
Abstract
Abstract
Background:
The pathophysiologic features of acute respiratory distress syndrome (ARDS) are attributed to neutrophil accumulation and over-activation. Low blood immunoglobulin G concentrations in septic shock patients are associated with higher risk of developing ARDS. This study showed the effects of intravenous immunoglobulin (IVIg) on neutrophil apoptosis and accumulation in the lung during murine endotoxemia.
Methods:
Male C57BL/6J mice were injected with saline or 7 mg/kg of lipopolysaccharide (LPS), and 3 h later also were injected with saline, IVIg 300 mg/kg, or IVIg 1000 mg/kg intraperitoneally. At 12 h after LPS injection, mice were sacrificed and peripheral blood and lungs were collected. The lung messenger ribonucleic acid expression (tumor necrosis factor-α [TNF-α], inducible nitric oxide synthase [iNOS], and intercellular adhesion molecule-1 [ICAM-1]) was determined using quantitative realtime reverse transcriptase-polymerase chain reaction. Lungs were immersed in 4% paraformaldehyde and then embedded in paraffin. Tissue slices were prepared and stained with naphthol AS-D chloroacetate esterase to detect neutrophils. The numbers of neutrophils (characterized by the segment number of their nuclei) were counted. Peripheral neutrophil apoptosis was detected by annexin V using flow cytometry and lung neutrophil apoptosis was detected by cleaved caspase-3 using immunohistochemistry.
Results:
The survival rates of the saline group, LPS group, and IVIg group were all 100%. Apoptosis of peripheral blood neutrophils was inhibited by LPS. Neutrophil accumulation in the lung was decreased by both IVIg 300 mg/kg and 1000 mg/kg. Segmented neutrophils were reduced by IVIg during endotoxemia. However, IVIg 300 mg/kg and 1000 mg/kg had no influence on the lung messenger ribonucleic acid expression of TNF-α, iNOS, or ICAM-1. Cleaved-caspase-3-positive neutrophils were increased in the IVIg 300 mg/kg group during endotoxemia. The 1000 mg/kg IVIG dose reduced the number of segmented neutrophils, but did not induce cleaved-caspase 3-positive neutrophils.
Conclusion:
A therapeutic IVIg dose can attenuate neutrophil accumulation and regulate neutrophil apoptosis in the lung during endotoxemia. It is possible that the pathways by which IVIG induces neutrophil apoptosis may differ depending on the IVIg concentration.
S
Neutrophils are believed to contribute to the pathophysiologic features of ARDS, such as exudation of protein-rich fluid across the alveolocapillary endothelial cell barrier [2]. Several pro-inflammatory cytokines, chemotactic factors, and adhesion molecules released or expressed by macrophages or endothelial cells induce migration of neutrophils to sites of inflammation [3]. Although neutrophils function as an innate immune defense, activated neutrophils phagocytose bacteria by releasing toxic reactive oxygen species and granule enzymes to protect the host from bacterial invasion. However, an excessive pro-inflammatory response from neutrophils can cause catastrophic collateral damage to host tissues [3]. Therefore, timely apoptosis and removal of neutrophils are crucial for minimizing tissue injury by reducing inflammatory reactions derived from sustained activated neutrophils.
Intravenous immunoglobulin (IVIg) containing the pooled immunoglobulin G (IgG) isolated from plasma has been used as an efficient anti-inflammatory therapeutic regimen in a growing number of autoimmune diseases [4]. Intravenous immunoglobulin enables maintenance of adequate antibody concentrations to prevent infections and confers passive immunity. Intravenous immunoglobulin is administrated often to immune-deficient patients who have decreased or abolished antibody production capabilities. Although IVIg's mechanisms of action are not elucidated fully, it is known that IVIg blocks the Fc-receptor, inhibits pro-inflammatory cytokine production [5], and participates in antigen-antibody reactions in sepsis [6]. A recent clinical study showed that septic shock patients with low-IgG concentrations have a higher tendency to develop ARDS associated with higher mortality [7]. Hagiwara et al. reported that administration of high-dose (1,000 mg/kg) IVIg reduced mortality and improved pulmonary pathology in a rat sepsis model via reduction of high mobility group box protein 1 (HMGB1) [8]. Takeshita et al. reported that IVIg induced LPS-stimulated neutrophil apoptosis in vitro [9]. Thus, clinical/experimental studies reveal that IVIg may be a useful adjuvant anti-inflammatory treatment during sepsis, as it modulates inflammation. Accordingly, we hypothesized that IVIg regulates equipped neutrophil apoptosis and attenuates the accumulation of apoptosis-delayed neutrophils in the lung, possibly leading to inhibition of ARDS during sepsis. To test this hypothesis, we investigated whether IVIg can ameliorate endotoxin-induced ARDS and reduce neutrophil accumulation. Also, we elucidated the involved mechanisms of IVIg's anti-inflammatory effects by focusing on the regulation of neutrophil apoptosis in the lung during systemic inflammation in mice.
