
Abstract
Abstract
Background:
Secretory immunoglobulin A (sIgA) increases in the airways of humans and mice after injury to protect against infection. The pro-inflammatory cytokines tumor necrosis factor (TNF)-α, interleukin (IL)-1β, and IL-6 are linked molecularly to sIgA production and secretion and are required for sIgA increases in the airway after injury in a mouse model. We investigated the injury effect on airway and serum concentrations to determine the source of the cytokines involved in the airway IgA response.
Methods:
In the first experiment, TNF-α, IL-1β, and IL-6 concentrations in bronchoalveolar lavage (BAL) fluid and serum obtained from 11 ventilated trauma patients within 30 h of admission were compared with those in eight elective surgical patients. In the second experiment, male ICR mice received no injury (n = 7) or injury with sham celiotomy and neck incisions (n = 8) with sacrifice of all animals at 8 h for BAL fluid and serum cytokine measurements by enzyme-linked immunosorbent assay.
Results:
Injured patients had significantly higher BAL fluid and serum TNF-α, IL-1β, and IL-6 concentrations, with greater increases in the BAL fluid than in the serum. Injured mice had significantly increased BAL fluid concentrations of TNF-α, IL-1β, and IL-6 without significant changes in serum TNF-α or IL-1β. Serum IL-6 increased significantly.
Conclusions:
Injury significantly increases human and mouse airway TNF-α, IL-1β, and IL-6. Increases are greater in the airway than in serum, implying a local rather than a systemic stress response to injury.
An important first immunologic defense against pneumonia occurs at the mucosal border within the lung airways [6]. Although multiple components defend this border, the strategic molecule of mucosal immunity is secretory immunoglobulin A (sIgA) [7]. This protein binds pathogens at the mucosal border and prevents their attachment to the mucosa and tissue invasion, thereby protecting the host from pneumonia [8, 9].
Recently, we observed an acute increase in bronchoalveolar lavage (BAL) fluid concentrations of sIgA in intubated trauma patients within 30 h of injury [10]. This airway response appears to constitute an innate pulmonary defense mechanism, as low sIgA concentrations increase bacterial adherence and the risk of pneumonia in intubated patients [11]. We also showed that this airway sIgA response occurs in a mouse model of controlled injury, with peaks in airway sIgA at 8 h after injury and return to baseline by 24 h [10]. We subsequently studied several potential mechanisms involved in this innate airway sIgA increase in our mouse injury model.
Tumor necrosis factor (TNF)-α, interleukin-1 (IL)-1β, and IL-6 are three commonly studied pro-inflammatory cytokines that increase shortly after injury [12, 13]. Several investigators showed that pro-inflammatory cytokine concentrations increase in BAL specimens and correlate with both the risk of adult respiratory dysfunction syndrome (ARDS) and its pathogenesis after trauma [14–17]. These pro-inflammatory cytokines also are likely to be involved in the protective innate sIgA increase after injury. Both TNF-α and IL-1β increase polymeric immunoglobulin receptor (pIgR) in vitro and in vivo [18–20]. This receptor specifically transports IgA across the epithelium via transcytosis after dimeric IgA, produced by plasma cells, binds to the pIgR molecule expressed on the basolateral surface of the epithelium. Cleavage of this molecule on the luminal side of the epithelium releases sIgA into the airway [21]. Interleukin-6 causes terminal differentiation of B cells to IgA-secreting plasma cells [22, 23]. We recently showed in our murine injury model that blockade of either TNF-α or IL-1β effectively eliminates (TNF-α) or reduces (IL-1β) the innate increase in IgA after injury [24]. Not surprisingly, systemic injection of TNF-α, IL-1β, and IL-6 into mice together (but not alone) reproduced this response without any other injury [25].
