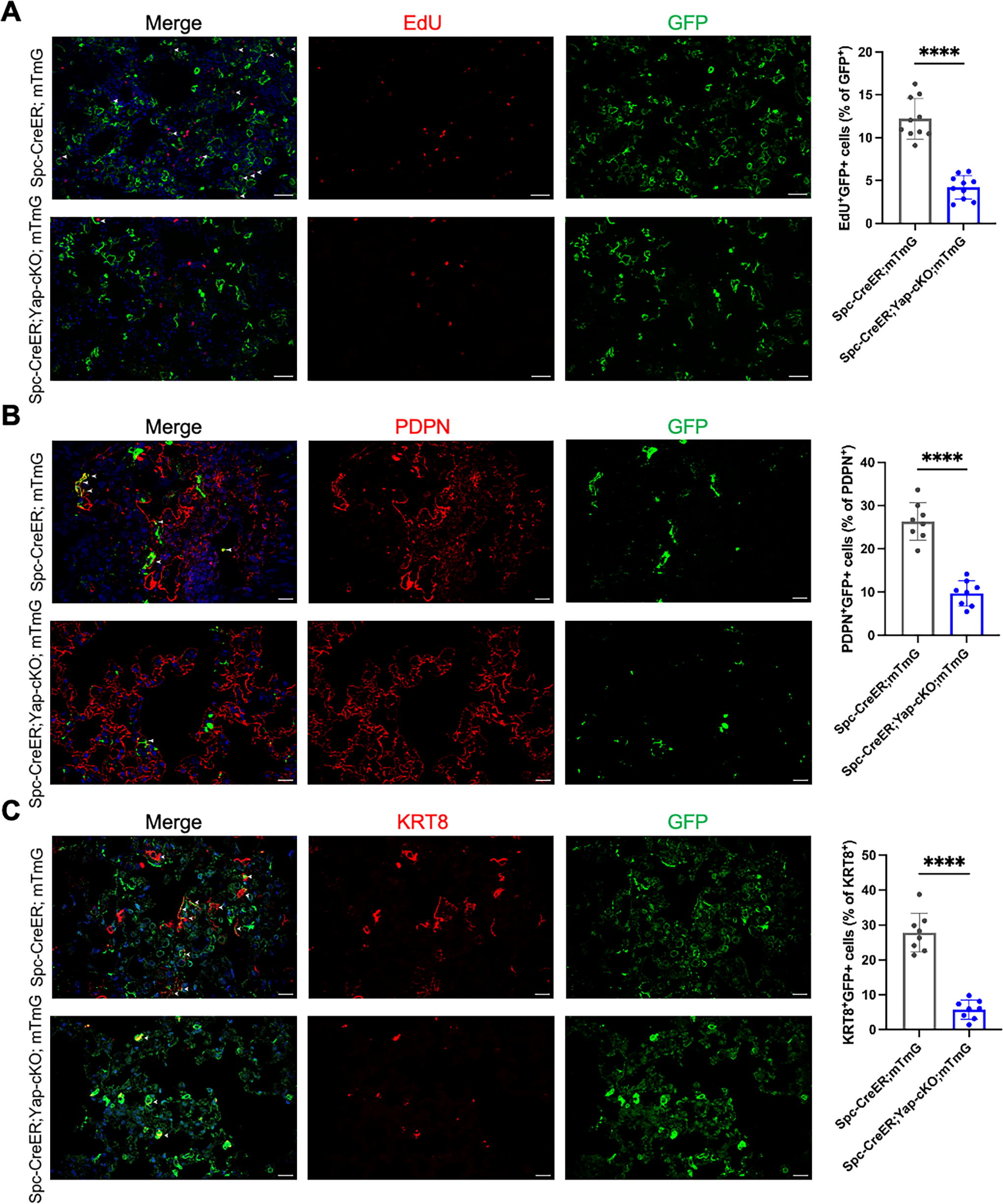
Yap deficiency in AEC2 impaired AEC2 stemness and alveolar regeneration. (
YAP controlled AEC2 stemness by modulating the regulators of cell proliferation and differentiation
We next investigated the mechanism via which YAP regulated alveolar regeneration. Proliferation markers, including Ccnd1, Ccne1, Ccng1, Cdk1, Ki67, and Top2a, were downregulated in Spc-CreER;Yap-cKO;mTmG mice compared to controls, indicating decreased cell proliferation in the mutant lung (Fig. 4A). To examine the regulation of YAP on AEC2 stemness directly, we derived alveolar organoids from primary AEC2 isolated from Spc-CreER;mTmG and Spc-CreER;Yap-cKO;mTmG mice. The alveolar organoids derived from the mutant AEC2 exhibited attenuated growth and decreased size, indicating impaired AEC2 stemness in the mutant (Fig. 4B–D). Moreover, genes associated with regulating cell cycle, AEC2 differentiation, and AEC1 signature were downregulated (Fig. 4E and F), consistent with the in vivo findings.