Materials and Methods
Reagents
Lipopolysaccharide (LPS: Escherichia coli O111:B4) and bovine serum albumin (BSA) were purchased from Sigma (St. Louis, MO). Intravenous immunoglobulin (IVIg) was supplied by Japan Blood Products Organization (Osaka, Japan).
Mouse acute endotoxemia model
Male seven- to nine-week-old C57BL/6Jcl (wild type; WT) mice were purchased from CLEA Japan (Tokyo, Japan). All animal experiments were conducted in accordance with the Institutional Guidelines for Animal Experimentation and Animal Care at Kobe University. Diethyl ether was used for inhalation anesthesia in all animal experiments. Mice were injected with 7 mg/kg body weight of LPS or saline intraperitoneally (i.p.), followed by saline, or 300 or 1,000 mg/kg IVIg (1 mL of volume) via i.p. route. To avoid the antigen-antibody reaction of IVIg with LPS, IVIg was injected three hours after LPS injection. The survival rate was determined for 24 h after LPS injection using 10 mice in each group. The other mice (six mice in each group, except for the LPS+IVIg 300 mg/kg group [n=7]) were sacrificed by cardiac puncture 12 h after LPS injection and their lungs were collected. The left lower lobes were fixed with 4% paraformaldehyde for histopathologic examination. The right lower lobes were soaked in TRIzol (Invitrogen, Carlsbad, CA), snap frozen, and stored at −80°C until molecular analysis.
Lung histology
For histopathologic examination, the trachea was cannulated with an indwelling needle, then 0.5 mL of 4% paraformaldehyde in phosphate buffered saline (PBS) was carefully instilled to fix alveolar morphology. Whole lungs were immersed in the 4% paraformaldehyde for 24 h, dehydrated through graded-alcohol dilutions, and then embedded in paraffin. Tissue slices (3 micron) were prepared and stained with naphthol AS-D chloroacetate esterase stain (Sigma). Described briefly, 1 mL solution A (Fast Red violet LB Base solution mixed with 4% sodium nitrite) and 2 mL solution B (naphtol AS-D chloroacetate in dimetilfolmaldehyde) were mixed with 100 mL PBS (pH7.4) at 37°C. The de-paraffinized section reacted with the solution for one hour at 37°C. The nuclei were stained by hematoxylin-eosin (Sigma). The numbers of positive cells (neutrophils) were counted in each of the three fields per mouse under a microscope using ×200 magnification.
Tissue slices (3 micron) were prepared and stained with hematoxylin-eosin (Sigma). The numbers of neutrophils (characterized by the segment number of their nuclei) were counted in each of the three fields per mouse under a microscope using×1,000 magnification.
Immunohistochemistry
Tissue slices (3 micron) were obtained and deparaffinized, hydrated, and processed. Described briefly, for antigen retrieval, slides were placed in 1 mmol/L EDTA buffer (pH 8.0) and heated in an autoclave for 30 min. Endogenous peroxidase was blocked by 10% hydrogen peroxide for 10 min. The sections were incubated overnight with anti-mouse cleaved caspase-3 antibody (rabbit polyclonal, 1:8000; Cell Signaling, Beverly, MA). The sections were then allowed to react with Histofine Simple Stain Mouse MAX PO(R) (Nichirei Bioscience, Tokyo, Japan) for 30 min. Finally, the reaction was revealed using 3,3′-diaminobenzidine tetrahydrochloride (DAB) (Dako Japan, Tokyo, Japan), counterstained with hematoxylin-eosin, and coverslipped with Entellan (Sigma).