Although these inflammatory cytokines clearly play some role in the airway sIgA response to injury, it remained unclear whether systemic factors, local pulmonary factors, or both controlled the sIgA response in the mouse model. It also remained unclear whether similar patterns of inflammatory cytokines occur in humans after trauma, which prompted us to reexamine the serum and BAL response of these cytokines in the samples obtained from the severely injured patients in our published study [10] and compare the results with new data acquired using our murine injury model. We sought to determine if the airway response was a localized reaction or driven by a systemic response to injury. Additionally, we examined whether the murine injury model correlated with the human clinical response. We hypothesized that although the lung responds to systemic signals, the innate sIgA response remained a local reaction in both mice and human beings. We also hypothesized that the murine injury response mimicked and accurately reflected the human response. This would provide additional evidence that the murine model reliably defines the mechanisms involved in this human immunologic injury response.
Patients, Materials, and Methods
The University of Wisconsin-Madison Human Subjects Institutional Review Board granted approval for the clinical projects. The Animal Care and Use Committee of the University of Wisconsin–Madison and Middleton Veterans Administration Hospital, Madison, approved all animal experimental protocols.
Human Protocol
Informed consent was obtained for patients ≥18 years of age who were admitted from June 2004 to June 2007 to the Trauma Intensive Care Unit and were expected to be intubated and survive for a minimum of four to five days. Bronchoalveolar lavage for sample collection was performed within 30 h of admission. Patients were sedated with propofol (Diprivan®; AstraZeneca, Wilmington, DE) or midazolam (Versed®; Bedford Labs, Bedford, OH) and paralyzed with cisatracurium (Nimbex®, Abbott Laboratories Chicago, IL) during sample collection.
As a negative control, some patients undergoing elective outpatient surgery from July 2006 to May 2007 who required general anesthesia and intubation granted consent for study participation. Exclusion criteria were: (1) Age <18 years; (2) pregnant women, prisoners, and institutionalized or mentally impaired individuals; (3) patients with a history of asthma or documented airway irritability; (4) patients with a history of pulmonary resection; and (5) patients receiving cardiac medications for dysrhythmia.
Lavage Sample Collection
A flexible fiberoptic bronchoscope was advanced into the bronchus intermedius on the right and just distal to the upper lobe take-off on the left. Sixty milliliters of sterile saline was injected followed by aspiration into a sterile trap within 5 sec. An attempt was made to collect a minimum of 10 mL per sample. All samples were taken directly to the laboratory for analysis. Samples were transferred to sterile tubes and centrifuged at 3,000 rpm for 10 min at 4°C to remove mucus and debris. The supernatant liquid was decanted into another sterile tube and stored at 4°C until analyzed.
Serum Collection
One 3-mL sample of venous blood was collected at the time of each BAL and spun at 3,000 rpm for 10 min at 4°C. The serum was retained for measurement of urea nitrogen and selected cytokines.
Determination of human epithelial lining fluid volume
The volume of epithelial lung fluid (ELF) recovered was determined using published techniques predicated on free urea diffusion into the lung. The amount of urea in the BAL samples was determined by the urease method (Sigma-Aldrich, St. Louis, MO). Briefly, 50 microliters of BAL sample was incubated with 0.5 mL of urease reagent for 20 min at room temperature, followed by the addition of 1 mL each of phenol nitroprusside and sodium hypochlorite. The reaction was read immediately at 570 nm, and the amount of urea was calculated from a standard curve that is linear from 3 to 75 mg/dL. The plasma urea concentration was obtained using the same method except that the sample volume was 10 microliters. The volume of the ELF was calculated as follows:
Quantitative analysis of TNF-α, IL-1β, and IL-6
Concentrations of TNF-α, IL-1β, and IL-6 in BAL fluid and serum samples were determined in picograms/milliliter using a sandwich enzyme-linked immunosorbent assay (ELISA) for each of the cytokines (R&D Systems, Minneapolis, MN). Briefly, separate 96-well plates were coated with capture antibody diluted in phosphate-buffered saline (PBS) at 4.0 micrograms/mL for TNF-α and IL-1β and 2.0 mcg/mL for IL-6. Plates were incubated overnight at 4°C, then washed three times and blocked with Reagent Diluent (1% bovine serum albumin [BSA] in PBS) 300 microliters/well and incubated at room temperature for 1 h (R&D Systems). After the plates had been washed three times, sample or standard (R&D Systems) 100 microliters/well was added, and the plates were incubated for 1 h at room temperature. Plates were washed three times, and the respective detection antibody in Reagent Diluent (TNF-α 250 ng/mL, IL-1β 300 ng/mL, IL-6 200 ng/mL) 100 microliters/well was added and incubated 1 h at room temperature. After the plates were washed three times, streptavidin-horseradish peroxidase diluted in RD 1:200 was added at 100 microliters/well, and the specimen was incubated for 20 min at room temperature in the dark. Plates were again washed three times, and substrate solution (1:1 hydrogen peroxide and tetramethylbenzidine) 100 microliters/well was added, after which the plates were incubated for 20 min at room temperature in the dark. The reaction was stopped by adding 2N H2SO4 50 microliters/well, and absorbance was read at 450 nm in a Vmax Kinetic Microplate Reader (Molecular Devices, Sunnyvale, CA). The mass amounts of TNF-α, IL-1β, or IL-6 were calculated by plotting their absorbance values on their respective standard curves, which were calculated using a four-parameter logistic fit with SOFTmax PRO software (Molecular Devices).