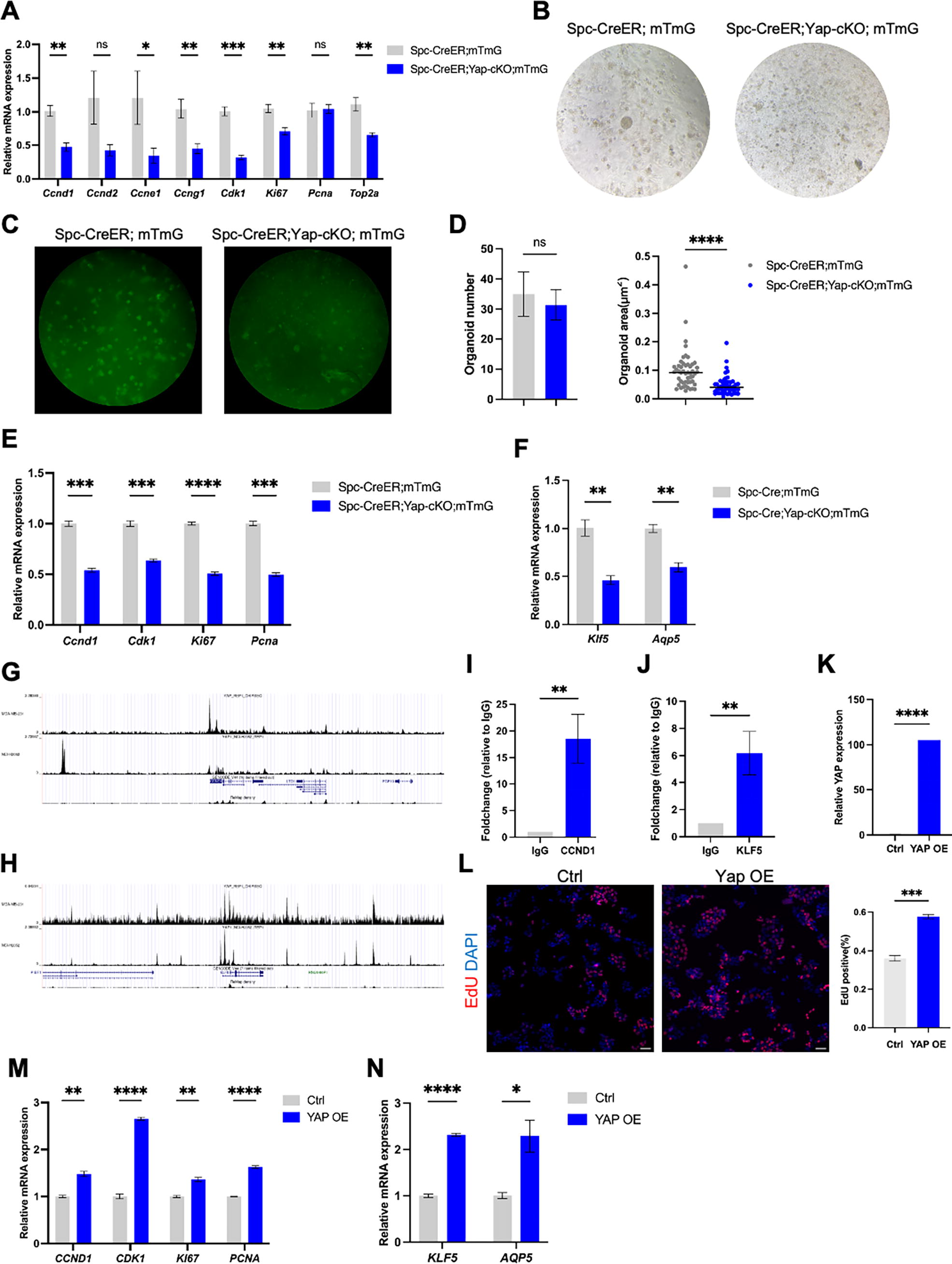
YAP stimulated cell proliferation thus prompted AEC2 stemness. (
Next, we asked whether YAP modulated the expressions of these genes through direct transcriptional regulation. CCND1 is a well-known regulator of cell cycle progression and KLF5 is a protein that regulates AEC1 differentiation. 23 Interestingly, our investigation revealed the presence of conserved TEA Domain Transcription Factor (TEAD) binding motifs within the promoter regions of Cyclin D1 (CCND1) and KLF Transcription Factor 5 (KLF5), situated approximately 2.5 kb and 2.8 kb upstream of their respective transcriptional start sites (Fig. 4G and H). Furthermore, bioinformatic interrogation of publicly available YAP chromatin immunoprecipitation (ChIP)-seq datasets yielded compelling evidence for YAP occupancy at the TEAD binding sites within the CCND1 and KLF5 promoters in NCI-H2052 and MDA-MB 231 cells (Fig. 4G and H). It was verified that the enrichment of YAP within the promoters of CCND1 and KLF5 in A549 cells through ChIP (Fig. 4I and J). Collectively, these observations suggest a direct role for YAP in the transcriptional regulation of genes critical for AEC2 proliferation and differentiation.