Cells were counted under a microscope in a blinded manner. In fields showing a more intense reaction, all cells were counted (×400 magnification, five fields per each mouse, 200–300 cells per field) and the percentage of cleaved caspase-3-positive cells were calculated.
RNA extraction, cDNA synthesis, and RT-PCR
Total ribonucleic acid (RNA) was extracted from lungs with TRIzol reagent according to the manufacturer's instructions. The RNA concentration and purity were determined by absorbance at 260 and 280 nm. One mcg of total RNA extracted from each lung was reverse-transcribed to yield single stranded complementary deoxyribonucleic acid (cDNA) using iScript cDNA Synthesis Kits (Biorad Hercules, CA) according to the manufacturer's instructions. Quantitative real-time polymerase chain reaction (RT-PCR) to determine expression of messenger RNAs (mRNAs) for inflammatory molecules (tumor necrosis factor-alpha [TNF-α], inducible nitric oxide synthase [iNOS], inter-cellular adhesion molecule-1 [ICAM-1]) in the lungs was performed with MyiQ (Biorad). A total of 2 mcL of cDNA obtained by reverse transcription was used for amplification in a final volume of 25 mcL containing 12.5 mcL of 2×SYBR Green Real-time PCR Master Mix (TOYOBO, Osaka, Japan) and 200 nM of specific forward and reverse primers. Relative expression of the genes was calculated using the ddCt method after normalization to the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) level. The primer sequences and annealing temperatures are listed in Table 1.
ICAM-1=inter-cellular adhesion molecule 1; TNF-α=tumor necrosis factor-α; iNOS=inducible nitric oxide synthase; GAPDH=glyceraldehyde-3-phosphate dehydrogenase.
Cell staining and flow cytometric analysis
To prevent non-specific antibody binding, peripheral blood (1×106 cells per reaction) was incubated with CD16/32 antibody (1:50) diluted in PBS containing 1% bovine serum albumin (PBS-BSA buffer) for 5 min at 4°C. After rinsing with PBS-BSA buffer, the cells were incubated for 1 h at 4°C with FITC-conjugated anti-Gr-1 antibody (1:1,000) and PE-conjugated anti-B220 antibody (1:1,000) in PBS-BSA buffer. To detect apoptotic cells, the cells were incubated for 10 min at room temperature with APC-conjugated annexin V in 100 mcL of annexin V staining buffer (0.14 M NaCl and 2.5 mM CaCl2) and then analyzed by flow cytometry on a FACSCaliber (Becton Dickinson, San Jose, CA).
Statistical analysis
Data were expressed as mean±standard error (SE). All experiments were analyzed using the Tukey-Kramer post-hoc test and differences were considered significant at p<0.05.
Results
Mouse survival rates
The survival rates of the saline group, LPS group, and IVIg group were all 100% at 24 h after injection (data not shown).
IVIg-induced apoptosis of neutrophils in peripheral blood
To determine neutrophil apoptosis in peripheral blood, we performed flow cytometry using anti-Gr-1 (neutrophil) antibody and annexin V (apoptosis). The frequency of neutrophil apoptosis in the peripheral blood was found to be significantly lower in the LPS group than in the saline group (Fig. 1), indicating that neutrophil apoptosis was extremely inhibited by LPS. The IVIg group tended to induce neutrophil apoptosis in the peripheral blood compared to the LPS group (Fig. 1), but not significantly. The results indicated that LPS decreased circulating neutrophil apoptosis and may have induced the infiltration of neutrophils in distant organs, such as the lung.

Intravenous immunoglobulin-induced peripheral blood neutrophil apoptosis during endotoxemia. To evaluate neutrophil apoptosis in peripheral blood, annexin V-positive neutrophils were measured by flow cytometory. At 12 h after saline or lipopolysaccharide (LPS) injection, peripheral blood was collected. Lipopolysaccharide reduced neutrophils apoptosis significantly.