Animal Protocol
Male five-to-seven-week-old Institute of Cancer Research mice were purchased from Harlan (Indianapolis, IN) and housed in the Animal Research Facility of the William S. Middleton Memorial Veterans Hospital, an American Association for Accreditation of Laboratory Animal Care-approved conventional facility. Mice were allowed to acclimate for one week with free access to standard chow (PMI Nutritional International, St. Louis, MO) and water under controlled conditions of temperature and humidity with a 12:12 hour light:dark cycle.
Injury
Animals were anesthetized with an intraperitoneal ketamine (100 mg/kg) and acepromazine (5 mg/kg) mixture. The skin was disinfected using 75% ethanol, and two incisions were created. First, a 3.0-cm celiotomy incision was made, and the small intestine was gently eviscerated and returned immediately to the peritoneal cavity. The incision was closed in two layers with three simple interrupted 4-0 silk sutures per layer. Second, a 1.5-cm ventral neck incision was made and blunt dissection carried down to the pretracheal plane. This incision was closed with a single layer of two simple interrupted 4-0 silk sutures. Animals were allowed to recover and were then sacrificed 8 h (n = 8) after injury by exsanguination from a left axillary artery transection. Prior to sacrifice, awake animals received additional anesthesia (as much as half of the original dose) until the righting reflex was lost. Another group of animals was sacrificed without injury to serve as controls (n = 8).
Blood was collected from the left axillary artery transection site for the serum sample. For the BAL specimen, a tracheotomy was created, and 1 mL of normal saline was injected with an 18-gauge catheter distally and aspirated.
Quantitative analysis of TNF-α, IL-1β, and IL-6
Concentrations of TNF-α, IL-1β, and IL-6 were measured in pg/mL in BAL fluid and serum using solid-phase sandwich enzyme-linked imunosorbent assays (ELISAs) (BD Biosciences, Bedford, MA). Briefly, separate 96-well plates were coated with anti-mouse TNF-α, IL-1β, or IL-6 in a 1:250 dilution in 0.1 M sodium carbonate coating buffer (pH 9.5) 100 microliters/well and incubated overnight at 4°C. Plates were washed three times and blocked with 200 microliters of PBS with 10% fetal bovine serum (FBS) for 1 h at room temperature. One hundred microliters of BAL fluid, serum, or cytokine standard (BD Biosciences) was added, and the plates were incubated for 2 h at room temperature. The diluent was PBS with 10% FBS. Plates were washed five times, and 100 microliters of a 1:250 dilution of the secondary antibody, either biotinylated anti-mouse TNF-α or IL-1β, was added, after which the plates were incubated 1 h at room temperature. After the plates had been washed five times, streptavidin-horseradish peroxidase conjugate was added, and the mixture was incubated 30 min at room temperature. For IL-6, a 1:250 dilution of the secondary antibody was used; however, this was mixed with the streptavidin-horseradish peroxidase, done in one step, and allowed to incubate for 1 h. Plates were then washed seven times, and 100 microliters of the substrate solution (tetramethylbenzidine and hydrogen peroxide) was added; the mixture was then incubated for 30 min at room temperature in the dark. The reaction was stopped by adding 50 microliters of 2N H2SO4, and the absorbance was read at 450 nm in a Vmax Kinetic Microplate Reader (Molecular Devices). The mass amounts of TNF-α, IL-1β, or IL-6 were calculated by plotting their absorbances on their respective standard curves, which were calculated using a four-parameter logistic fit with SOFTmax PRO.