To buttress the hypothesis of YAP’s direct influence on AEC2 stemness, YAP was overexpressed in A549 cells, a model closely resembling human AEC2 cells. YAP overexpression promoted cell proliferation, indicated by increased EdU-positive cells, and elevated expression of CCND1, CCNG1, CDK1, KI67, and PCNA genes (Fig. 4K–M). Moreover, the expression levels of governing AEC2 differentiation and AEC1 signature genes, KLF5 and AQP5, were also upregulated upon YAP overexpression in A549 cells (Fig. 4N). These findings provide robust support for the notion that YAP acts as a key regulator of alveolar regeneration by orchestrating both the proliferation and differentiation of AEC2.
Discussion
The urgent need for novel pharmaceutical targets to address pulmonary fibrosis underscores the significance of elucidating the pathogenesis of this debilitating disease, which is intricately linked to the impaired repair capacity of AEC2. Previous investigations have elucidated the role of TAZ in promoting alveolar regeneration by regulating AEC2 stemness, while the contribution of YAP remained unclear. 14 However, our study sheds light on the regulatory role of YAP, alongside TAZ, in ameliorating bleomycin-induced fibrosis by directly modulating AEC2 stemness through the regulation of cell cycle progression and AEC1 signature genes, thereby fostering alveolar regeneration. This highlights the therapeutic potential of YAP activation within the alveolar epithelium as a promising avenue for treating pulmonary fibrosis.
However, the safety considerations surrounding YAP-based interventions for organ regeneration and tissue repair necessitate careful evaluation due to its dual role in disease pathogenesis. 24 Although YAP activation has been associated with promoting organ regeneration and tissue repair, 25,26 its overactivation has been implicated in the progression of malignant diseases, including tumorigenesis 27 and organ fibrosis. 28 For instance, overactivation of YAP in mice leads to liver cancers including hepatocellular carcinoma and cholangiocarcinoma. 29 To mitigate these potential adverse sequelae, YAP overexpression should be meticulously controlled to achieve a modest level of activation. This can be accomplished by utilizing a weak promoter such as Sftpc promoter, instead of strong ones like the CMV promoter. On the other hand, while its activation in the epithelium promotes regeneration and thus alleviates fibrosis, YAP expression in lung mesenchyme exacerbates pulmonary fibrosis, 30 presenting a significant challenge in the context of selective YAP modulation in AEC2 cells and concurrent inhibition in fibroblasts.
Addressing this challenge requires innovative strategies, such as targeted YAP overexpression specifically within AEC2 cells utilizing adeno-associated viruses (AAV). Recently, AAV technology has been employed for targeted therapy by modulating gene expression in specific cell types. 31,32 It has been reported that overexpression of YAP in the heart using AAV9 stimulates cardiac regeneration in adult mice following myocardial infarction. 33 Moreover, Konkimalla et al. have elucidated specific expression of genes in alveolar epithelium via AAV in the lung. 34 This approach holds promise in achieving selective YAP activation while minimizing adverse effects on fibroblast-mediated fibrotic processes, thus offering a potential solution to the complexities associated with YAP modulation in pulmonary fibrosis.
In summary, our investigation into the role of YAP in alveolar regeneration and its implications for IPF pathogenesis underscores the therapeutic potential of YAP activation within AEC2 cells. Our findings contribute to the development of novel therapeutic strategies aimed at addressing the unmet clinical needs of patients with IPF. Further research efforts are warranted to elucidate the intricacies of YAP-mediated signaling pathways and refine targeted treatment approaches for this debilitating disease.
Footnotes
Acknowledgments
The authors would like to thank Qianting Yang and Dr. Zheng Zhang for providing the human pulmonary fibrosis sample, and Dr. Pao-Tien Chuang for providing the Yap-flox mice.
Ethics Approval
The study involving human samples was approved by the Medical Ethics Committee of the Sun Yat-sen University under protocol number 10-2021 and informed written consent was obtained from all the participants. All the animal studies were approved by the Experimental Animal Ethics Committee of Sun Yat-sen University under protocol number SYSU-IACUC-MED-2021-B0076.
Availability of Data and Material
The data that support the findings of this study are available from the corresponding author Chuwen Lin upon request.
Author Disclosure Statement
The authors have no conflicts of interest to declare.
Funding Information
This work was funded by the Natural Science Foundation of Guangdong (grant #2024A1515011087), Shenzhen Science and Technology Innovation Committee grants (
Supplementary Material
Supplementary Data S1
Supplementary Figure S1
Supplementary Table S1
Supplementary Table S2