IVIg-attenuated neutrophil accumulation in the lung
Neutrophil accumulation was evaluated by naphthol AS-D chloroacetate esterase stain using paraffin sections. The number of neutrophils significantly increased after i.p. LPS (Fig. 2B and E). Intravenous immunoglobulin attenuated neutrophil accumulation significantly during endotoxemia (Fig. 2C, D, and E). To determine neutrophil age, we checked the nuclei form of accumulated neutrophils by hematoxylin and eosin stain using paraffin sections. Band neutrophils and segmented neutrophils with two to four nuclear lobes increased after i.p. LPS (Fig. 3). Intravenous immunoglobulin at either 300 or 1,000 mg/kg did not influence the number of band neutrophils. However, IVIg decreased the number of segmented neutrophils with two to four nuclear lobes significantly (Fig. 3).

Intravenous immunoglobulin (IVIg)-attenuated neutrophil accumulation in the lung during endotoxemia. To evaluate neutrophil accumulation in the lung, naphthol AS-D chloroacetate esterase staining was performed using lung paraffin sections. At 12 h after saline or lipopolysaccharide (LPS) injection, lungs were collected.
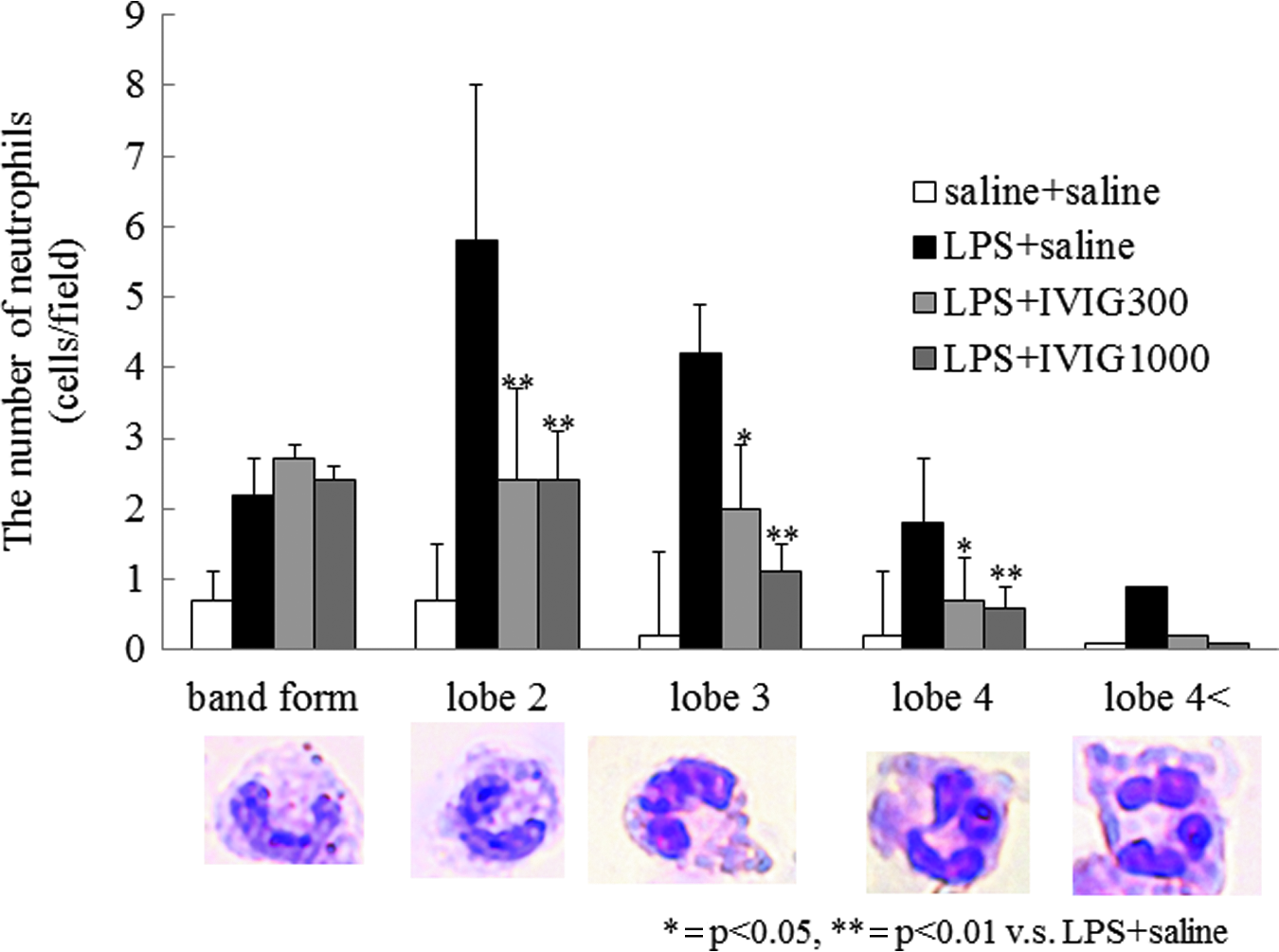
Intravenous immunoglobulin (IVIg) did not influence the number of band-form neutrophils whereas IVIg reduced the number of segmented neutrophils during endotoxemia. The numbers of neutrophils were counted in a high power field (×1,000) by microscopy. Averages were then calculated in three fields/mouse. Intravenous immunoglobulin reduced the lipopolysaccharide (LPS)-induced increase in the total number of neutrophils and the number of segmented neutrophils (2–4 nuclear lobes), however, IVIg did not influence the number of band-form neutrophils. Data show mean±standard error (SE). *p<0.05; **p<0.01 vs. saline+saline group by the Tukey-Kramer post hoc test. Color image is available online at www.liebertpub.com/sur