Statistical analysis
The Shapiro-Wilk test was performed on all data sets from the human protocol to determine if the data were normally distributed. Comparison of TNF-α, IL-1β, and IL-6 concentrations (pg/mL of ELF) between the trauma samples and control patient samples for BAL fluid and serum as well as BAL fluid compared with serum were performed via the Wilcoxon-Mann-Whitney test with α = 0.05. Values are presented as means ± standard error of the mean.
The Shapiro-Wilk test was performed on all data sets from the animal protocol to determine if there was normal distribution. The TNF-α, IL-1β, and IL-6 data from each treatment group were compared using the Student t-test with α = 0.05. Numerical results are presented as means ± standard error of the mean.
Results
Human Protocol
Table 1 shows the demographics, mechanisms of injuries, operations performed during the first week, and smoking histories for the 11 trauma patients. Table 2 depicts this information for the eight elective surgery (control) patients. There were no significant differences between the two groups with regard to age or sex. Five of the trauma patients had a smoking history, whereas only one elective surgery patient had a smoking history.
B = bilateral; DAI = diffuse axonal injury; EDH = epidural hematoma; ex-fix = external fixation; ex-lap = exploratory laparotomy; fx = fracture; hemo = hemorrhagic; ICP = intracranial pressure; IPH = intraparenchymal hemorrhage; IVCF = inferior vena cava filter; ISS = Injury Severity Score; J tube = jejunostomy feeding tube; lac = laceration; lap = laparotomy; MCC = motorcycle crash; MVC = motor vehicle crash; ORIF = open reduction/internal fixation; PEG = percutaneous endoscopic gastrostomy; PTX = pneumothorax; pulm = pulmonary; SAH = subarachnoid hemorrhage; SB = small bowel; SDH = subdural hemorrhage; T bone = temporal bone; TibFib = tibia and fibula.
IVC = inferior vena cava.
Compared with the control patients, trauma patients had significantly higher BAL fluid concentrations of TNF-α (670.8 ± 280 pg/mL vs. 21.3 ± 5.7 pg/mL; p < 0.006), IL-1β (3,634.0 ± 1,092.6 pg/mL vs. 21.6 ± 9.6 pg/mL; p < 0.001), and IL-6 (5,449.8 ± 1,266.8 pg/mL vs. 829.4 ± 617.6 pg/mL; p < 0.001) (Fig. 1A). Trauma patients also showed significantly higher serum concentrations of all three cytokines: TNF-α (108.2 ± 27.1 pg/mL vs. 16.6 ± 13.3 pg/mL; p < 0.003), IL-1β (48.0 ± 14.6 pg/mL vs. 5.3 ± 4.1 pg/mL; p < 0.004), and IL-6 (260.0 ± 57.3 pg/mL vs. 2.0 ± 0.8 pg/mL; p < 0.001) (Fig. 1B). Control patients had significantly higher BAL fluid than serum concentrations of IL-1β (p < 0.01) and IL-6 (p < 0.01), with no significant difference between the TNF-α concentrations (p = 0.20). The trauma patients also had significantly higher concentrations of IL-1β (p < 0.01) and IL-6 (p < 0.01) in BAL fluid than in serum, whereas TNF-α showed a trend toward a higher concentration in BAL fluid than in serum (p = 0.12). However, the quantitative increase in BAL concentrations of all three cytokines was much greater than in the serum. The human data were not normally distributed according to the Shapiro-Wilk test.

Human cytokine response to trauma. (
Animal Protocol
Injury significantly increased BAL fluid concentrations of TNF-α (104.1 ± 15.8 pg/mL vs. 30.5 ± 6.8 pg/mL; p < 0.002), IL-1β (261.4 ± 39.0 pg/mL vs. 85.5 ± 15.8 pg/mL; p < 0.003), and IL-6 (145.4 ± 25.3 pg/mL vs. 43.3 ± 7.9 pg/mL; p < 0.005) (Fig. 2A). Injury also significantly increased the serum concentrations of IL-6 (738.2 ± 155.2 pg/mL vs. 0.0 ± 0.0 pg/mL; p = 0.0001) whereas TNF-α did not (Fig 2B). Serum IL-1β was not detectable in any animals.
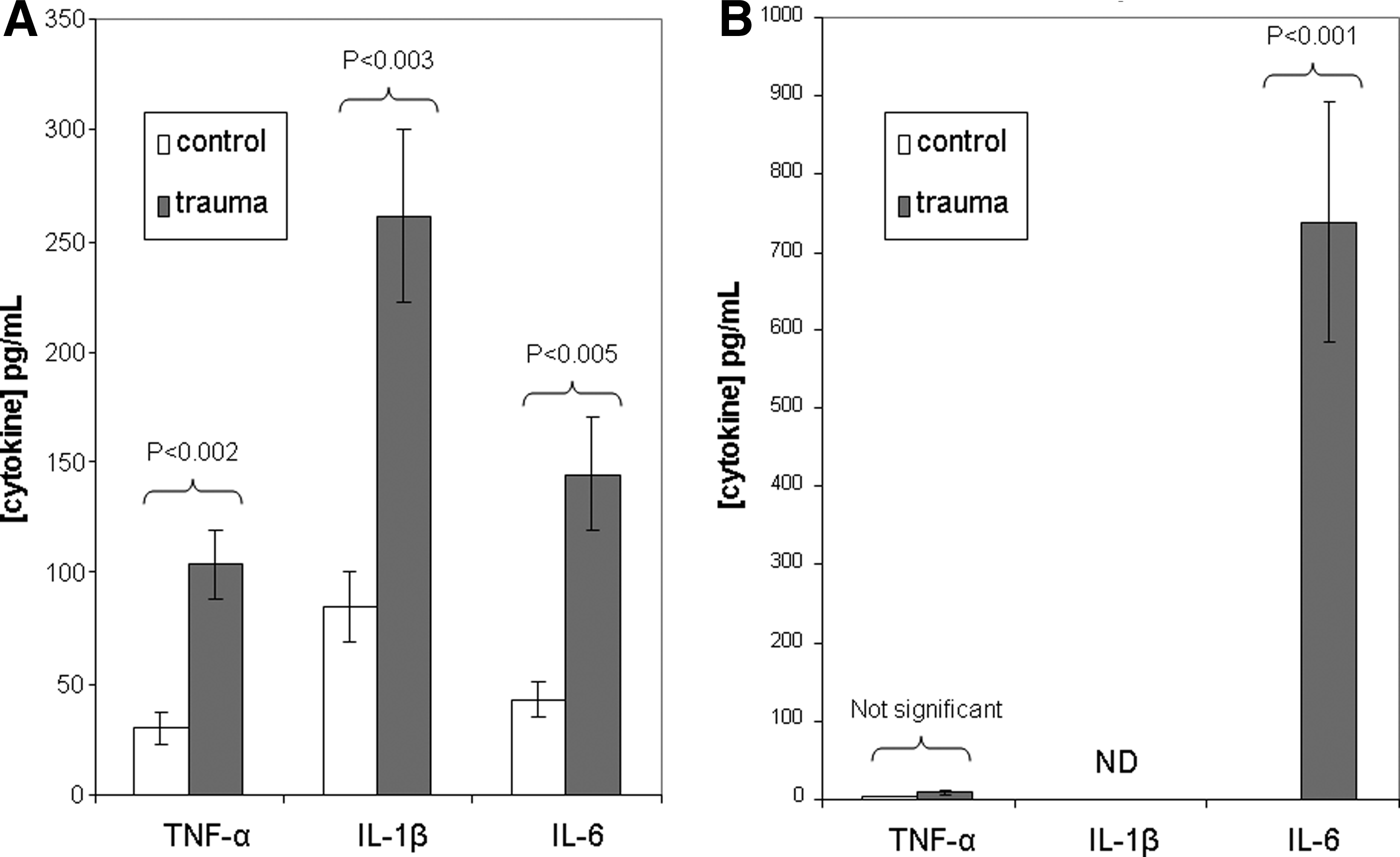
Murine cytokine response to trauma. (
The concentrations of TNF-α and IL-6 in BAL fluid were significantly greater than serum concentrations in uninjured controls (p < 0.01 for both). These differences remained in the injured animals (p < 0.01 for both); however, BAL fluid concentrations of TNF-α and IL-1β increased significantly compared with control animals, whereas serum concentrations did not. However, serum concentrations of IL-6 increased significantly after injury and significantly exceeded the BAL fluid concentrations. The mouse data were normally distributed according to the Shapiro-Wilk test.