Intravenous immunoglobulin did not influence the mRNA expression of TNF-α, ICAM-1, or iNOS in the lung. The mRNA of TNF-α, iNOS, and ICAM-1 were quantified by real-time quantitative RT-PCR to evaluate the lung inflammation or cell adhesion. Lipopolysaccharide increased lung TNF-α and iNOS mRNA expression significantly (data not shown). Intravenous immunoglobulin at the doses of 300 mg/kg and 1,000 mg/kg did not influence TNF-α concentrations. IVIg tended to reduce mRNA expression of iNOS in a dose-dependent manner but there were no significant differences (data not shown). Intravenous immunoglobulin tended to increase ICAM-1 mRNA expression but there were no significant differences in ICAM-1 mRNA expression during endotoxemia (data not shown).
IVIg-induced apoptosis of neutrophils without inducing apoptosis of lung epithelial cells
Apoptosis in the lung was assessed by counting cleaved caspase-3-positive (apoptotic) cells and negative (non-apoptotic) cells in five fields (×400) per mouse. Neutrophil and epithelial apoptotic cells were differentiated morphologically using×1,000 magnification. The percentages of apoptotic lung epithelial cells were 1.8%, 1.8%, and 2.0% in the LPS, IVIg 300 mg/kg, and IVIg 1,000 mg/kg groups, respectively (data not shown). The percentages of apoptotic neutrophils in the lung were 2.1%, 3.2%, and 1.6% in the LPS (Fig. 4A), IVIg 300 mg/kg (Fig. 4B), and IVIg 1,000 mg/kg (Fig. 4C) groups, respectively, indicating that neutrophil apoptosis was significantly increased in the IVIg 300 mg/kg group compared to the LPS group (Fig. 4D).