Discussion
Airway sIgA concentrations increase in both humans and mice after injury. This response presumably represents a mechanism to protect the lung from early infection during stress, as sIgA normally binds bacteria to prevent their attachment to the epithelium and invasion of tissue. The multi-factorial response involves IgA production in the lung parenchyma and transport into the lumen. This work focuses on these factors and strongly implies a local pulmonary, cytokine-driven mechanism. It provides additional evidence that data obtained from the mouse model of injury are relevant to the human injury response.
On a molecular level, TNF-α, IL-1β, and IL-6 affect both the production and the transport of IgA across the epithelial border. Interleukin-6 causes terminal differentiation of B-cells to IgA-secreting plasma cells at mucosal sites [22, 23]. In vitro experiments show that TNF-α and IL-1β stimulate pIgR transcription via the nuclear factor-kappa B (NF-κB) pathway [18, 19, 26]. The pIgR, expressed on the basolateral surface of the epithelium in the lung, binds IgA and transports it to the apical side, where enzymatic cleavage releases IgA into the airway lumen along with a remnant secretory component, resulting in sIgA [21]. Therefore, transport of IgA consumes pIgR in a 1:1 ratio [27]. We showed previously that lung concentrations of pIgR and lung tissue concentrations of IgA in mice remain constant after injury despite increases in airway sIgA [28]. This indicates upregulation of pIgR production after injury and a potential role for TNF-α, IL-1β, and IL-6 in the airway IgA increase after injury.
Trauma patients have significantly higher airway concentrations of sIgA within 30 h of injury, a response that was reproducible in our mouse injury model [10]. Experimentally, we demonstrated a critical role of TNF-α, IL-1β, and IL-6 in this murine response, as TNF-α blockade eliminates the airway sIgA response, whereas blockade of IL-1β reduces it [24]. In addition, intraperitoneal injection of recombinant TNF-α, IL-1β, and IL-6 together (but not separately) reproduces the airway IgA increase [25]. Because intraperitoneal cytokine administration generates a systemic response and because the pro-inflammatory cytokines increase systemically after injury, it remained unclear whether the normal in vivo airway sIgA response to injury resulted from systemic or local factors, prompting reevaluation of our trauma patient data and the conduct of new experiments in the murine model. Our data imply a localized rather than a systemic response to generate the airway sIgA increase in both humans and mice. In humans, injury increases both serum and BAL fluid concentrations of TNF-α, IL-1β, and IL-6 compared with control patients, but concentrations of these cytokines in BAL fluid specimens from injured patients had increased more than the concentrations in serum samples drawn simultaneously. This result implies that the increases in BAL fluid after trauma could not occur simply from lung permeability changes and leakage of serum cytokines into the airway. Because BAL fluid concentrations of TNF-α, IL-1β, and IL-6 increased to a greater extent than those in the serum, we believe this reflects a localized airway response. It does not eliminate the possibility of active transport of these cytokines from the serum to the airway following injury; however, we believe this to be unlikely.
Similar to the human data, murine injury increased BAL fluid concentrations of TNF-α, IL-1β, and IL-6 compared with uninjured controls. However, the magnitude of the increase in BAL fluid cytokines relative to serum concentrations was blunted compared with our human trauma population, where these concentrations increased dramatically compared with control patients. This may be attributable to the limited nature of the injury in the murine model compared with the severe injuries sustained in our trauma population, as recruitment was limited to patients expected to be intubated for at least five days, and the Injury Severity Scores were high. Similar to the human response, injury significantly increased the serum IL-6 concentrations in the mouse, with a trend toward increased serum TNF-α. However, IL-1β was not detectable in the serum of the control or injury mice. Similar to the human beings, uninjured mice had significantly higher pre-injury concentrations of TNF-α, IL-1β, and IL-6 in BAL fluid compared with serum. Following injury, BAL fluid concentrations of TNF-α and IL-1β increased much more than the serum concentrations, just as in the trauma patients. A single difference between the human and mouse response to injury was seen with IL-6, as the concentration increased significantly more in mouse serum than in the BAL fluid. Overall, however, the mouse response to injury mimics the human response, with significant increases in BAL fluid concentrations of TNF-α and IL-1β that exceed serum increases, IL-6 being the lone exception.
Although we have demonstrated an important role for TNF-α, IL-1β, and IL-6 in the protective airway sIgA response to injury, substantial evidence links these pro-inflammatory cytokines to pulmonary pathology. The increases in TNF-α, IL-1β, and IL-6 in the serum after injury or trauma are associated with an acute systemic reaction resulting in hemodynamic changes, muscle catabolism, fever, migration of leukocytes into a variety of tissues, and increased production of acute-phase proteins by the liver [12, 13, 29, 30]. Clinically, an exaggerated response of pro-inflammatory cytokines in the serum after trauma has been associated with SIRS, sepsis, and multiple organ dysfunction (MOD) [3, 31-33]. Concentrations of IL-6 in the serum after injury correlate with many of these outcomes and serve as a marker of systemic inflammation [34]. The degree of airway increase in some pro-inflammatory cytokines after injury has been correlated with the development of ARDS and pneumonia [15, 17, 35]. Keel et al. found that BAL fluid concentrations of IL-1β (and IL-8) increased significantly in trauma patients suffering significant chest trauma, and many of these patients went on to develop ARDS, whereas BAL fluid concentrations of TNF-α, IL-1β, and IL-6 correlated with the development of pneumonia [36]. Tumor necrosis factor-α and IL-1β also appear to stimulate a lung response to ischemia–reperfusion injury, which can occur with the hypotension that follows significant trauma [37].
Although our data imply that local pulmonary cytokine release maintains the sIgA response, it remains unknown what triggers the response initially. Systemic administration of TNF-α, IL-1β, and IL-6 together can precipitate the airway sIgA response in mice, suggesting that systemic release of these cytokines may be the trigger, as their blockade eliminates or reduces the response in mice [25]. Unfortunately, because of the delays inherent in the early treatment of trauma patients and in obtaining informed consent, early BAL fluid samples remain unavailable, and we depend on the animal models to provide those insights. To draw any conclusion, it is essential that models reflect the human response to the extent the changes can be studied in humans. Our work supports our belief that the mouse model reflects the human response and that part of the initial systemic inflammatory response after injury stimulates the local airway phenomenon.
Our clinical and experimental work supports the concept that airway sIgA plays an important role in preventing pneumonia. Injury and systemic inflammation stimulate airway sIgA increases and involve the pro-inflammatory cytokines TNF-α, IL-1β, and IL-6. The greatly elevated BAL fluid concentrations of these cytokines indicate that a localized pulmonary response rather than systemic changes likely maintains a prolonged sIgA response, whereas systemic cytokine release initiates the mucosal immune reaction to injury. There are strong similarities between the human and murine responses, and these similarities allow extrapolation of murine injury data to the human condition.
Footnotes
Acknowledgments
We sincerely thank Glen Leverson and Yin Wan of the University of Wisconsin Department of Surgery for support with statistical analysis and interpretation.
This research is supported by National Institute of Health Grant R01 GM53439. This material also is based on work supported in part by the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Service. The contents of this article do not represent the views of the Department of Veterans Affairs or the United States Government.
Author Disclosure Statement
The authors have no commercial associations or conflicts of interests associated with this work.
Presented at the American Society of Parenteral and Enteral Nutrition (A.S.P.E.N.)/Clinical Nutrition Week, New Orleans, Louisiana, February 1–4, 2009.