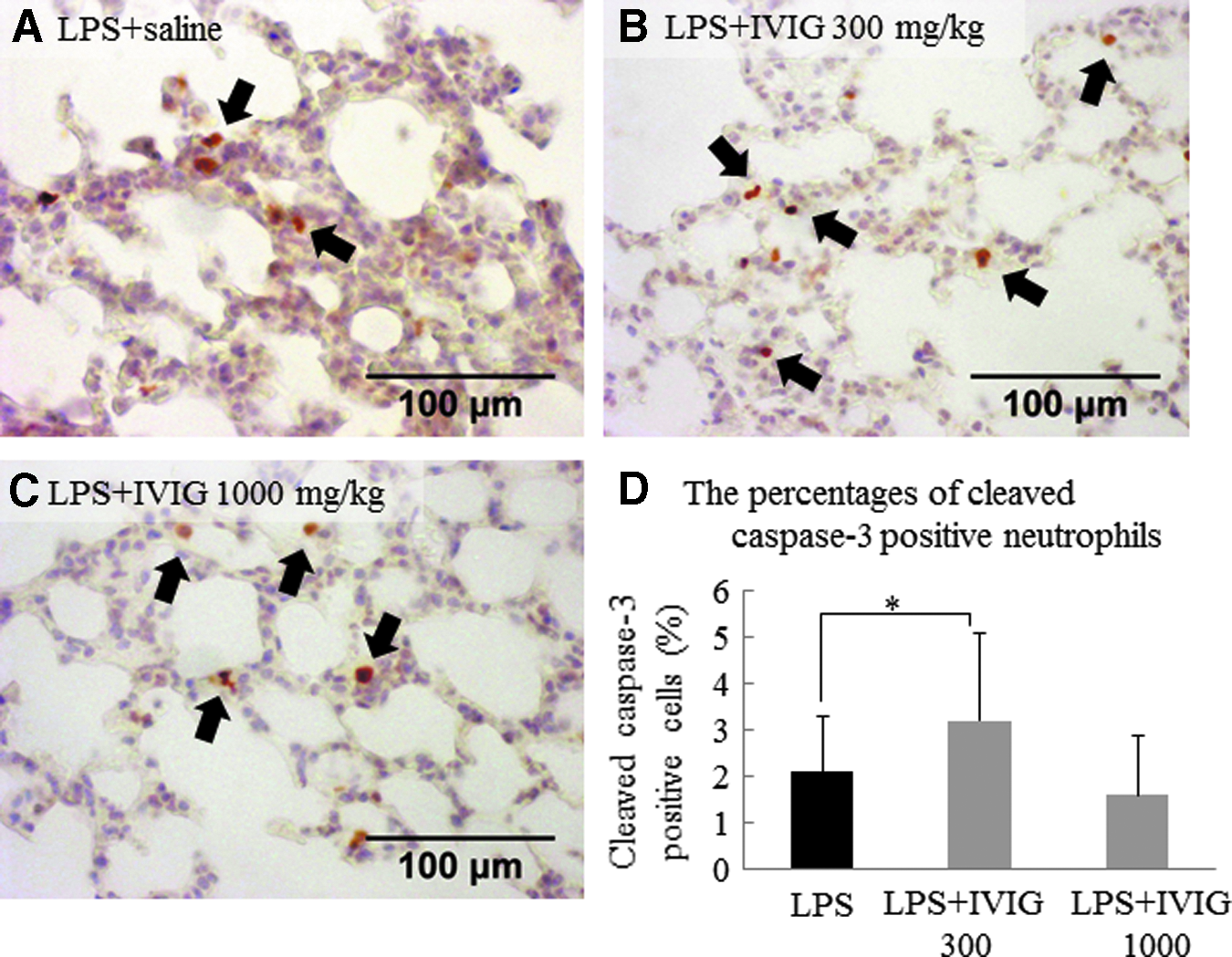
Intravenous immunoglobulin (IVIg) 300 mg/kg induced neutrophil apoptosis via caspase-3 during endotoxemia. To evaluate neutrophil apoptosis in the lung, cleaved caspase-3 staining was performed using lung paraffin section. At 12 h after saline or lipopolysaccharide (LPS) injection, lungs were collected.
Discussion
This study demonstrated that IVIg attenuated LPS-induced neutrophil accumulation and prevented delayed-neutrophil apoptosis in the lung during mouse endotoxin-induced inflammation. Our results suggest that IVIg attenuated neutrophil accumulation with timely apoptosis of neutrophils without inhibiting host defensive inflammatory mediators in the lung. These findings may suggest that IVIg prevents neutrophil-mediated inflammatory injury of the lung during endotoxemia.
The number of neutrophil nucleus lobes increases with cell age; young neutrophils have band-form nuclei or two segmented nuclear lobes; middle-aged neutrophils have three or four nuclear lobes; and old neutrophils have more than four nuclear lobes [10]. In non-inflammatory situations, most circulating neutrophils are young or middle-aged, as older neutrophils are removed through regulated apoptosis. However, neutrophil life span is prolonged by delaying apoptosis in the presence of multiple inflammatory mediators. The present study showed that LPS increased the number of young, middle-aged, and old neutrophils in the lung, suggesting recruitment of neutrophils from bone marrow and impaired regulation of neutrophil apoptosis. Administration of both 300 mg/kg and 1,000 mg/kg IVIg decreased the number of neutrophils with two to four nuclear lobes but did not influence the number of band-form nuclear cells. These findings suggest that IVIg decreased the number of neutrophils accumulated in the lung by inducing neutrophil apoptosis, but did not inhibit the release of neutrophils from bone marrow during endotoxin-induced systemic inflammation. In fact, the cleaved-caspase 3-positive neutrophils were significantly increased in the IVIG 300 mg/kg group.
As mentioned above, cleaved-caspase 3-positive neutrophils (apoptotic neutrophils) were significantly increased following administration of IVIg 300 mg/kg, while the high 1,000 mg/kg IVIg dose did not alter apoptosis. However, the numbers of neutrophils with two to four nuclei lobes were significantly reduced and neutrophil apoptosis in the peripheral blood was significantly induced by IVIg 1,000 mg/kg. The present study suggests that IVIg at a 1,000 mg/kg dose may induce caspase-independent death in neutrophils. Neutrophils have sialic acid-binding immunoglobulin-like lectin 9 (Siglec9), which is activated by IVIg, resulting in both caspase-dependent and caspase-independent pathways of neutrophil death [11]. In addition, granulocyte macrophage-colony stimulating factor (GM-CSF)-delayed neutrophil apoptosis was induced by IVIg doses under 10 mg/mL, whereas the high dose of IVIg (more than 10 mg/mL) induced caspase-independent signals leading to neutrophil death via the Siglec9 pathway [11]. Thus, the 1,000 mg/kg IVIg dose may have induced LPS-delayed neutrophil apoptosis via a caspase-independent pathway involving Siglec9.
The reported anti-inflammatory effects of IVIg have indicated that it reduced serum or plasma cytokine concentrations in vivo [8,12,13] IVIg therapy requires high doses (1,000–2,000 mg/kg) of IVIg because a minor population of IgG crystallizable fragments (Fcs) with glycans terminating in α2,6 sialic acids (sFc) is an important factor in anti-inflammatory effects [14,15]. The sFc target is composed of myeloid regulatory cells expressing the lectin dendritic cell-specific ICAM-3 grabbing non-integrin (DC-SIGN). In the lung, some alveolar macrophages express DC-SIGN (16), and DC-SIGN-positive alveolar macrophages are induced by Mycobacterium tuberculosis, but not by LPS [17]. High-dose (1,000 mg/kg) IVIg contains more sFc fragments than low-dose IVIg but the number of target cells in the lung is not sufficient to attenuate the inflammatory mediators in the lung. This also could be one explanation for the findings that high-dose IVIg had less influence on the inflammatory events in the present study. Thus, IVIg may have more influence on other organs such as the liver or the lymph nodes than on lung because there are more target cells in these organs.
A limitation of this study was that the alterations of inflammatory mediators by IVIG administration were not demonstrated. The peak time of TNF-α is 1 h to 3 h after LPS stimuli [18]. However, this study was performed with an emphasis on the elucidation of the effect of post-administration of IVIg because patients with sepsis are also administrated IVIg after the peak of inflammation. Thus, the time point at which this study was performed made it difficult to investigate the direct influence of IVIg on inflammatory mediators such as TNF-α mRNA expression.
In conclusion, IVIg at doses of 300 mg/kg and 1,000 mg/kg prevented LPS-delayed neutrophil apoptosis and neutrophil accumulation in the damaged lung due to endotoxemia. Although further studies are needed to support our data, IVIg may be a useful therapeutic drug to prevent neutrophil-mediated ARDS during sepsis. Also, it is possible that the pathways by which IVIg induces neutrophil apoptosis may differ depending on the IVIg concentration.
Footnotes
Author Disclosure Statement
No competing financial interests exit.